










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.46 no.3 Bogotá July/Sept. 2018
https://doi.org/10.1097/cj9.0000000000000059
Artículo de revisión
Implicaciones anestésicas de las distrofias musculares
a Instituto Roosevelt. Bogotá, Colombia
b Cuarto año, residente de anestesiología y reanimación, Fundación Universitaria de Ciencias de la Salud. Bogotá, Colombia.
Introducción:
Las distrofias musculares son un grupo de enfermedades genéticas que se caracterizan por compromiso en la síntesis o regeneración de las proteínas contráctiles del musculo. Aunque pertenecen al mismo grupo de enfermedades tienen características muy diferentes en su presentación clínica y en su origen genético. Estas enfermedades se clasifican como huérfanas debido a que tienen una incidencia muy baja en la población general, pero representan un enorme reto anestésico, especialmente en la población pediátrica.
Objetivo:
Describir los principales aspectos clínicos de las distrofias musculares, su etiología, implicaciones anestésicas y principales complicaciones que pueden ocurrir durante el perioperatorio.
Metodología:
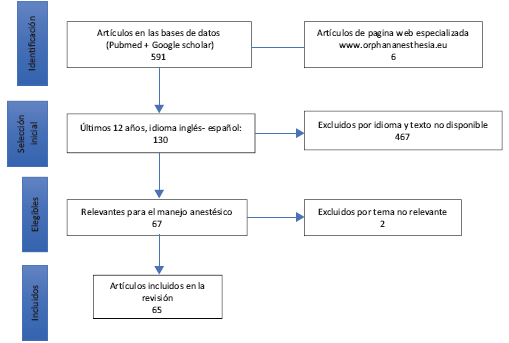
Se presenta un artículo de revisión basado en una búsqueda sistemática de la literatura para una revisión descriptiva, donde la principal fuente de información son los reportes de caso obtenidos en las bases de datos de pubmed, google académico y páginas web especializadas en enfermedades raras, con el propósito de describir las principales implicaciones anestésicas de este grupo de enfermedades.
Resultados:
Se obtuvo un total de 65 referencias bibliográficas las cuales fueron seleccionadas por los autores de acuerdo con la relevancia del tema para la revisión final.
Conclusión:
Las distrofias musculares son un grupo heterogéneo de enfermedades que comparten una etiología común que es la lesión directa en la fibra muscular con un compromiso sistémico progresivo. Se diferencian en su presentación clínica, origen genético y riesgos anestésicos que son principalmente complicaciones cardiovasculares por arritmias malignas, rabdomiolisis aguda desencadenada por fármacos utilizados en la anestesia y falla respiratoria perioperatoria.
Palabras clave: Distrofias Musculares; Distrofia Miotónica; Distroia Muscular de Duchenne; Miotonía Congénita; Anestesia
Introduction:
Muscular dystrophies are a group of genetic diseases characterized by the compromised synthesis or regeneration of the muscle contractile proteins. Although they belong to the same group of diseases, they have different characteristics in their clinical presentation and in their genetic origin. These diseases are classified as orphan as they have a low incidence among the general population, but represent a huge anesthetic challenge, particularly among the pediatric population.
Objective:
To describe the main clinical aspects of muscular dystrophies, their etiology, anesthetic implications, and the major complications that may occur during the perioperative management.
Methodology:
A review article is discussed based on a systematic search of the literature to produce a descriptive review. The main source of information is case reports obtained from databases as PubMed, Google Scholar, and websites specialized in rare diseases, to describe the main anesthetic implications of muscular dystrophies.
Results:
A total of 65 references were identified by the authors in accordance with the relevance of the topic for the final review.
Conclusion:
Muscular dystrophies are a heterogeneous group of diseases that share a common etiology due to direct injury of the muscle fiber with a progressive and systemic compromise. Each type of muscular dystrophy is different in terms of its clinical presentation, genetic origin, and anesthetic risks which are mainly cardiovascular complications due to malignant arrhythmias, acute rhabdomyolysis triggered by drugs used in anesthesia, and perioperative respiratory failure.
Keywords: Muscular Dystrophies; Myotonic Dystrophy; Muscular Dystrophy; Duchenne; Myotonia Congenita; Anesthesia
Introducción
Las distrofias musculares son un grupo de más de 30 enfermedades genéticas que se caracterizan por compromiso en la síntesis o en la regeneración de las proteínas contráctiles lo que produce debilidad y degeneración progresiva de los músculos esqueléticos. Aunque pertenecen al mismo grupo de enfermedades tienen características muy diferentes en su presentación clínica y en su origen genético. La incidencia de estas enfermedades varía entre 1 caso por cada 10.000 a 100.000 nacidos vivos.1,2 Aunque son enfermedades muy raras, representan un enorme reto anestésico, sobre todo en la población pediátrica.
El objetivo de esta revisión es describir los principales aspectos clínicos de las distrofias musculares, la etiología, las implicaciones anestésicas y las principales complicaciones que pueden ocurrir durante el perioperatorio.
Metodología
Se consultaron las bases de datos de pubmed, google académico y el sitio web de orphan-anesthesia, con los términos MeSH para cada enfermedad y el termino MeSH: anestesia (Tabla 1). Se seleccionaron las publicaciones de los últimos 12 años, en idioma inglés o español, que estuvieran disponibles en texto completo y relevantes con el manejo anestésico. Los artículos fueron seleccionados por los autores principales (P.E.M. y A.M.B.) de acuerdo con el objetivo de la revisión. La selección final y la clasificación de los artículos se describe en la Tabla 2 yen la Figura 1. Debido al tipo de artículos encontrados, que en su mayoría son reportes y series de casos, se decidió no hacer análisis de GRADE para clasificar la evidencia.
