











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Muscular dystrophies are a set of more than 30 genetic diseases characterized by a compromised synthesis or regeneration of contractile proteins, which causes weakness and progressive degeneration of the skeletal muscles. Although they belong to the same group of diseases, they have very different characteristics in their clinical presentation and in their genetic origin. The incidence of these diseases varies around 1 case per every 10,000 to 100,000 life births.1,2 Although these are very rare diseases, they represent a huge anesthetic challenge, particularly among the pediatric population.
The purpose of this review is to describe the major clinical aspects of muscular dystrophies, the etiology, the anesthetic implications, and the most relevant complications that may arise over the perioperative period.
Methodology
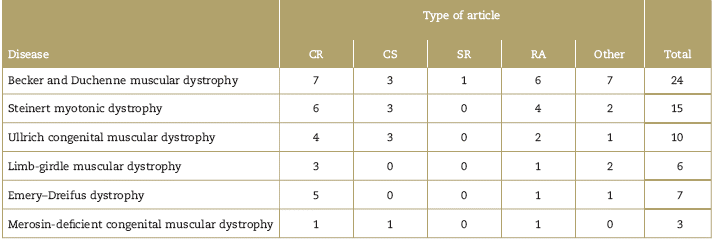
PubMed, Google academic, and the web page of orphan-anesthesia databases were consulted, using the MeSH terms for each disease and the term MeSH: anesthesia (Table 1). The publications of the last 12 years in English or Spanish available as full text and relevant for anesthetic management were selected. The articles were chosen based on the principal authors (P.E.M. and A.M.B.), in accordance with the purpose of the review. The final selection and classification of the articles is described in Table 2 and Fig. 1. Due to the type of articles identified- mostly reports and case series-the decision was made not to make Grading of Recommendations Assessment, Development and Evaluation (GRADE) analyses to classify the evidence.
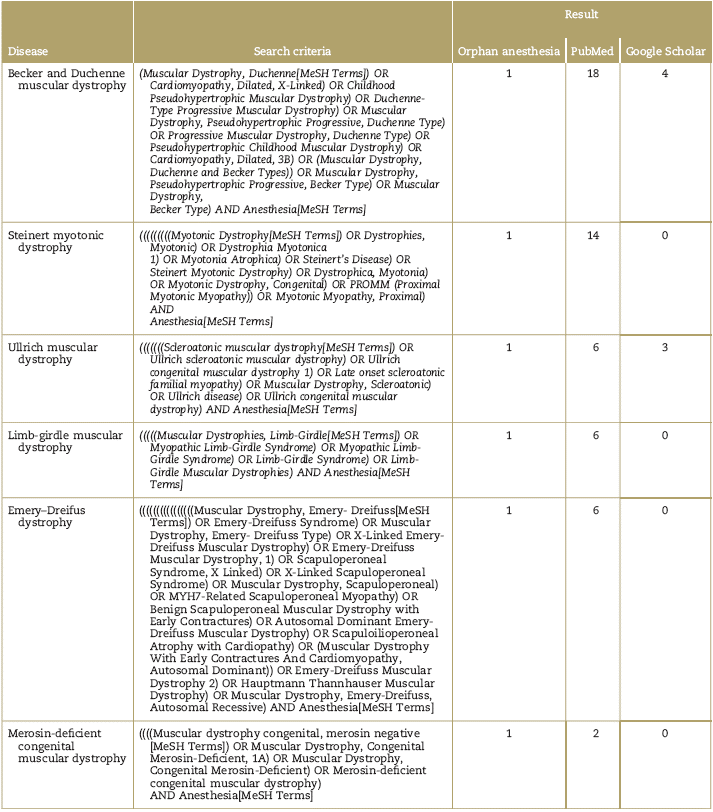
Table 2 Classification of the selected articles in accordance with the type of publication
CR=case report; CS=case series; Other=consensus, expert recommendations, editorials, letters to the editor or correspondence; RA=review article’ RS=systematic review.
Source: Authors.
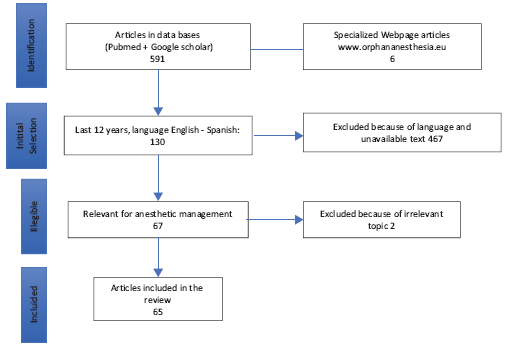
Muscular dystrophies description
Dystrophinopathies
This classification includes Becker muscular dystrophy and Duchenne muscular dystrophy. They are the same disease in terms of their genetic origin: the dystrophy gene; however, they differ in terms of the dystrophin deficit which is almost complete in Duchenne's disease, but only partial in Becker's disease. This is why they have a very different clinical presentation.
