










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.37 no.4 Bogotá Oct./Dec. 2009
REVIEW ARTICLES
Anaesthesia for Liver Transplantation in Fulminant Hepatic Failure A practical approach
Anestesia para trasplante hepático en hepatitis fulminante
Joel Avancini Rocha Filho, MD,* Ricardo Souza Nani, MD,* Maria José Carvalho Carmona, MD,* Mauricio Vanegas Ballesteros, MD,** Luiz Augusto Carneiro D’Albuquerque, MD,**
* Departamento de Anestesiología, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo –
Brasil
E-mail: joelrocha@me.com
** Departamento de Trasplante y Cirugía de Hígado, Hospital das Clínicas da Faculdade de Medicina da Universidade de
São Paulo – Brasil
Recibido: septiembre 18/2009. Aceptado: diciembre 11/2009
SUMMARY
Fulminant hepatic failure (FHF) is defined by sudden onset of encephalopathy, coagulopathy and jaundice in an otherwise normal individual. Fulminant hepatic failure results in progressive multi-organ failure with a dramatic impact in the brain. Severe cerebral edema is a frequent finding that ultimately lead to intracranial hypertension and death. The management of patients with FHF is aimed mainly in prevent or reversing increased intracranial pressure associated with support treatment for other failing organs. The definitive treatment for patients with FHF is liver transplantation. This article aims to present a practical approach to anesthesia care and intraoperative management of patients with FHF.
Key Words: Fulminant Hepatic failure, Liver failure, Intracranial Hypertension, Liver Transplantation, Anesthesia (Source: MeSH, NLM)
RESUMEN
La falla hepática fulminante (FHF) es definida como el comienzo súbito de encefalopatía, coagulopatía e ictericia en un individuo que se encontraba en condiciones normales de salud. La falla hepática fulminante termina en un deterioro progresivo de múltiples órganos con un impacto dramático en el cerebro.
El edema cerebral severo es un hallazgo frecuente que, finalmente, conlleva a hipertensión intracraneal y a la muerte. El manejo de pacientes con FHF apunta principalmente a prevenir o reversar el incremento de la presión intracraneal, asociada con el tratamiento de soporte para la falla de otros órganos. El tratamiento definitivo para pacientes con FHF es el trasplante de hígado. Este artículo tiene como objetivo presentar una aproximación práctica al cuidado anestésico e intraoperativo para el manejo de pacientes con FHF.
Palabras Clave: Fallo hepático agudo, insuficiencia hepática, hipertensión intracraneal, trasplante de higado, anestesia (Fuente: DeCS, BIREME)
INTRODUCTION
Fulminant hepatic failure (FHF) is a serious affection characterised by the development of encephalopathy and coagulopathy during the 8 weeks following the appearance of jaundice, in a patient without previous hepatic compromise. The most common causes are viral hepatitis (VHB in most cases), drug-induced hepatitis (paracetamol, halothane, isoniazide, riphampicin, AINES, sulphonamides, flutamide, valproic acid, carbamazepine, ecstasy) and exogenous intoxication (trichloroethylene, tetrachloroethane, fungi [Amanita phaloides]). There is spontaneous recovery of hepatic function in 70% of grade 1 and 2 hepatic encephalopathy and less than 20% in grades 3 and 4. Mortality approaches 80% without hepatic transplantation and its indication is based on King’s College Hospital criteria (Table 1).(1)

Intracranial hypertension (IH), the main cause of mortality in these patients (responsible for 50% to 80% of deaths) and secondary to cerebral oedema, is the central process responsible for the hepatic encephalopathy. (2-4) The clinical manifestation of hepatic encephalopathy varies from confusion to coma and has a direct relationship with cerebral oedema’s intensity and the evolution of FHF severity. Extra-hepatic manifestations rapidly evolve at the same time, characterised by circulatory hyperdynamism with growing dependence on α-adrenergic pharmacological support, ARF, coagulopathy and hypoglycaemia. Without liver transplantation progression occurs toward clinical deterioration, cerebral hypoperfusion and multiorgan failure.