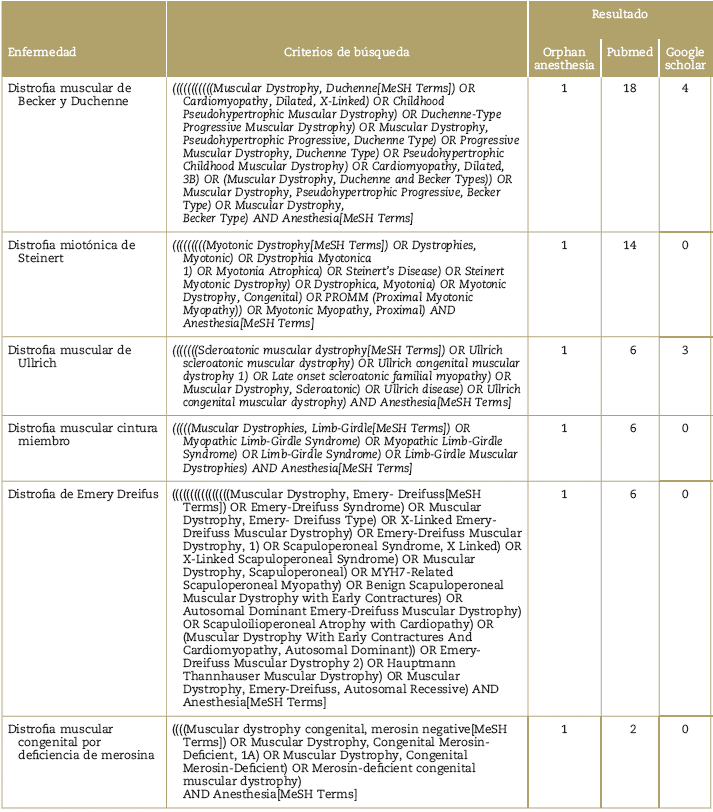
Tabla 2 Clasificación de los artículos seleccionados, de acuerdo con el tipo de publicación
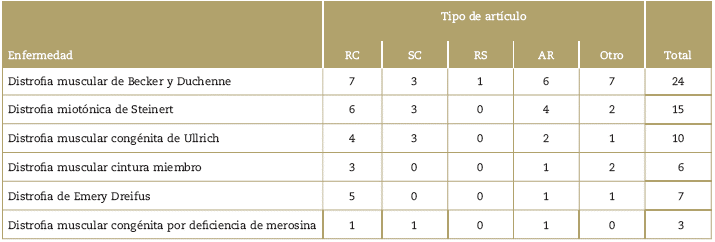
RC = Reporte de caso, SC = Serie de casos, RS = Revisión sistemática, AR = Artículo de revisión, Otro = consensos, recomendaciones de expertos, editoriales, cartas al editor o correspondencia.
Fuente: Autores.
Descripción de las distrofias musculares
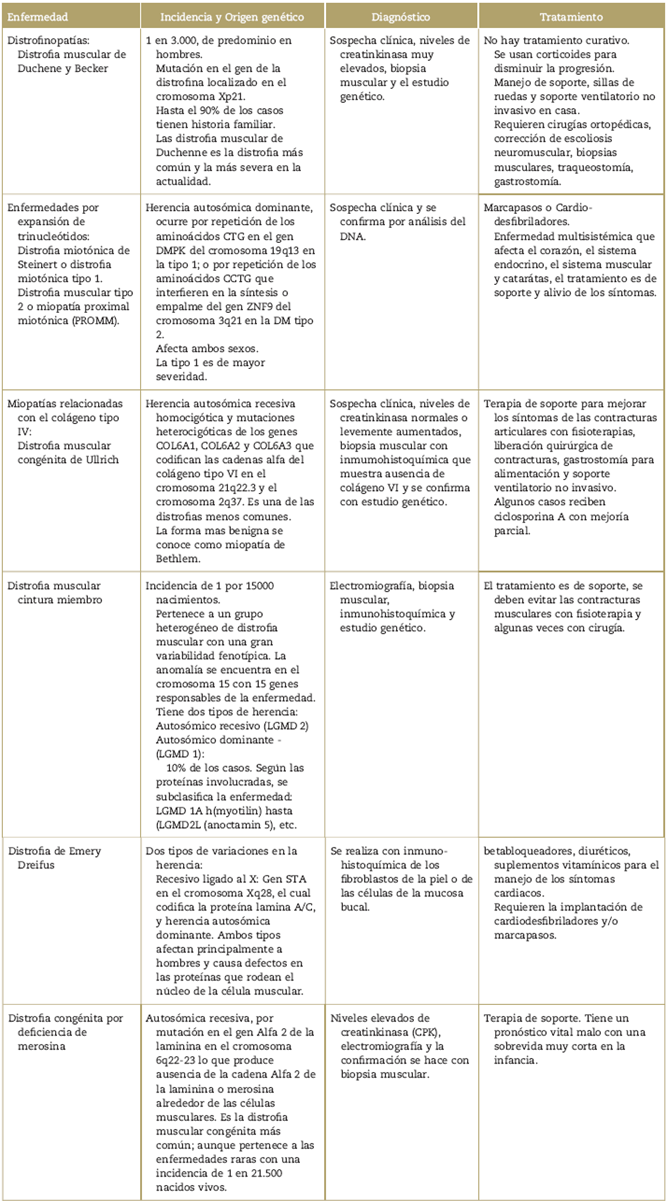
Distrofinopatías
En esta clasificación están la distrofia muscular de Becker y la distrofia muscular de Duchenne. Son la misma enfermedad desde su origen genético: el gen de la distrofina, pero se diferencian en el déficit de distrofina el cual es casi completo en la enfermedad de Duchenne, pero es sólo parcial en la enfermedad de Becker; por esta razón tienen una presentación clínica muy diferente una de otra.
