










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.36 no.4 Bogotá Oct./Dec. 2008
Intubación fibróptica y manejo anestésico de un paciente pediátrico con Síndrome de
Freeman-Sheldon
Juliana M. Mendoza Villa1, ]uan D. Marín Gavina2, Piedad Echeverri3, DianaP. Medina Meza4, María F. Rivera5
1 Anestesióloga y docente de la Clínica Universitaria Bolivariana. Anestesióloga de la IPS Universitaria León XIII. Medellín. Colombia, jmmv80@gmail.com
2 Anestesiólogo de la Clínica el Rosario. Medellín. Colombia.
3 Anestesióloga Pediatra. Hospital de la Misericordia. Bogotá. Colombia.
4 Anestesióloga Hospital Universitario San Vicente de Paúl. Medellín. Colombia.
5 Anestesióloga del Hospital la María y la IPS Universitaria León XIII. Medellín. Colombia.
RESUMEN
Presentamoo el caso de un niño ñon síndrome de Freeman-Sheldon programado para la correccióc quirúrgica de su microstomia. El niño presentabn la característica de "cara de silvador"y deformidades de artrogríposis en manos y pies. Se expone el manejo anestésico y de la vía aéred
Palabras clave: Síndrome de Freeman-Sheldon (SFS). Síndrome de ecara en silbido". Vía aérea difícil en pediatría. Artrogríposis.
SUMMARY
We describe e case of a child with typical clinicalfeaturea of Freeman-Sheldon syndrome (FSS) presentedfor elective surgical correction of microstomia. The anaesthetic cnd airtuay problems encountered erediscussed.
Key words: Freeman-Sheldon syndrome. Whistlingface syndrome. Arthrogryposis, Airway managemant.
El síndrome de Freeman-Sheldon es una miopatía congénita generalizada que cursa con deformidades fundamentalmente en cara, manos y pies. Estos pacientes desde pequeños son sometidos a numerosas intervenciones quirúrgicas tendientes a mejorar su calidad de vida, y constituyen un reto para el anestesiólogo debido a las dificultades en obtener un acceso intravenoso, en el manejo de la vía área y por el riesgo de hipertermia maligna.
CASO CLÍNICO
Niño de 15 meses con diagnóstico de síndrome de Freeman-Sheldon, programado para corrección quirúrgica de microstomia. Con antecedente quirúrgico de herniorrafia inguinal a los 9 meses de edad y manejo anestésico con máscara laríngea (número 2), anestesia total intravenosa con propofol (10 mg^ kg1 h) y remifentanil (0,4 ug-1 kg 1 min), sin complicaciones.
Durante la evaluación preanestésica se evidencia un peso de 10.5 kilogramos, una apertura oral de 1 cm, raíz nasal ancha con orificios nasales estrechos, fittrumlsargp, microstomia, micro-retrognatia, paladar ojival y depresión central en mentón (mentón en forma de H) característico del síndrome (Ver figura 1). Además, presenta, frente prominente, epicanto, cuello corto y ancho, extremidades superiores cortas con contractura en flexión del codo, desviación ul-nar de las manos con contractura en flexión de los dedos, pies equinovaros e hipertonía generalizada de predominio axial con limitación de flexo-extensión cervical. No presenta comorbilidades cardiopulmo-nares ni reflujo gastro-esofágico. No se evidencia lenguaje verbal ni patrón de marcha.