Disorders concerning the functions of a diseased liver’s synthesis may be avoided to a certain point by the infusion of fresh frozen plasma, whilst metabolic function insufficiency responsible for cerebral oedema and extra hepatic manifestations of acute hepatic failure still do not have satisfactory support measures. Technological perspectives for the support and treatment of acute hepatic insufficiency are constantly being developed, ranging from hepatic dialysis systems (biological and non-biological) to xenotransplant (transplant between individuals from different species); however, early intervention in carrying out the orthotropic transplant of the liver is the only definitive current therapeutic option available for these patients.(1, 5-10)
The patient
Cardiovascular
Circulatory hyperdynamism is a cardiovascular alteration which is characteristic of acute hepatic insufficiency. This haemodynamic profile is directly proportional to the degree of hepatic dysfunction, being constituted by reduced systemic vascular resistance, increased cardiac index, arterial hypotension and tachycardia. Circulatory alteration is possibly due to an excess of vasoactive substances (mainly vasodilators) which are not duly cleansed from the circulation by hepatic affection (i.e. nitric oxide, c GMP, glucagon, ferritin and VIP).(11, 12) Such predominance of arteriolar vasodilatation in acute hepatic insufficiency is accompanied by increased sympathetic activity and activation of the renin-angiotensin- aldosterone system, providing an increase in total blood volume; however, due to its poor distribution, central volemy frequently becomes reduced. Vasodilatation increases peripheral arteriovenous shunt, thereby raising the content and saturation of oxygen from mixed venous blood with consequent reduction of arteriovenous oxygen difference.
2. Pulmonary
Hypoxaemia (PaO2<70 mmHg, room air) is a frequent sign, mainly due to intrapulmonary shunt caused by abnormally vasodilated pulmonary arterioles and compromise of hypoxic pulmonary vasoconstriction. Controlling CO2 levels and protecting the airways constitute the main indications for early tracheal intubation. (13, 14)
3. Coagulation
The liver is the major site for the synthesis of procoagulants and anticoagulants (except for factor VIII, tissue plasminogen activator [t-PA] and plasminogen activator inhibitor), and it is also the major site for cleansing activated coagulation factors, t-PA and fibrin degradation products (FDPs).(15, 16) Coagulopathy in advanced hepatic disease is due to reduced coagulation factor synthesis and function associated with a reduced number of platelets and compromised platelet aggregation. Thrombocytopenia in acute hepatic insufficiency is a frequent find and is directly related to bleeding. Prothrombin time (or INR) is an independent marker of the severity and prognosis in fulminant hepatic failure.
Renal
The first manifestation of renal compromise is the reduction of renal blood flow, secondary to neurohumoral stimulation due to increased sympathetic activity and activation of the reninangiotensin- aldosterone system. The aetiology of renal affection in acute hepatic insufficiency is a combination of hepatorenal ARF and acute tubular necrosis (ATN) and up to 75% of these patients need dialytic therapy. Continuous haemofiltration is preferred to the intermittent type due to a lesser compromise in systemic haemodynamics and intracranial pressure (ICP).(14)
Hepatorenal syndrome (HRS) is a functional ARF picture characterised by intense renal vasoconstriction without significant morphological lesion, particularly frequent in chronic hepatic disease, as well as occurring in acute disease. Pathogenesis is not full understood but includes central hypovolemy with reduced venous return due to splacnic vasodilatation, manifesting itself with oliguria (<500 ml/day), serum creatinine >1.5 mg/dL (clearance <40 mL/min) and low urinary sodium (<10 mEq/L).(17) Differential diagnosis includes prerenal ARF (which responds to volemic expansion) and ATN (which presents urinary sodium >10 mEq/L).(17) HRS is a serious, potentially reversible clinical picture with liver transplantation and demands adequate volemy maintenance for preventing prerenal aggression in such already compromised kidneys.(18)
5. Central nervous system
Hepatic encephalopathy is a neuropsychiatric syndrome characteristic of acute hepatic insufficiency. Encephalopathy is a sign of severity, its progression in acute hepatic insufficiency implies an aggravating prognosis and it can be reverted with the re-establishment of hepatic function or via a liver transplantation. Cerebral oedema is at the centre of the process responsible for hepatic encephalopathy.(3, 4) Ammonia ceases to become converted into urea by the liver in hepatic insufficiency, thereby leading to hyperammonaemia and low serum urea. In the case of hyperammonaemia, significant amounts of ammonia will be detoxified in astrocytes generating glutamine, an amino acid having a potent osmotic effect, generating glial oedema, alteration of interneural communication and potentializing γ-aminobutyric neuroinhibition (GABA).(19)
Clinical repercussion depends on the speed of increased ammonaemia and SNC’s adaptive ability, having clinical manifestations directly proportional to cerebral oedema intensity, ranging from mental confusion to decortication, decerebration and anisocoria (Table 2). Manoeuvres and/or drugs increasing intracranial pressure in all patients having encephalopathy must thus be avoided, independently of encephalopathy grade.