La enfermedad de Duchenne es la distrofia muscular más común y severa, la edad promedio de diagnóstico es de 3 - 5 años para la distrofia de Duchenne; y en la vida adulta, para la distrofia de Becker.3 Se presenta principalmente en el sexo masculino, sin embargo algunas mujeres portadoras del gen pueden presentar patología muscular o cardiaca.4-6
Los signos clínicos en el Duchenne son debilidad muscular progresiva severa que se inicia en la infancia (aproximadamente 18 meses de vida) y produce la pérdida definitiva de la marcha cerca de los 10 años de edad debido a una reorganización muscular patológica con infiltración grasa y aumento del tejido fibroso que reemplaza el tejido muscular.3
El curso progresivo de la distrofia de Duchenne compromete la función cardiaca y la función respiratoria por compromiso de los músculos respiratorios. La sobrevida general de estos pacientes es de 20 a 30 años y la muerte se produce por falla ventilatoria en el 90% de los casos y 10% por falla cardiaca.5,7,8 Después de los 30 años, más del 90% de los pacientes con Duchenne tienen compromiso cardiaco.5 Los hallazgos clínicos característicos son: debilidad progresiva de miembros inferiores, seudohipertrofia de los músculos gastronemios (evidente entre 2 - 6 años de edad), niveles altos de creatinkinasa, enfermedad pulmonar crónica con un patrón restrictivo por compromiso de los músculos respiratorios y laríngeos; y en la adolescencia hay signos de cardiomiopatía.5,8,9 Estos pacientes requieren cirugías frecuentes como corrección de escoliosis neuromuscular, cirugías ortopédicas, biopsias musculares y en estados avanzados requieren gastrostomías y traqueostomías.
La distrofia de Becker se caracteriza por debilidad muscular en miembros inferiores y pelvis, su progresión es mas lenta que la distrofia de Duchenne porque la deficiencia en la distrofina es parcial. Los síntomas habitualmente aparecen en la adolescencia y pueden desarrollar cardiomiopatía dilatada a la edad de 30 años (nunca antes de los 16 años).9 La sobrevida es mayor, entre 30 a 60 años y aunque tienen menos comorbilidades respiratorias,5,10 tienen alteraciones en el electrocardiograma (EKG) hasta en el 75% de los casos con un riesgo mayor de presentar falla cardiaca.8,11
El test de contractura muscular con cafeína-halotano puede resultar positivo, (sensibilidad de 95 - 97% y especificidad de 80 -90% para hipertermia maligna). No obstante, esta prueba da contracturas anormales en los pacientes con distrofia muscular de Becker y Duchenne con mayor incidencia de falsos positivos porque los miocitos comprometidos son dependientes del voltaje.6
Hasta el momento, no existen otros métodos diagnósticos para determinar la verdadera suceptibilidad de estos pacientes a la hipertermia maligna.
Distrofia miotónica de steinert
Enfermedad neuromuscular y multisistémica; se caracteriza por anormalidad en los canales de sodio y cloro, produce debilidad muscular distal, axial, faríngea y respiratoria asociada a arritmias cardiacas, cardiomiopatía, diabetes mellitus, alteración tiroidea y trastornos cognitivos.1
Se clasifica en dos tipos: la distrofia muscular (DM1) tipo 1 o distrofia muscular clásica (enfermedad de Steinert) de carácter congénito por alteración de la transcripción de los genes que codifican los canales de cloro, receptores de insulina, troponinas y receptores NMDA, tiene un inicio muy variable en la vida, con una presentación muy diversa que se puede manifestar entre los 10 a 40 años de edad;1 y la distrofia muscular tipo 2 (DM2) o miopatía miotónica proximal (PROMM), que se presenta habitualmente en los adultos y tiene menor riesgo anestésico que la tipo 1. La disfunción cognitiva, la cardiomiopatía y muerte súbita son mas comunes en la tipo 1.12-15
Miopatías relacionadas con el colágeno tipo VI
A este grupo de enfermedades pertenecen la distrofia muscular congénita de Ullrich y la distrofia muscular cintura-miembro.
La enfermedad de Ullrich es un desorden neuromuscular severo causado por mutaciones en los genes que codifican el colágeno VI que es una glicoproteína fundamental de la matriz extracelular que se encarga de mantener la integridad tisular y sirve como una red estructural que le brinda soporte a las células de todos los tejidos.16,17 Se manifiesta desde la etapa neonatal con hipotonía congénita y muchos pacientes no logran la marcha, presentan contracturas articulares proximales, hiperlaxitud distal con escoliosis rápidamente progresiva e inteligencia normal, función cardíaca normal y niveles de creatinkinasas normales o levemente elevados.17-19 La sobrevida habitual es hasta la segunda década de la vida y la muerte ocurre por insuficiencia respiratoria.20-22 Existe una forma más benigna de la enfermedad conocida como miopatía de Bethlem o distrofia muscular de progresión lenta.
La distrofia muscular cintura miembro es una enfermedad neuromuscular caracterizada por un desajuste entre la ruptura y la reparación de la fibra muscular. Tiene una incidencia de 1 caso en 1500 nacidos vivos. Se manifiesta como debilidad muscular proximal que compromete la cintura pélvica y la cintura escapular progresiva, elevación de los niveles de creatinkinasa y tiene un inicio de presentación muy variable, pues se puede presentar desde la infancia hasta la vida adulta.23,24 El diagnóstico es difícil de realizar porque la edad de inicio y la presentación clínica son muy variables. El diagnóstico final se realiza con electromiografía, sospecha clínica, biopsia muscular, inmunohistoquímica y genética. Según su origen genético se clasifica en: distrofia muscular cintura miembro tipo 1 (LGMD1) con transmisión autosómica dominante; y distrofia muscular tipo 2 (LGMD2) con trasmisión autosómica recesiva; también se clasifica según las proteínas involucradas como: miotilina, lamina A/C, caveolina 3, calpaina, disferlina, sarcoglicanos, entre otros.24 La distrofia tipo 1 tiene manifestaciones cardiacas como arritmias malignas que requieren la implantación de cardiodesfibriladores o marcapasos; y la tipo 2, desarrollan cardiomiopatía dilatada en el 50% de los casos.25
Distrofia muscular de Emery Dreifus
Caracterizada por una mutación genética que afecta la producción de una proteína de 34 kDa en la lámina A/C que forma parte de la membrana nuclear.26-28 Se caracteriza por contracturas articulares principalmente en tendón de Aquiles, codo y columna vertebral, posteriormente compromete el cuello y la parte inferior de la espalda produciendo un raquis rígido; también cursa con alteraciones en la conducción cardiaca y miocardiopatía dilatada aunque el grado de lesión cardiaca no se correlaciona con el de lesión muscular.29 Las contracturas en flexión aparecen a los 4-5 años de vida y la marcha puede conservarse con ayuda de aparatos.26,27,30,31 La expectativa de vida es aproximadamente de 46 años, el 12% mueren por falla cardiaca y el 46% presentan muerte súbita cardiaca.31
Aunque se ha reportado que aunque el test de contractura muscular in vitro es negativo para riesgo de hipertermia maligna, existen publicaciones que apoyan el uso de técnicas seguras como la anestesia total intravenosa.32 Se han reportado dificultades para la intubación y para la anestesia regional epidural y espinal en estos pacientes.