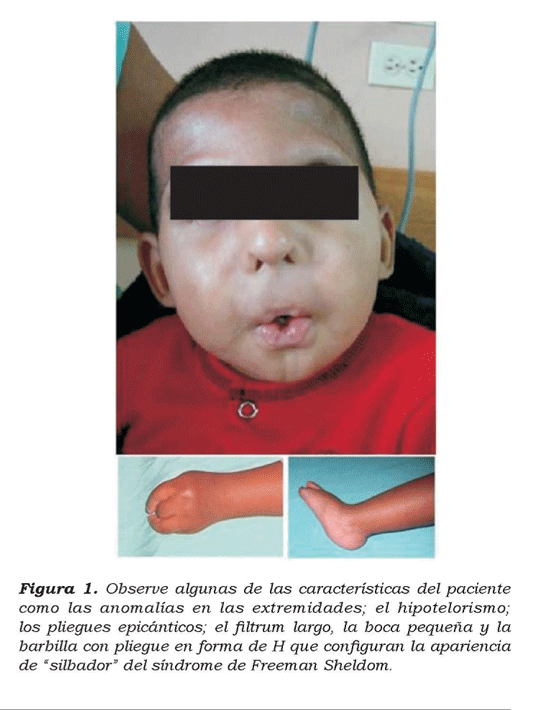
Debido a la acentuada microstomía y a la retrognatia, se considera una vía aérea difícil anticipada y se decide para el manejo inicial (Plan A) realizar un intento de laringoscopia. En caso de laringoscopia no exitosa se continuaría con una intubación nasal guiada por fibrobroncoscopio flexible, bajo inducción intravenosa y conservando la ventilación espontánea del paciente. Como manejo alterno (Plan B) se utilizaría una máscara laríngea "prosear número 2 y a continuación se despertaría al paciente Como plan de emergencia (Pkm C) disponíamos de equipopara ventilación jet
Para evitar el estrés del niño, éste fue preparado y acompañado por los padres a la consulta preanestésica. Llega a quirófano con un ayuno de 3 horas para líquidos claros y 6 horas para los demás alimentos. Una hora antes de cirugía, se realiza premedicación con ketamina 50 mg y midazolam 4 mg vía oral. Se usan parches con anestésico local en los posibles sitios de punción y, después de varios intentos, se canaliza vena(venocath No. 22). Se administra 0 2 me atropina I.V. y se traslada el paciente a quirófano
Previamente, se cambió la cal sodada del canister y se realizó lavado del circuito con oxígeno a 10 litros/minuto durante 10 minutos. Además, se realizó chequeo de la máquina para asegurar el cierre adecuado de vaporizadores.
Durante el procedimiento de intubación se administra midazolam 2,5 mg y bolos de fentanil 0,025 mcg, ketamina 0,25 mg y propofol 10 mg, titulados según respuesta. Se practica inicialmente una laringoscopia directa en donde se visualiza un Cormarck 4 (cuatro). Después de la aplicación de Afrín® y lidocaína en spray al 10% por ambas fosas nasales, se pasa un tubo endotraquel número 3.5 por fosa nasal derecha, el cual se usa como cánula nasofaríngea y se conecta a la manguera de anestesia para administrar oxígeno con una Fi02 de 100%. Se introduce el fibrocoscopio (DE 2.8 mm; Karl Storz) por fosa nasal izquierda, administrando lidocaína al 1% sin epinefrina 2 ce, por el canal de trabajo, consiguiéndose la intubación nasotraqueal, con un tubo sin balón número 4.0, al primer intento y sin complicaciones. (Ver figura 2)
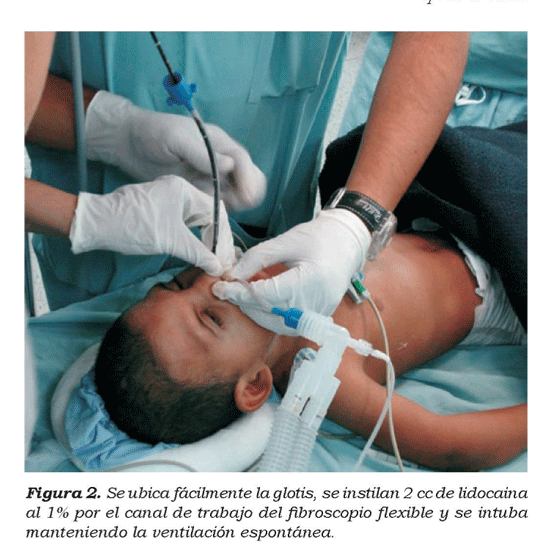
Durante el intraoperatorio se mantiene una fracción inspirada de oxígeno al 60% y se inicia anestesia total intravenosa, con infusión de remi-fentanil a 0.3 ug-1 kg-1 min y propofol a 10 mg-1 kg-1 h, titulando de acuerdo a la respuesta hemodinámica del paciente. Adicionalmente se administra dipirona 30 mg-1 kg-1 y morfina 50 ug-1 kg-1 para la analgesia postoperatoria.
Finalizada la intervención quirúrgica, se suspende la infusión de propofol y de remifentanil. El paciente inicia ventilación espontánea a los 5 minutos de suspenderse el opioide. Posteriormente recupera la conciencia y los reflejos de la vía aérea con un despertar tranquilo, momento en el cual se retira el tubo orotraqueal y se deja en observación por 6 horas, sin complicaciones.
DISCUSIÓN
En 1938 Freemany Sheldon describieron 2 niños con "distrofia craneocarpotarsal", microstomía, microrretrognatia, filtrum alargado con orificios nasales estrechos e hipertelorismo. Las alteraciones del rostro son características e hicieron que Burian en 1963 acuñara el término de "síndrome en cara de silbido". Estas se deben a fibrosis y tono muscular aumentado en los músculos faciales1
Su patrón hereditario no esta claro. La mayoría de los casos son de tipo autosómico dominante, pero también se han descrito casos de tipo autosómico recesivo2. Algunos autores lo consideran una forma de artrogriposis miopática con ubicación en el cromosoma 113.