In patients having refractory intracranial hypertension greater than 50 mmHg associated with less than 40 mmHg cerebral perfusion pressure for more than 2 hours liver transplantation may not be a therapeutic option.(1, 20)
III. Intraoperative period
1. Anaesthesia
Anaesthetic induction may be normally done after conventional non-invasive monitoring, except in patients suffering from encephalopathy grade 3/4 or when using vasoactive drugs.
The risk which is always present of pulmonary aspiration during induction indicates intubation accompanied by a rapid sequence of drugs and Sellick’s manoeuvre, after administering propofol or barbituric associated with opioid and neuromuscular blockade with succinylcholine. The use of inhalatory anaesthetics must be avoided in maintaining anaesthesia, mainly in patients with reduced cerebral compliance and in those depending on α-adrenergic pharmacological hemodynamic support. Anaesthesia must be complemented by opioids and neuromuscular blockers (NMB) having less compromised pharmacokinetics in advanced hepatic disease, such as fentanyl or sufentanil, associated with atracurium or, preferentially, cisatracurium due to it releasing less histamine (both being organ-independent elimination NMB).
In patients suffering hepatic encephalopathy due to fulminant hepatic failure all efforts must be concentrated on maintaining cerebral perfusion pressure above 50 mmHg (CPP=MAP-ICP) to avoid cerebral hypoperfusion and less than 65 mmHg to avoid cerebral hyperaemia.(21, 22) Orotracheal intubation must be promptly carried out during the pre-operative period in patients suffering grade 3 encephalopathy for protecting airways and so that hyperventilation can be available early on as part of the therapeutic arsenal. An ICP monitoring catheter must be placed of those in whom evolution to grade 4 is expected, this being fundamental in managing such patients.(23-25)
Patients having encephalopathy grades 3 and 4 are considered to be at greater risk for cerebral ischemia during the intraoperative period. Extra-hepatic alterations generally improve immediately with the explant of the native organ, the cerebral oedema has a slower resolution and ICH peaks can occur even after reperfusion of the graft and during the immediate postoperative period, in spite of the graft’s good functioning.(26) Phase I and graft reperfusion are periods during which increased ICP and reduced cerebral perfusion most frequently occur.( 26, 27) The head up position (20°) during the whole procedure and the intermittent use of manitol (20% - 0.5 to 1.0 g/kg in 20 minutes), thiopental (125 mg in bolus) and hyperventilation whenever ICP rises above 25 mmHg are the recommended cerebral protection strategy. (14, 21, 28)
Moderate hypothermia (33.5°C) may prevent increases in ICP during liver transplantation in patients having ICH who are resistant to conventional therapy.(27, 29, 30) Its protectioninducing effects seem to be strengthened by reduced production and response in organs secondary to the toxic products released by the necrotic liver.(29) However, the increased risk of cardiac depression, arrythmias, blood hypocoagulation and reduced renal function must be considered regarding its neuroprotective effect.
2. Monitoring and equipment needed
Liver transplantation is a high complexity procedure in which the results are dependent on technological advances for avoiding multifactor hemodynamic instability and the metabolic alterations characterising the severity of fulminant hepatic failure. Invasive monitoring must include, at least, one arterial line and two venous accesses lines having a calibre greater than 8 Fr (one for attaching a Swan-Ganz catheter) (Table 3).(21)

Monitoring intracranial pressure (ICP) provides essential information during the perioperative liver transplantation period in patients suffering from fulminant hepatitis. The ICP monitoring catheter for these patients must be placed after correcting coagulation alterations (<1.5 INR and >50,000 cell /mm3 platelets) and should preferably be localised in an intraventricular position for reducing the risk of intracranial haemorrhage.
Support equipment must be immediately available and constantly supervised; it can be classified into four groups as described in Table 4.