Distrofia muscular congénita por deficiencia de merosina (DMC-DM)
Pertenece al grupo de las distrofias musculares congénitas, y se da por un defecto en la proteína merosina que rodea las fibras musculares. Se caracteriza por debilidad muscular progresiva severa con hipotonía desde el nacimiento, múltiples contracturas, dificultades para la deglución, escoliosis, enfermedad pulmonar restrictiva severa, compromiso cardiaco que varía entre bloqueos de rama y cardiomiopatía dilatada y niveles elevados de creatinkinasa.2,33,34
Las principales características clínicas de las distrofias musculares se describen en la Tabla 3.35-40
Implicaciones anestésicas de las distrofias musculares
Valoración preanestésica
Los pacientes con distrofia muscular generalmente requieren cirugías ortopédicas a temprana edad, corrección de escoliosis, biopsias musculares, liberación de tendones o transferencias tendinosas.3,41 En el caso de las distrofias que causan mayor afectación cardiaca como la distrofia miotónica de Steinert, Emery Dreifus, y cintura miembro, requerirán anestesia para implantación de cardiodesibriladores y marcapasos1,30,31,36,42 debido al alto riesgo de muerte súbita por fibrilación ventricular, taquicardia ventricular o bloqueos cardiacos completos;26 y en etapas avanzadas, requieren traqueostomías, gastrostomías y procedimientos terapéuticos o diagnósticos como tomografías y resonancias magnéticas.2
Se recomienda una evaluación integral del estado funcional del paciente con énfasis en los órganos de mayor afectación para cada tipo de distrofia. Se deben evaluar los criterios de vía aérea difícil que se presentan con frecuencia en este tipo de pacientes, así como descartar ulceras de presión por posiciones crónicas.
En la Tabla 4 se describen los exámenes de laboratorios y pruebas complementarias prequirúrgicas que se recomiendan solicitar en los pacientes con distrofia muscular que son programados para cirugía. Los exámenes más frecuentes que se recomiendan son: niveles de creatinkinasa, hemograma, pruebas de coagulación, ecocardiograma, electrocardiograma y pruebas de función pulmonar.
Tabla 4 Exámenes de laboratorio y pruebas complementarias recomendadas en la evaluación preanestésica de los pacientes con distrofias musculares programados para cirugía
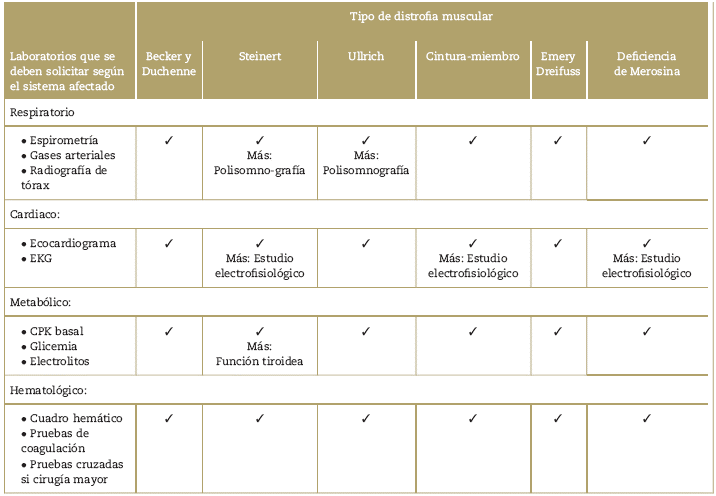
Fuente: Autores.
Se debe evaluar la capacidad vital forzada (CVF) y el volumen espiratorio forzado en el primer segundo (VEF1) en decúbito supino y sentado. Se considera que si el valor de la CVF es menor al 50% de lo predicho, el paciente tiene alto riesgo de necesitar ventilación no invasiva postoperatoria; y si es inferior al 30% indica que el paciente es de alto riesgo y requeriría soporte ventilatorio invasivo postoperatorio.17,19,20,38,39 En ocasiones puede ser necesario realizar estudios adicionales para valorar la función diafragmática, pues si esta alterada requerirá iniciar el soporte ventilatorio mecánico no invasivo antes de la cirugía.
Los criterios de severidad y de alto riesgo perioperatorio son:
CVF < 25%, PEP < 30%, FE VI < 0,5
PEP: Presión espiratoria pico, FE VI: Fracción de eyección del ventrículo izquierdo.39
Debido a la incidencia de arritmias malignas y muerte súbita en estos pacientes se recomienda realizar estudios electrofisiológicos, sobretodo en las distrofias de cintura miembro, enfermedad de Steinert y Emery Dreifuss.12
Manejo anestésico
El manejo anestésico de los pacientes con distrofias musculares es especialmente difícil por el riesgo de rabdomiolisis que desencadena hiperpotasemia y paro cardíaco, arritmias malignas, aumento de la debilidad muscular, problemas con el manejo de la vía aérea y reagudización de la insuficiencia respiratoria. Es importante resaltar que el riesgo anestésico depende del tipo de cirugía, del estado clínico de cada paciente y de la progresión de la enfermedad, las recomendaciones que se describen a continuación son generales pero no superan el correcto juicio clínico en cada paciente específico.
La monitorización completa de estos pacientes requerirá electrocardiograma, presión arterial no invasiva e invasiva, capnografía, pulsoximetría, temperatura, diuresis, parámetros ventilatorios y monitorización de la relajación neuromuscular.