El espectro clínico del síndrome es amplio. La contractura de los músculos esqueléticos conduce a un cuello corto y posición cefálica de la laringe. El compromiso de la musculatura oro y nasofaríngea puede producir obstrucción crónica de la vía aérea
superior. Secundario a esto, puede desarrollarse hipertensión pulmonar. Además, pueden tener alteraciones de la caja torácica y enfermedad pulmonar restrictiva. Puede existir también espina bífida oculta. La incidencia de enfermedad cardiaca congénita es similar a la de la población general4.
Desde temprana edad estos pacientes requieren un elevado número de cirugías para mejorar su funcionamiento y aspecto5 6. Pueden necesitar varias correcciones faciales de la microstomia, de problemas oculares y nasales; cirugía antirreflujo o gastrostomía; corrección de problemas hemiarios de la pared abdominal y de problemas ortopédicos como la artrogriposis y luxación congénita de cadera, etc.
La presencia de una miopatía de base implica una susceptibilidad aumentada a la hipertermia maligna y existen varios reportes de caso que evidencian esta complicación o espasmo maseterino tras la exposición a agentes potencialmente desencadenantes7 n. También hay un caso reportado de síndrome neuroléptico maligno en un niño con SFS que recibió metoclopramida12, por lo que debe tenerse presente este efecto adverso cuando se administran antido-paminérgicos.
Es importante la preparación de la máquina de anestesia cambiando la cal sodada, removiendo o sellando los vaporizadores y "lavándola" con un flujo de gases frescos a 10 litros/minuto por mínimo 5 minutos. Debe evitarse los anestésicos volátiles y la succinilcolina. Son seguros el oxido nitroso, los opiáceos, losbarbitúricos, el etomidato, elpropofol, la ketamina y los relajantes musculares no despolarizantes^. La anestesia local y regional también es segura para los personas con susceptibilidad a hipertermia maligna, pero tales procedimientos en los pacientes con SFS pueden ser técnicamente difíciles14.
En la población pediátrica la técnica de elección para efectuar intubación endotraqueal cuando existe una Vía Aérea Difícil Anticipada es la fibros-copia flexible con el paciente bajo inducción con halogenado, manteniendo ventilación espontánea. Sin embargo, por el riesgo de hipertermia maligna, optamos por seguir una técnica intravenosa pura. En algunos centros, que cuentan con fácü disponibilidad al dantroleno, usan como primera opción los halogenados con monitoria continua de la temperatura del paciente1516, pero hay que tener en cuenta la alta morbi-mortaMad de la hipertermia maligna.
En la evaluación pre-anestésica es fundamental hacer un completo examen de la vía aérea constatando su permeabilidad y la severidad de las malformaciones. Algunos recomiendan hacer exploraciones complementarias, como radiografía lateral de cabeza y cuello o fibroscopia rinofaríngea17, sin embargo el valor predictor de estas ayudas no ha sido bien establecido.
En general, es recomendable efectuar un ensayo de intubación endotraqueal previo. Sin embargo, en la mayoría de casos, la microstomia, la retrognatia, el cuello corto y la rigidez axial, imposibilitan esta técnica, por lo que siempre se debe tener a la mano otros planes de manejo18 19.
En la literatura existen varios reportes de casos describiendo el uso de máscara laríngea en estos pacientes y algunos autores han recomendado el dispositivo como una opción de manejo en procedimientos cortos y sin relajante neuromuscular20, 21. Sin embargo, el método de elección para asegurar una vía aérea difícil anticipada es el tubo endotraqueal y los dispositivos supraglóticos solo se deberían usar como planes de rescate de la vía aérea o como adyuvantes para realizar intubación fibróptica 22-23. Específicamente en los lactantes, el dispositivo2 suprilótico que mejor desempeño ha demostrado es la máscara laríngea proseal, generando mejores condiciones de ventilación y mayor tasa de éxito en la colocación cuando se compara con los demás dispositivos24.
Finalmente, siempre hay que tener a la mano un acceso quirúrgico, como punción cricoidea para ventilación jet, por si todo lo anterior falla y se presenta un caso de no intubación y no ventilación.
CONCLUSIONES
El síndrome de Freeman Sheldon en una enfermedad congénita rara con consideraciones muy importantes desde el punto de vista anestésico como son las dificultades para el manejo de la vía aérea, la susceptibilidad a desarrollar hipertermia maligna en el perioperatorio y la dificultad para asegurar un acceso venoso.