Coagulation is routinely monitored during liver transplantation for reducing bleeding and the amount of haemocomponents administered, which is associated with increased perioperative morbimortality in liver transplantation.(31, 32) Monitoring by means of traditional coagulation tests (prothrombin time, activated partial thromboplastin, thrombing time, fibrinogen, dimer D) or by thromboelastography (TEG) and platelet count must indicate whether there is coagulopathy, rapidly identify the cause and suitable therapy, thereby optimising the use of haemocomponents. The difficulty of making a suitable evaluation and the time spent in carrying out the battery of conventional coagulation exams hampers their intraoperative implementation and favours incorporating TEG which, in fact, is the only test providing information about coagulation and fibrinolysis via a single blood analysis. It is easy to carry out and interpret, even allowing an in vitro analysis of therapeutics.(33) The overall coagulation profile may be qualitatively and quantitatively interpreted in terms of hypo, normal or hypercoagulability state and also allows an analysis of the degree of lysis (Figure 1). Our intraoperative strategy is based on TEG together with platelet number count.
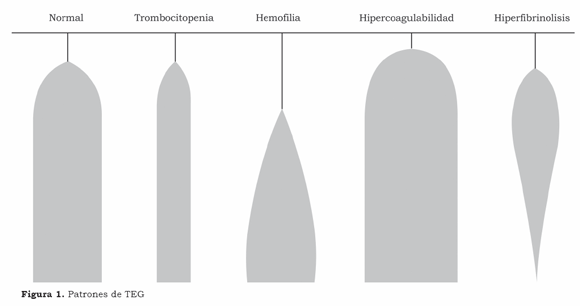
Blood coagulation is thus monitored by clinical and laboratory analysis; the use of blood components is limited to dealing with clinically important disturbances of coagulation, particularly when these have been confirmed by the laboratory analysis.
3. Transplant
Surgery (for didactic purposes) consists of three phases. The first or pre-anhepatic phase begins with anaesthetic induction, includes the hepatectomy and ends with hepatic vascular exclusion and the removal of the native organ. Phase 2 or the anhepatic phase extends from hepatic vascular exclusion to revascularisation of the graft. Phase 3 or the neo-hepatic phase begins with hepatic reperfusion and re-establishing venous return to the heart and ends with closing the abdominal wall.(21)
3.1. Pre-anhepatic phase
Phase 1 is generally the surgery’s most laborious phase, bleeding with all its consequences (hypovolemy, hypocalcaemia, hypothermia and transfusions) being the most common complication. Our intraoperative volemic reposition protocol consists of maintaining 25% to 30% haematocrite throughout all transplant phases.
Hypocalcaemia (ionised calcium <1.13 mmol/L) frequently occurs during this phase, mainly during the administration of haemocomponents. There is reduced circulatory cleansing of citrate present in the haemocomponents in hepatic disease (which is rapidly metabolised by the normal liver), making these patients particularly sensitive to citrate poisoning. Hypocalcaemia is typically manifested with myocardial depression (arterial hypotension, increased filling pressures) and lengthening of the QT interval may be detected in the electrocardioscope. The treatment consists of administering calcium chloride (10 mg/Kg) or calcium gluconate (30 mg/Kg).
Serum ionic magnesium levels also become reduced by the citrate infusion rate and hypomagnesaemia is associated with myocardial depression and cardiac arrythmias.(34) Even though intraoperative magnesium determination may not be frequent, myocardial depression and arrythmia clinical pictures following polytransfusion and reperfusion resistant to conventional therapeutics (mainly in patients with preoperative hypomagnesaemia) indicate magnesium reposition.
The risk of hypoglycaemia is always present in hepatic insufficiency due to reduced deposits of hepatic glycogen, compromised activation of glyconeogenesis and reduced insulin cleansing by the diseased liver in acute hepatic insufficiency; intensive monitoring must be carried out during transplant phases 1 and 2.(35, 36)
Hypopotasaemia is common in fulminant hepatitis, independent of renal compromise; however, aggressive corrections anticipating the hypercalemic period accompanying hepatic reperfusion must be avoided. Intraoperative dialysis must be considered early on during surgery in oliguric patients suffering from hyperpotasaemia.
3.2. Anhepatic phase
The most significant haemodynamic alterations in liver transplantation occur at the beginning and end of this phase, generally lasting around 60 minutes. In spite of circulatory hyperdynamism possibly improving with the removal of the native organ, alterations in venous return secondary to hepatectomy at the beginning of this phase, together with haemodynamic alterations in reperfusion at the end of it make this period the one having the most agitated haemodynamic profile during the surgery.