Se recomienda la administración de soluciones glucosadas que disminuyen el riesgo de rabdomiolisis e hiperpotasemia aguda,43 se deben evitar períodos prolongados de ayuno y considerar la proilaxis de la broncoaspiración con antagonistas H2, metoclopramida y citrato de sodio.44-46
La anestesia general intravenosa total (TIVA) es beneficiosa en estos pacientes.3 El propofol es el hipnótico mas utilizado para la inducción y mantenimiento de la anestesia en pacientes con distrofia muscular de Duchenne.47 Si existe afectación cardiaca se pueden utilizar otras alternativas como etomidato y tiopental que han sido utilizados con éxito en pacientes con distrofias miotónicas.46 Los anestésicos volátiles no se utilizan en la mayoría de reportes de caso debido a que pueden desencadenar miotonías y crisis de rabdomiolisis; el óxido nitroso es el único agente inhalado que se utiliza entre el 34 -78% de estos resportes.3,48 No obstante, se ha reportado el uso de sevofluorane en la inducción anestésica en pacientes con distrofia muscular sin complicaciones asociadas; los eventos reportados de rabdomiolisis asociados a exposiciones a agentes inhalados tienen en común dos situaciones: primero, el inicio del evento adverso se presenta en el área de recuperación con el movimiento de los pacientes; y segundo, es más frecuente en niños menores de 8 años, en quienes la enfermedad es menos severa, pero la membrana celular muscular es mas inestable y vulnerable a desencadenar fenómenos de rabdomiolisis, incluso con la actividad muscular normal.45,48,49 Por esta razón, algunos autores concluyen que la evidencia no es lo suficientemente fuerte para relacionar los agentes inhalados con eventos adversos como la rabdomiolisis y que dicha asociación es multifactorial, pero que existen técnicas anestésicas mas seguras.48,49 En la distrofia miotónica no están contraindicados los halogenados, pero pueden aumentar la debilidad muscular.1,12,50 En la experiencia del autor se recomienda usar la inducción inhalatoria para facilitarla canalización de un acceso intravenoso y cambiar inmediatamente a una anestesia total intravenosa o sedación asociada a anestesia regional. Esta estrategia acorta el tiempo de exposición a anestésicos inhalados lo que teóricamente disminuye la probabilidad de contracturas musculares y rabdomiolisis.
Los opioides como el remifentanil y el fentanil han demostrado ser seguros en estos pacientes.1
La ketamina y el midazolam en bajas dosis se han reportado como opciones seguras para la sedo-analgesia asociadas a técnicas regionales (espinal o epidural) en estos pacientes.47,51,52 Es importante resaltar que el inicio de acción de los relajantes musculares no despolarizantes es mayor, su comportamiento es errático con un tiempo de duración mayor y un alto riesgo de parálisis residual por lo que es indispensable la monitorización de la función neuromuscular.9,53
El relajante muscular no despolarizante más frecuentemente utilizado es el rocuronio,47,48 que tiene la posibilidad de neutralizar su acción farmacológica mediante el uso de un reversor específico. Aunque el uso de sugamadex en pacientes con trastornos neuromusculares no se ha establecido completamente y su papel en pacientes con distrofias y cardiomiopatía dilatada no está completamente conocido, se ha reportado su uso en pacientes con distrofias y cardiomiopatía dilatada;10 a una dosis de 4mg/kg (dosis descrita para adultos) con recuperación del 100% de la función neuromuscular con mínimos eventos adversos cardiovasculares.54 Por el contrario, la neostigmina puede desencadenar miotonías agudas, rabdomiolisis, arritmias malignas y falla cardiaca.10,54 El riesgo de hipertemia maligna es similar al de la población general.55
La anestesia regional espinal, caudal o epidural se puede utilizar con seguridad y éxito en estos pacientes, aunque se han descrito casos de cotracturas miotónicas en bloqueo neuroaxial incompleto y es técnicamente más difícil de realizar por las anormalidades en la columna vertebral.3,42,55-58
Los pacientes programados para cirugía mayor deben recibir terapia antifibrinolítica con ácido tranexámico, y técnicas de ahorro sanguíneo, así como estrategias de calentamiento con fluidos calientes y mantas térmicas para prevenir la hipotermia que puede desencadenar contracturas mioclónicas, y aumentar el sangrado.47,58 Estos pacientes requieren monitorización completa con presión arterial no invasiva, capnografía, cardioscopio, pulsoxímetro, presión de la vía aérea, temperatura, sonda vesical, catéter venoso central y línea arterial.3,59 Es importante resaltar que la fluidoterapia en estos pacientes se debe hacer con cristaloides que no contengan potasio y que no hay contraindicaciones absolutas para el uso de coloides.60
Alertas especiales para el manejo anestésico de los pacientes con distrofias musculares
Los pacientes con distrofia muscular tienen contraindicación absoluta para recibir relajantes musculares despolarizantes como la succinilcolina y se debe tener precaución con en el uso de anestésicos inhalados que pueden desencadenar crisis de rabdomiolisis.8,47,55 La FDA (Food and Drug Administration) en 1992 expidió una alerta en donde la succinilcolina debe evitarse en pacientes con historia de miopatías o hipotonías congénitas no claras. Los anestésicos inhalados pueden ser usados con precaución en niños con distrofias y se debe monitorizar los niveles de potasio, los niveles de creatinkinasa prequirúrgicos y la función cardiaca.9
Manejo postoperatorio y complicaciones perioperatorias