Aunque en la mayoría de los reportes de caso se utiliza la máscara laríngea como primer abordaje de la vía aérea en procedimiento cortos y de bajo riesgo, siempre se debe tener presente que la forma más segura de enfrentar una vía aérea difícil anticipada cuando se requiere anestesia general, consiste en la intubación orotraqueal con planes claros de manejo adaptados a cada paciente y con un plan de extubación despierto.
BIBLIOGRAFÍA
1. McKusick VA. Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. 12th ed. Baltimore: Johns Hopkins University Press. 1998. [ Links ]
2. Carakushansky G, et al. Recessive type of Freeman-Sheldon syndrome. Journal of Paediatric 2001; 77 (5): 425-430 [ Links ]
3. Bamshad, M, et al. "A Revised and Extended Classification of Distal Arthrogryposis". Ameriean Journal of Medieal Geneties 1996; 65: 277-281. [ Links ]
4. Infosino A. Pediatric upper airway and congenital anomalies. Anesthesiology Clin N Am 2002; 20: 747-766 [ Links ]
5. Calderón JL, Taoube K. Freeman Sheldon syndrome: clinical manifestations and anesthetic and surgical management. An Esp Pediatr 2002; 56: 175-179. [ Links ]
6. Kimie Ohyama, et al. Freeman-Sheldon syndrome: case management from age 6 to 16 years. Cleft palate-craniofacial journal 1997; 34 (2): 151-153. [ Links ]
7. Brownell AKW. Malignant hyperthermia: Relationship to other diseases. Br J Anaesth 1988; 60: 303-308. [ Links ]
8. Laishley RS, Roy W. Freeman-Sheldon syndrome: report of three cases and the anaesthetic implications. Can Anaesth Soc 1986; 33 (3): 388-93 [ Links ]
9. Sobrado CG, et al. Síndrome de Freeman Sheldon: Rigidez muscular generalizada tras inducción anestésica. Rev Esp Anestesiol Reanim, 1994; 41: 182-184. [ Links ]
10. Jones R, Dolcourt JL. Muscle rigidity following halothane anesthesia in two patients with Freeman-Sheldon syndrome. Anesthesiology 1992; 77: 599-600. [ Links ]
11.Richard S, et al. Freeman-Sheldon syndrome: report of three case and the anesthetic implication. Can Anaesth Soc J 1986; 33: 388-393. [ Links ]
12.Stein MH, et al. Neuroleptic Malignant Syndrome Induced by Metoclopramide in an Infant with Freeman-Sheldon Síndrome. International Anesthesia Research Society 2006: 103 (3): 786-787. [ Links ]
13.Gerald AG, et al. Malignant Hyperthermia. Müler's Anesthesia. Sexta edición. Volumen uno. Capítulo 29. Pagina 1185. [ Links ]
14.Duggar RGJr, et al. Whistling face syndrome: general anesthesia and early postoperative caudal analgesia. Anesthesiology 1989; 70: 545-7. [ Links ]
15.Kim JS, et al. Awake nasotracheal intubation using fiberoptic bronchoscope in a pediatric patient with Freeman-Sheldon syndrome. Pediatric Anesthesia. 15(9):790-792, September 2005. [ Links ]
16. Agritmis A, et al. Anesthetic management of a patient with Freeman-Sheldon syndrome. Pediatric Anesthesia 2004; 14: 874-877 [ Links ]
17. Yamamoto S, etal. Anesthetic management of a patient with Freeman-Sheldon syndrome. Masui 1994; 43: 1748-1753. [ Links ]
18.Suanes Cabello A, et al. Síndrome de Freeman-Sheldon. Aportación de un caso. An Esp Pediatr 1991; 34: 247-249. [ Links ]
19.Namiki M, et al. Anesthetic management of a patient with Freeman-Sheldon syndrome. Masui 2000; 49: 901-902. [ Links ]
20. Albert Chen, et al. Anesthesia for Freeman-Sheldon Syndrome Using a Folded Laryngeal Mask Airway. Anesth Analg 2005; 101: 606-15 [ Links ]
21.Cruickshanks GF, et al. Anesthesia for Freeman-Sheldon syndrome using a laryngeal mask airway. Can J Anaesth 1999;46:783-787 [ Links ]
22.Munro HM, et al. Freeman-Sheldon (whistling face) syndrome. Anaesthetic airway management. Paediatr Anaesth 1997; 7: 345-348. [ Links ]
23.Brambrink AM, Braun U. Airway management in infants and children. Best Practice & Research Clinical Anaesthesiology 2005; 19(4): 675-697. [ Links ]
24.Goldmann K, et al. The Size VA ProSeal Laryngeal Mask Airway in Infants: A Randomized, Crossover Investigation with the Classic Laryngeal Mask Airway. Anesth Analg 2006; 102:405-10. [ Links ]