This phase begins with pinching off the vena porta (flow: 1,300 mL/min) and (depending on the operative technique) also interrupting the lower vena cava’s blood flow, being haemodynamically characterised by reduced cardiac index, increased peripheral vascular resistance and low cardiac filling pressures.
Hepatectomy can be done by two surgical techniques (piggyback technique or by the conventional technique which is haemodynamic differentiated by pinching off the lower vena cava), thereby affecting the degree of compromise of cardiac venous return during the whole anhepatic phase. The conventional technique requires the lower vena cava to be totally pinched off for hepatic extraction, thus reducing cardiac preload by 50%. The vena cava remains almost intact during graft implant in the piggyback technique, thereby providing better hemodynamic stability during this period. Choosing one technique and/or another depends on both preference of the surgical team and technical difficulties occurring during hepatectomy.
Hypocalcaemia, hypomagnesaemia, hypothermia and hypoglycaemia become more intense and occur more frequently during phase 2 with the total absence of hepatic function. Fibrinolysis may have its clinical picture aggravated during this phase due to the absence of hepatic depuration of t-PA. Patients suffering from advanced hepatic affection and prolonged phase 2 are those in whom laboratory exams more commonly reveal hyperfibrinolysis with an important clinical manifestation. Their therapy must be performed with 50 mg/Kg aminocaproic acid or 10 mg/Kg tranexamic acid for clinical control of hyperfibrinolysis until the graft functions.
The main objective during the anhepatic phase is to prepare the patient for hepatic reperfusion. Considering this to be the moment during transplant where hemodynamic alterations occur with greater frequency and intensity, it is recommended to increase the inspired O2 fraction (FIO2) to 1.0, five minutes before reperfusion.
3.3. Neohepatic phase - reperfusion
Hepatic reperfusion (i.e. restoring hepatic blood flow) is a critical moment during liver transplantation and the most frequent period of hemodynamic instability.(37-40) In patients suffering from fulminant hepatic failure, this period is of high risk for cerebral ischemia.(26-28)
Haemodynamic response occurs due to the sudden release of acidotic, hypothermic and hyperpotasaemic blood in the circulation which is rich in vasoactive substances and toxic agents released by both the graft during ischemia and reperfusion as well as those accumulated in splancnic circulation during pinching off the vena porta.(41)
From the hemodynamic point of view, this period is characterised by a fall in mean arterial pressure and systemic vascular resistance with increased cardiac index, usually declining in one or two hours.(39)
Post-reperfusion syndrome (PRS) is an acute, transitory hemodynamic cardiovascular collapse phenomenon which happens following liver revascularisation, leading to reduced arterial pressure, systemic vascular resistance and myocardial contractility accompanied by increased filling pressures, pulmonary vascular resistance (manifest by increased mean pulmonary arterial pressure)(42) and bradycardia,( 43) which could suddenly evolve to cardiac arrest.(44) PRS occurs in around 30% of liver transplants and classically has a hemodynamic pattern characterised by a fall in mean arterial pressure of more than 30% of pre-reperfusion values, lasting for more than one minute and occurring during the first five minutes of reperfusion.( 39) Other groups define PRS as a fall in mean arterial pressure to values lower than 60 mmHg in adults and 50 mmHg in children during the first minutes of reperfusion.(38) Donors and marginal grafts (high degree of steatosis, history of cardiac arrest, depending on increased α-adrenergic pharmacological support, hypotension and older than 50 years) and prolonged ischemia time constitute risk factors for developing reperfusion syndrome and hyperpotasaemia during graft reperfusion.(12, 45)
The syndrome tends to be more severe during the first minute of reperfusion and generally stops being critical after the 5th minute. Mean arterial pressure less than 60 mmHg is preferentially treated with adrenalin in 50 mg bolus and an infusion of liquids orientated by venous pressures. The need for continuous adrenergic support is decided by the evolution of hemodynamic data.
Prophylaxis is currently the only way of attenuating the hemodynamic effects of reperfusion during the intraoperative period and is based on rigorous hemodynamic control by means of invasive monitoring associated with metabolic and hydroelectrolytic control during the periods preceding reperfusion.