Los pacientes con distrofias musculares deben tener una recuperación anestésica en unidades de vigilancia intensiva especialmente en pacientes sintomáticos, tras cirugía abdominal mayor o si la alteración muscular es severa.57 Se debe garantizar una buena analgesia porque disminuye las complicaciones respiratorias. Sin embargo, se debe considerar que estos pacientes son mas sensibles a los efectos de los opiáceos (sistémicos y neuroaxiales) por lo que presentan mayor riesgo de depresión respiratoria, exacerban la paresia gastrointestinal y aumentan el riesgo de reflujo, aspiración y alteración ventilatoria. Si existe depresión respiratoria tras la reversión de la relajación neuromuscular se debe considerar posponer la extubación 24 a 48 horas después, o considerar el uso de ventilación mecánica no invasiva.55
Los antinflamatorios no esteroideos (AINES) se deben utilizar con precaución porque también pueden desencadenar crisis de rabdomiolisis.43 Se ha reportado el uso paracetamol intravenoso o rectal,61,62 anestesia regional epidural y bloqueos de plexo nervioso guiado por ultrasonido; así como bloqueos paravertebrales para procedimientos en el tórax.61,63,64
Los pacientes con distrofia muscular tiene alto riesgo de apnea y muerte tras la extubación en las 24 horas posteriores a la cirugía.55
En los estadios iniciales de la enfermedad con compromiso leve y marcha conservada se puede utilizar la sedación y la analgesia con benzodiacepinas y opioides a bajas dosis; pero los pacientes en estados avanzados y con compromiso respiratorio, se debe evitar el uso de opioides o sedantes en el posoperatorio.3,45 En caso de ser necesarios, se deben titular cuidadosamente y mantener al paciente bajo vigilancia estricta en unidad de cuidado intensivo.11,64
Para la extubación se recomienda hacerlo con el paciente despierto y si fue necesario utilizar relajantes musculares no despolarizantes se debe garantizar su reversión completa para evitar la relajación residual.4,10,44,64
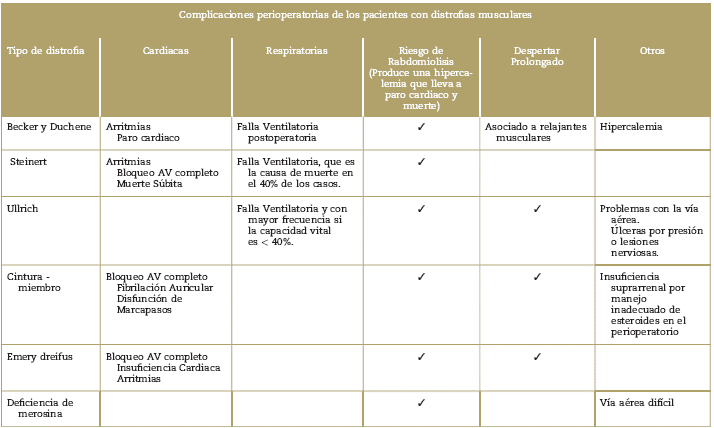
Independiente del agente anestésico que se utilice, los pacientes con distrofias tienen un riesgo elevado de complicaciones como: falla respiratoria, rabdomiolisis, arritmias, paro cardiaco y reacciones parecidas a la hipertermia maligna que necesitan un tratamiento sintomático agudo, pero que no resuelven con dantrolene.44,46,55,63,64 Los pacientes con distrofias se pueden clasificar según el riesgo perioperatorio de acuerdo con la escala MIRS (muscular impairment rating scale); el riesgo es intermedio en el grado MIRS 3; y muy alto en los grados 4 y 5 (ver Tabla 5).65 El riesgo quirúrgico también se aumenta en cirugía mayor y en los pacientes a quienes se les administró relajante muscular. En la Tabla 6 se describen las complicaciones perioperatorias más frecuentes para cada distrofia muscular.
Tabla 5 Escala MIRS (Muscular Impairment Rating Scale) para clasificar el compromiso muscular de la distrofia muscular65

Fuente: Autores.
Limitaciones
El objetivo de este artículo es hacer una revisión de las implicaciones anestésicas de las distrofias mas comunes basado en una búsqueda sistemática en la literatura. No obstante, debido a la calidad de la evidencia, que se basa en reportes de caso, series de casos y revisiones de tema, se presenta la información en forma de revisión descriptiva. Son necesarios estudios que aporten mayor nivel de evidencia aunque probablemente esto sea difícil por la baja frecuencia de este tipo de patología, para demostrar las estrategias anestésicas más seguras en los pacientes con distrofias musculares. Hasta el momento sólo disponemos de la experiencia compartida de los autores y de las instituciones que se dedican a la atención de pacientes con enfermedades raras; cabe resaltar que gran parte de los estudios disponibles son series de casos en pacientes pediátricos
Conclusión
Las distrofias musculares son un grupo de enfermedades congénitas heterogéneas con un origen genético diferente y con unas manifestaciones clínicas muy variables. Todas tienen implicaciones anestésicas muy importantes que ponen en alto riesgo perioperatorio a los pacientes por lo que requieren un conocimiento preciso para anticiparse y hacer un manejo específico de cada caso. Las principales complicaciones de las distrofias musculares son la falla ventilatoria, las arritmias cardiacas malignas, la hipercalemia, la rabdomiolisis, la inestabilidad cardiovascular y la muerte súbita. Aunque estas enfermedades no parecen estar relacionadas con hipertermia maligna, si tienen en común que debe evitarse el uso de los relajantes musculares despolarizantes y se debe preferir el uso de técnicas anestésicas intravenosas o anestesia regional para disminuir el riesgo de estas complicaciones, las cuales pueden estar asociadas al uso de los anestésicos inhalatorios. Se recomienda hacer una valoración prequirúrgica cuidadosa, identificar el estado general de cada paciente, el riesgo quirúrgico y tener un enfoque multidisciplinario que garantice la mejor atención perioperatoria que disminuya las complicaciones relacionadas con la enfermedad y el manejo anestésico. Es indispensable evaluar el riesgo / beneficio de cualquier procedimiento quirúrgico y anestésico en los pacientes con distrofia muscular antes de realizar su intervención.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
REFERENCIAS
1. Hoppe K, Reyher C, Jurkatt-Rott K, et al. Distrofia miotónica 1 y 2. 2014; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/186-distrofia-miot0nica-1-y-2.html. [ Links ]
2. Jimenez N, Linston D. Distrofia muscular congénita por deficiencia de merosina. 2013; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/132-distrofia-muscular-congénita-por-deficiencia-de-merosina.html. [ Links ]