Hyperpotasaemia is the electrolytic alteration having the most serious consequences during reperfusion, making it obligatory that prophylactic measures for maintaining serum potassium levels stay below 4.0 mEq/L during the anhepatic phase. A potassium level greater than 5.0 mEq/L during anhepatic phase constitutes a risk factor for developing hyperpotasaemia during reperfusion(46) and must therefore be treated aggressively.(35, 47)
Electrocardioscopically, hyperpotasaemia is manifest by sharp, symmetrical T-waves and, in the most serious cases, by lengthening of the QRS complex, which could evolve to ventricular tachycardia or VF. Calcium and sodium bicarbonate administration are the most efficient measures for treating hyperpotasaemia secondary to reperfusion.
Coagulopathy occurring early during the neohepatic phase is due to an exacerbated and transitory increase in t-PA activity, increase in proteolytic activity, heparin or heparinoid factors release from the graft, hypothermia and acidosis, resulting in a clinical picture of hyperfibrinolysis and hypocoagulation.(48-51)
Coagulopathy (if the implanted liver functions suitably) is restricted to the first hour of reperfusion and usually does not require intervention.( 50)
The growing need for hemodynamic pharmacological support, serious and prolonged hyperfibrinolysis, hypocalcaemia, persistent metabolic acidosis, refractary hypothermia, hypoglycaemia and oliguria are classical signs of graft dysfunction.(37, 39)
The fact that hemodynamic, metabolic, coagulation, electrolytic and acid-based reperfusion alterations become self-limited typically characterises a graft’s good functioning (Table 5).

REFERENCIAS
1. Punch JD. Bridges to transplantation. Anesthesiol Clin North America. 2004 Dec;22(4):863-9.
2. Vaquero J, Chung C, Cahill ME, Blei AT. Pathogenesis of hepatic encephalopathy in acute liver failure. Semin Liver Dis. 2003 Aug;23(3):259-69.
3. Donovan JP, Schafer DF, Shaw BW, Jr., Sorrell MF. Cerebral oedema and increased intracranial pressure in chronic liver disease. Lancet. 1998 Mar 7;351(9104):719-21.
4. Haussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema? J Hepatol. 2000 Jun;32(6):1035-8.
5. Bacher A, Zimpfer M. Hot topics in liver intensive care. Transplant Proc. 2008 May;40(4):1179-82.
6. Debray D, Yousef N, Durand P. New management options for end-stage chronic liver disease and acute liver failure: potential for pediatric patients. Paediatr Drugs. 2006;8(1):1-13.
7. Han MK, Hyzy R. Advances in critical care management of hepatic failure and insufficiency. Crit Care Med. 2006 Sep;34(9 Suppl):S225-31.
8. Ekser B, Gridelli B, Tector AJ, Cooper DK. Pig liver xenotransplantation as a bridge to allotransplantation: which patients might benefit? Transplantation. 2009 Nov 15;88(9):1041-9.
9. Galvao FH, Pompeu E, de Mello ES, da Costa Lino Costa A, Mory E, Dos Santos RM, et al. Experimental multivisceral xenotransplantation. Xenotransplantation. 2008 May;15(3):184-90.
10. Mitzner SR, Stange J, Klammt S, Koball S, Hickstein H, Reisinger EC. Albumin dialysis MARS: knowledge from 10 years of clinical investigation. ASAIO J. 2009 Sep-Oct;55(5):498-502.
11. Mushlin PS, Gelman S. Anesthesia and the Liver, em: Barash PG, Cullen BF, Stoelting RK - Clinical Anesthesia, 4ª edição, Filadélfia, Lippincott Williams & Wilkins, 2001;1067-101.
12. Steadman RH. Anesthesia for liver transplant surgery. Anesthesiol Clin North America. 2004 Dec;22(4):687- 711.
13. Krowka MJ, Porayko MK, Plevak DJ, Pappas SC, Steers JL, Krom RA, et al. Hepatopulmonary syndrome with progressive hypoxemia as an indication for liver transplantation: case reports and literature review. Mayo Clin Proc. 1997 Jan;72(1):44-53.
14. Lai WK, Murphy N. Management of acute liver failure. Continuing Education in Anaesthesia, Critical Care & Pain. 2004;4(2):40-3.
15. Kang Y, Lewis JH, Navalgund A, Russell MW, Bontempo FA, Niren LS, et al. Epsilon-aminocaproic acid for treatment of fibrinolysis during liver transplantation. Anesthesiology. 1987 Jun;66(6):766-73.
16. Petrovich CT. An approach to the patient who may have a bleeding disorder. ASA Refresher Courses in Anesthesiology. 2004;32(236):1-5.
17. Moller S, Bendtsen F, Henriksen JH. Pathophysiological basis of pharmacotherapy in the hepatorenal syndrome. Scand J Gastroenterol. 2005;40:491- 500.
18. Iwatsuki S, Popovtzer MM, Corman JL, Ishikawa M, Putnam CW, Katz FH, et al. Recovery from “hepatorenal syndrome” after orthotopic liver transplantation. N Engl J Med. 1973 Nov 29;289(22):1155-9.
19. Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999 Mar;29(3):648-53.
20. Mukherjee KK, Chhabra R, Khosla VK. Raised intracranial pressure in hepatic encephalopathy. Indian J Gastroenterol. 2003 Dec;22 Suppl 2:S62-5.
21. Rocha Filho JA, Rocha JPS, Nani RS. Anestesia para transplante hepático. In: Cangiani LM, Posso IP, Poterio GMB, editors. Tratado de Anestesiologia. São Paulo: Atheneu; 2006. p. 1899-909.
22. Jalan R. Intracranial hypertension in acute liver failure: pathophysiological basis of rational management. Semin Liver Dis. 2003 Aug;23(3):271-82.
23. Ellis A, Wendon J. Circulatory, respiratory, cerebral, and renal derangements in acute liver failure: pathophysiology and management. Semin Liver Dis. 1996 Nov;16(4):379-88.
24. Wendon J, Lee W. Encephalopathy and cerebral edema in the setting of acute liver failure: pathogenesis and management. Neurocrit Care. 2008;9(1):97- 102.
25. Rabadan AT, Spaho N, Hernandez D, Gadano A, de Santibanes E. Intraparenchymal intracranial pressure monitoring in patients with acute liver failure. Arq Neuropsiquiatr. 2008 Jun;66(2B):374-7.
26. Detry O, Arkadopoulos N, Ting P, Kahaku E, Margulies J, Arnaout W, et al. Intracranial pressure during liver transplantation for fulminant hepatic failure. Transplantation. 1999 Mar 15;67(5):767-70.
27. Jalan R, Davies NA, Damink SW. Hypothermia for the management of intracranial hypertension in acute liver failure. Metab Brain Dis. 2002 Dec;17(4):437-44.
28. Filho JA, Machado MA, Nani RS, Rocha JP, Figueira ER, Bacchella T, et al. Hypertonic saline solution increases cerebral perfusion pressure during clinical orthotopic liver transplantation for fulminant hepatic
1. Punch JD. Bridges to transplantation. Anesthesiol Clin North America. 2004 Dec;22(4):863-9. [ Links ]
2. Vaquero J, Chung C, Cahill ME, Blei AT. Pathogenesis of hepatic encephalopathy in acute liver failure. Semin Liver Dis. 2003 Aug;23(3):259-69. [ Links ]
3. Donovan JP, Schafer DF, Shaw BW, Jr., Sorrell MF. Cerebral oedema and increased intracranial pressure in chronic liver disease. Lancet. 1998 Mar 7;351(9104):719-21. [ Links ]
4. Haussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema? J Hepatol. 2000 Jun;32(6):1035-8. [ Links ]
5. Bacher A, Zimpfer M. Hot topics in liver intensive care. Transplant Proc. 2008 May;40(4):1179-82. [ Links ]
6. Debray D, Yousef N, Durand P. New management options for end-stage chronic liver disease and acute liver failure: potential for pediatric patients. Paediatr Drugs. 2006;8(1):1-13. [ Links ]
7. Han MK, Hyzy R. Advances in critical care management of hepatic failure and insufficiency. Crit Care Med. 2006 Sep;34(9 Suppl):S225-31. [ Links ]
8. Ekser B, Gridelli B, Tector AJ, Cooper DK. Pig liver xenotransplantation as a bridge to allotransplantation: which patients might benefit? Transplantation. 2009 Nov 15;88(9):1041-9. [ Links ]