3. Munster T. Distrofia muscular de Duchenne. 2011; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/131-distrofia-muscular-de-duchenne.html. [ Links ]
4. De Boer HD, Van Esmond J, Booij L, et al. Reversal of rocuronium-induced profound neuromuscular block by sugammadex in Duchenne muscular dystrophy. Paediatr Anaesth 2009;19:1226-1228. [ Links ]
5. Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg 2009;109:1043-1048. [ Links ]
6. Hayes J, Veyckemans F, Bissonnette B. Duchenne muscular dystrophy: an old anesthesia problem revisited. Paediatr Anaesth 2008;18:100-106. [ Links ]
7. Cheuk DK, Wong V, Wraige E, et al. Surgery for scoliosis in Duchenne muscular dystrophy. Cochrane Database Syst Rev 2007;24 1:CD005375. [ Links ]
8. Segura LG, Lorenz JD, Weingarten TN, et al. Anesthesia and Duchenne orBeckermuscular dystrophy: reviewof117 anesthetic exposures. Paediatr Anaesth 2013;23:855-864. [ Links ]
9. Driessen JJ. Neuromuscular and mitochondrial disorders: what is relevant to the anaesthesiologist? Curr Opin Anaesthesiol 2008;21:350. [ Links ]
10. himauchi T, Yamaura K, Sugibe S, et al. Usefulness of sugammadex in a patient with Becker muscular dystrophy and dilated cardiomyopathy. Acta Anaesthesiol Taiwan 2014;52:146-148. [ Links ]
11. Lerman J. Perioperative management of the paediatric patient with coexisting neuromuscular disease. Br J Anaesth 2011;107 (suppl 1):i79-i89. [ Links ]
12. Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth 2013;23:794-803. [ Links ]
13. Catena V, Del Monte DD, Rubini A, et al. Anesthesia and myotonic dystrophy (Steinert's syndrome). The role of total intravenous anesthesia with propofol, cisatracurium and remifentanyl. Case report. Minerva Anestesiol 2007;73:475-479. [ Links ]
14. Kirzinger L, Schmidt A, Kornblum C, et al. Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. Eur J Neurol 2010;17:842-845. [ Links ]
15. Weingarten TN, Hofer RE, Milone M, et al. Anesthesia and myotonic dystrophy type 2: a case series. Can J Anaesth 2010; 57:248-255. [ Links ]
16. Haliloglu G, Topaloglu H. Ullrich congenital muscular dystrophy. Iran J Child Neurol 2011;5:1-13. [ Links ]
17. Yonekawa T, Nishino I. Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism(s). J Neurol Neurosurg Psychiatry 2015;86:280-287. [ Links ]
18. Bozorgmehr B, Kariminejad A, Nafissi S, et al. Ullrich congenital muscular dystrophy (UCMD): clinical and genetic correlations. Iran J Child Neurol 2013;7:15-22. [ Links ]
19. Park Y, Park MS, Sung DH, et al. Ullrich congenital muscular dystrophy possibly related with COL6A1 p.Gly302Arg variant. Ann Rehabil Med 2014;38:292-296. [ Links ]
20. Prottengeier J, Shammas K, Smith J. Collagen VI-related myopa-thy. 2015; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/231-collagen-vi-related-myopathy.html. [ Links ]
21. Miscione MT, Bruno F, Ripamonti C, et al. Body composition, muscle strength, and physical function of patients with Bethlem myopathy and Ullrich congenital muscular dystrophy. Sci World J 2013;2013:152684. [ Links ]
22. Kang PB, Morrison L, Iannaccone ST, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015; 84:1369-1378. [ Links ]
23. Lopez-Alvarez A, Roman-Fernandez A. Limb-girdle muscular dystrophy. 2015; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/200-limb-girdle-muscular-dystrophy.html. [ Links ]
24. Allen T, Maguire S. Anaesthetic management of a woman with autosomal recessive limb-girdle muscular dystrophy for emergency caesarean section. Int J Obstet Anesth 2007;16:370-374. [ Links ]
25. Richa FC. Anaesthetic management of a patient with limb-girdle muscular dystrophy for laparoscopic cholecystectomy. Eur J Anaesthesiol 2011;28:72-73. [ Links ]
26. Kim OM, Elliott D. Elective caesarean section for a woman with Emery-Dreifuss muscular dystrophy. Anaesth Intensive Care 2010;38:744-747. [ Links ]
27. Shende D, Agarwal R. Anaesthetic management of a patient with Emery-Dreifuss muscular dystrophy. Anaesth Intensive Care 2002;30:372-375. [ Links ]
28. Ozer Y, Medical A. An anaesthetic approach to a case with Emery-Dreifuss muscular dystrophy. J Neurol Sci Turk 2005;22: 195-199. 26. [ Links ]
29. Funnell A. Recomendaciones para anestesia en pacientes que sufren de Distrofia muscular de Emery-Dreifuss (DMED). 2014; [Cited 2017 Feb 6]. Available at: http://www.orphananesthesia.eu/en/component/docman/doc_download/129-distrofia-muscular-de-emery-dreifuss-dmed.html. [ Links ]
30. Aldwinckle RJ, Carr AS. The anesthetic management of a patient with Emery-Dreifuss muscular dystrophy for orthopedic surgery. Can J Anaesth 2002;49:467-470. [ Links ]
31. Funnell A, Morgan J, McFadzean W. Anaesthesia and orphan disease: management of cardiac and perioperative risks in a patient with Emery-Dreifuss muscular dystrophy. Euro J Anesthesiol 2012;29:593-604. [ Links ]
32. Schuster F, Wessig C, Schimmer C, et al. In vitro contracture test results and anaesthetic management of a patient with Emery Dreifuss muscular dystrophy for cardiac transplantation. Case Rep Anesthesiol 2015;2012:349046. [ Links ]
33. Jimenez N, Song K, Lynn AM. Hemodynamic instability during prone spine surgery in a patient with merosin-deficient congenital muscular dystrophy. Paediatr Anaesth 2013;23:294-296. [ Links ]
34. Scrivener TA, Ross SM, Street NE, et al. A case series of general anesthesia in children with laminin alpha2 (merosin)-deficient congenital muscular dystrophy. Paediatr Anaesth 2014;24: 464-465. [ Links ]
35. Apiliogullari S, Oc B, Kara I, et al. Unilateral spinal anesthesia in a pediatric patient with Duchenne muscular dystrophy: a case report. Paediatr Anaesth 2013;23:1106-1107. [ Links ]
36. Pickard A, Lobo C, Stoddart PA. The effect of rocuronium and sugammadex on neuromuscular blockade in a child with congenital myotonic dystrophy type 1. Paediatr Anaesth 2013; 23:871-873. [ Links ]
37. Vandenberghe W, Jacobs TF, Plasschaert FS, et al. Anesthesia and perioperative management for a patient with Ullrich syndrome undergoing surgery for scoliosis. Acta Anaesthesiol Belg 2010; 61:43-47. [ Links ]
38. Carrasco-Marina ML, Quijano-Roy S, Iglesias-Escalera G, et al. Ullrich congenital muscular dystrophy. The usefulness of muscular magnetic resonance imaging in its diagnosis. Rev Neurol 2015;61:44-46. [ Links ]
39. Khirani S, Dabaj I, Amaddeo A, et al. The value of respiratory muscle testing in a child with congenital muscular dystrophy. Respirol Case Rep 2014;2:95-98. [ Links ]
40. Anta Redondo D, Ruiz Lopez JJ, Gredilla Diaz E, et al. Anesthesia for cesarean section in a patient with limb-girdle muscular dystrophy. Rev Esp Anestesiol Reanim 2008;55:651-652. [ Links ]
41. Miles F, Dare T. Scoliosis repair in a teenager with Duchenne's muscular dystrophy: who calls the shots? Paediatr Anaesth 2009;19:1022-1024. [ Links ]
42. Bisinotto FM, Fabri DC, Calcado MS, et al. Anesthesia for video-laparoscopic cholecystectomy in a patient with Steinert disease. Case report and review of the literature. Rev Bras Anestesiol 2010;60:105-110. [ Links ]
43. Hopkins PM. Anaesthesia and the sex-linked dystrophies: between a rock and a hard place. Br J Anaesth 2010;104: 397-400. [ Links ]
44. Caliskan E, Sener M, Kocum A, et al. Duchenne muscular dystrophy: how I do it? Regional or general anesthesia? Paediatr Anaesth 2009;19:624-625. [ Links ]
45. Van Obbergh LJ, Corteel J, Papadopoulos J, et al. Anesthesia for a child suffering from a deletion in the Xp21 loci resulting in Duchenne disease, glycerol kinase deficiency and congenital adrenal hypoplasia. Paediatr Anaesth 2011;21:1085-1087. [ Links ]
46. Ferschl M, Moxley R, Day JW, et al. Practical suggestions for the anesthetic management of a myotonic dystrophy patient. Myotonic dystrophy foundation toolkit. 2013;73-80. [Cited 2017 Feb 6]. Available at: www.mda.org.nz/media/2496/Anesthesia_Guidelines_2015.pdf. [ Links ]
47. Muenster T, Mueller C, Forst J, et al. Anaesthetic management in patients with Duchenne muscular dystrophy undergoing orthopaedic surgery: a review of 232 cases. Eur J Anaesthesiol 2012;29:489-494. [ Links ]
48. Schwartz D. Regarding: muscular dystrophy and the safety of inhalational agents. Paediatr Anaesth 2007;17:96 author reply 96-97. [ Links ]
49. Yemen TA, McClain C. Muscular dystrophy, anesthesia and the safety of inhalational agents revisited; again. Pediatr Anesth 2006;16:105-108. [ Links ]
50. Veyckemans F. Can inhalationagents be used in the presence ofa child with myopathy? Curr Opin Anaesthesiol 2010;23:348-355. [ Links ]
51. Baticon Escudero PM, Marcos Vidal JM, Ramos Fernandez R. Incomplete axillary block and sedation with ketamine in a patient with myotonic dystrophy. Rev Esp Anestesiol Reanim 2008;55: 60-61. [ Links ]
52. El-Dawlatly A, Aldohayan A, Nawaz S, et al. Anesthetic management of a patient with myotonic dystrophy for laparoscopic cholecystectomy: a case report. Middle East J Anaesthesiol 2008;19:1135-1140. [ Links ]
53. Muenster T, Schmidt J, Wick S, et al. Rocuronium 0.3mgkg-1 (ED95) induces a normal peak effect but an altered time course of neuromuscular block in patients with Duchenne's muscular dystrophy. Paediatr Anaesth 2006;16:840-845. [ Links ]
54. Stourac P, Krikava I, Seidlova J, et al. Sugammadex in a parturient with myotonic dystrophy. Br J Anaesth 2013;110:657-658. [ Links ]
55. Birnkrant DJ, Panitch HB, Benditt JO, et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest 2007;132: 1977-1986. [ Links ]
56. Sivathondan D. Myotonic dystrophy and pain management of a patient undergoing total abdominal hysterectomy in a metropolitan general hospital. Anaesth Intensive Care 2006; 34:506-509. [ Links ]
57. Chuang MC, Duggan LV, Van Heest RD, et al. Laparoscopic cholecystectomy under spinal anesthesia in a patient with limb-girdle muscular dystrophy. Can J Anaesth 2013;60:1276-1277. [ Links ]
58. Klompe L, Lance M, Van der Woerd D, et al. Anaesthesiological and ventilatory precautions during cardiac surgery in Steinert's disease. J Card Surg 2007;22:74-75. [ Links ]
59. Kocabas S, Yedicocuklu D, Askar F, et al. Anesthetic management of a child with Duchenne muscular dystrophy undergoing correction of Fallot’s Tetralogy. Paediatr Anaesth 2008;18:448-450. [ Links ]
60. Vieito M, Plaja I, Vilaplana J, et al. Anesthesia with sevoflurane for tonsillectomy in a boy with Duchenne muscular dystrophy. Rev Esp Anestesiol Reanim 2006;53:437-441. [ Links ]
61. Vandepitte C, Gautier P, Bellen P, et al. Use of ultrasound-guided intercostal nerve block as a sole anaesthetic technique in a high-risk patient with Duchenne muscular dystrophy. Acta Anaesthesiol Belg 2013;64:91-94. [ Links ]
62. Errando CL, Perez-Caballero P. Anaesthetic management in patients with Duchenne muscular dystrophy. Eur J Anaesthesiol 2013;30:257. [ Links ]
63. Sinclair JL, Reed PW. Risk factors for perioperative adverse events in children with myotonic dystrophy. Paediatr Anaesth 2009;19: 740-747. [ Links ]
64. Kocum A, Sener M, Caliskan E, et al. Anesthetic management for a child with unknown type of limb-girdle muscular dystrophy. Pediatr Int 2010;52:e37-e38. [ Links ]
65. Mathieu J, Boivin H, Meunier D, et al. Assessment of a diseasespecific muscular impairment raiting scale in myotonic dystrophy. Neurology 2001;56:336-340. [ Links ]
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