9. Galvao FH, Pompeu E, de Mello ES, da Costa Lino Costa A, Mory E, Dos Santos RM, et al. Experimental multivisceral xenotransplantation. Xenotransplantation. 2008 May;15(3):184-90. [ Links ]
10. Mitzner SR, Stange J, Klammt S, Koball S, Hickstein H, Reisinger EC. Albumin dialysis MARS: knowledge from 10 years of clinical investigation. ASAIO J. 2009 Sep-Oct;55(5):498-502. [ Links ]
11. Mushlin PS, Gelman S. Anesthesia and the Liver, em: Barash PG, Cullen BF, Stoelting RK - Clinical Anesthesia, 4ª edição, Filadélfia, Lippincott Williams & Wilkins, 2001;1067-101. [ Links ]
12. Steadman RH. Anesthesia for liver transplant surgery. Anesthesiol Clin North America. 2004 Dec;22(4):687- 711. [ Links ]
13. Krowka MJ, Porayko MK, Plevak DJ, Pappas SC, Steers JL, Krom RA, et al. Hepatopulmonary syndrome with progressive hypoxemia as an indication for liver transplantation: case reports and literature review. Mayo Clin Proc. 1997 Jan;72(1):44-53. [ Links ]
14. Lai WK, Murphy N. Management of acute liver failure. Continuing Education in Anaesthesia, Critical Care & Pain. 2004;4(2):40-3. [ Links ]
15. Kang Y, Lewis JH, Navalgund A, Russell MW, Bontempo FA, Niren LS, et al. Epsilon-aminocaproic acid for treatment of fibrinolysis during liver transplantation. Anesthesiology. 1987 Jun;66(6):766-73. [ Links ]
16. Petrovich CT. An approach to the patient who may have a bleeding disorder. ASA Refresher Courses in Anesthesiology. 2004;32(236):1-5. [ Links ]
17. Moller S, Bendtsen F, Henriksen JH. Pathophysiological basis of pharmacotherapy in the hepatorenal syndrome. Scand J Gastroenterol. 2005;40:491- 500. [ Links ]
18. Iwatsuki S, Popovtzer MM, Corman JL, Ishikawa M, Putnam CW, Katz FH, et al. Recovery from "hepatorenal syndrome" after orthotopic liver transplantation. N Engl J Med. 1973 Nov 29;289(22):1155-9. [ Links ]
19. Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999 Mar;29(3):648-53. [ Links ]
20. Mukherjee KK, Chhabra R, Khosla VK. Raised intracranial pressure in hepatic encephalopathy. Indian J Gastroenterol. 2003 Dec;22 Suppl 2:S62-5. [ Links ]
21. Rocha Filho JA, Rocha JPS, Nani RS. Anestesia para transplante hepático. In: Cangiani LM, Posso IP, Poterio GMB, editors. Tratado de Anestesiologia. São Paulo: Atheneu; 2006. p. 1899-909. [ Links ]
22. Jalan R. Intracranial hypertension in acute liver failure: pathophysiological basis of rational management. Semin Liver Dis. 2003 Aug;23(3):271-82. [ Links ]
23. Ellis A, Wendon J. Circulatory, respiratory, cerebral, and renal derangements in acute liver failure: pathophysiology and management. Semin Liver Dis. 1996 Nov;16(4):379-88. [ Links ]
24. Wendon J, Lee W. Encephalopathy and cerebral edema in the setting of acute liver failure: pathogenesis and management. Neurocrit Care. 2008;9(1):97- 102. [ Links ]
25. Rabadan AT, Spaho N, Hernandez D, Gadano A, de Santibanes E. Intraparenchymal intracranial pressure monitoring in patients with acute liver failure. Arq Neuropsiquiatr. 2008 Jun;66(2B):374-7. [ Links ]
26. Detry O, Arkadopoulos N, Ting P, Kahaku E, Margulies J, Arnaout W, et al. Intracranial pressure during liver transplantation for fulminant hepatic failure. Transplantation. 1999 Mar 15;67(5):767-70. [ Links ]
27. Jalan R, Davies NA, Damink SW. Hypothermia for the management of intracranial hypertension in acute liver failure. Metab Brain Dis. 2002 Dec;17(4):437-44. [ Links ]
28. Filho JA, Machado MA, Nani RS, Rocha JP, Figueira ER, Bacchella T, et al. Hypertonic saline solution increases cerebral perfusion pressure during clinical orthotopic liver transplantation for fulminant hepatic [ Links ]