Duchenne's disease is the most common and severe muscular dystrophy; the mean age of diagnosis is 3 to 5 years for Duchenne's dystrophy, whereas for Becker's dystrophy is adulthood.3 It presents mostly in males; however, some women carriers of the gene may present muscular or cardiac pathology.4-6
The clinical sign in Duchenne's disease is severe progressive muscular weakness that begins during childhood (approximately around 18 months of age), leading to a definite loss of gait at around 10 years of age, due to a pathological muscular reorganization with fatty infiltration and increased fibrous tissue that replaces the muscular tissue.3
The progressive course of Duchenne's dystrophy compromises the cardiac and respiratory function due to the involvement of the respiratory muscles. The overall survival of these patients is 20 to 30 years, and they die because of ventilatory failure in 90% of the cases and 10% due to heart failure.5,7,8 After 30 years, over 90% of the patients with Duchenne present cardiac involvement.5 The characteristic clinical findings are progressive weakness of the lower extremities, pseudohypertrophy of the gastrocnemious muscles (evident between 2 and 6 years of age), high levels of creatinkinase, chronic pulmonary disease featuring a restrictive pattern due to involvement of the respiratory and laryngeal muscles. During adolescence, there are signs of cardiomyopathy.5,8,9 These patients need frequent surgical procedures such as correction of neuromuscular scoliosis, orthopedic surgeries, muscular biopsies, and, in advanced stages, gastrostomies and tracheostomies.
Becker's dystrophy is characterized by muscular weakness of the lower extremities and the pelvis; its progression is slower than Duchenne's dystrophy as the dystrophin deficit is partial. Symptoms usually present during adolescence and patients may develop dilated cardiomyopathy at age 30 (never before 16 years old).9 The survival is longer, between 30 and 60 years, and although the patient experience less respiratory comorbidities,5,10 they present alterations in the electrocardiogram (EKG) in up to 75% of the cases, with a higher risk of developing heart failure.8,11
The muscle contracture test with caffeine halothane may be positive (sensitivity 95%-97% and specificity 80%-90%) for malignant hyperthermia. Nevertheless, this test yields abnormal contractures in patients with Becker and Duchnne muscular dystrophy, with higher incidence of false positives as the compromised myocytes are voltage dependent.6 Up to date, there are no other diagnostic methods to establish the genuine susceptibility of these patients to malignant hyperthermia.
Steinert myotonic dystrophy
This neuromuscular and multisystemic disease is characterized by abnormal sodium and chloride channels, produces distal, axial, pharyngeal, and respiratory muscle weakness associated with cardiac arrhythmias, cardiomyopathy, diabetes mellitus, thyroid dysfunction, and cognitive disorders.1
Steinert myotonic dystrophy is classified into 2 types: muscular dystrophy type 1 (MD1) or classical muscular dystrophy (Steinert disease) of congenital nature due to alteration of the transcription of the genes encoding for chloride channels, insulin receptors, troponins, and N-methyl
De Aspartate (NMDA) receptors. It has a widely variable onset in life, with a very diverse presentation that may manifest between 10 and 40 years of age1; and type 2 muscular dystrophy (MD2) or proximal myotonic myopathy, that usually presents in adults and has a lower anesthetic risk than type 1. Cognitive dysfunction, cardiomyopathy, and sudden death are more common in type 1 merosin-deficient.12-15
Myopathies associated with collagen type VI
Ullrich's congenital muscular dystrophy and limb-girdle muscular dystrophy belong in this group of pathologies. Ullrich's disease is a severe neuromuscular disorder caused by mutations in the genes encoding for collagen VI, which is a fundamental glycoprotein of the extracellular matrix responsible for maintaining tissue integrityand provides a structural network to support cells in all tissues.16,17 It manifests since the neonatal period with congenital hypotony, and many patients are unable to walk, presenting proximal joint contractures, distal hyper-laxity with rapidly progressing scoliosis and normal intelligence, normal cardiac function, and normal or slightly elevated levels of creatinkinases.17-19 The usual survival extends to the second decade of life, and death occurs because of respiratory failure.20-22 There is a more benign form of the disease called Bethlem myopathy or slowly progressive muscular dystrophy.
Limb-girdle muscular dystrophy
This is neuromuscular disease characterized by a maladjustment between the rupture of the muscle fiber and its repair. Its incidence is 1 case per 1500 life births. It manifests as proximal muscle weakness involving the pelvic girdle and the progressive scapular girdle, elevation of the creatinkinase levels and has a widely variable onset as it may present from infancy to adulthood.23,24 The diagnosis is difficult to make as the age of onset and the clinical presentation are very variable. The final diagnosis is determined with electromyography, clinical suspicion, muscle biopsy, immunohistochemistry, and genetics. Based on its genetic origin, it is classified into limb-girdle MD1 with autosomal dominant transmission and MD2 with autosomal recessive transmission. It is also classified based on the proteins involved, such as myotilin, lamina A/C, caveolin 3, calpain, dysferlin, sarcoglycans, inter alia.24 Type 1 dystrophy exhibits cardiac manifestations such as malignant arrhythmias that require the implantation of cardiac defibrillators or pacemakers, and type 2, in which 50% of the cases develop dilated cardiomyopathy.25
Emery-Dreifus muscular dystrophy
Characterized by a genetic mutation affecting the production of a 34-kDa protein in the A/C lamina, which is part of the nuclear membrane.26-28 The condition presents with joint contractures, particularly the Achilles tendon, the elbow, and the spine; subsequently, there is neck and lower back involvement, leading to spinal stiffness. In addition, there are cardiac conduction alterations and dilated cardiomyopathy, although the extent of the cardiac lesions does not correlate with the extent of the muscular lesion.29 Flexure contractures develop around 4 to 5 years of age and gait may be preserved assisted with braces.26,27,30,31 Life expectancy is approximately 46 years and 12% of the patients die due to heart failure, whereas 46% present with sudden cardiac death.31
Although it has been reported that the in vitro muscle contracture test is negative for the risk of malignant hyperthermia, there are some publications in favor of the use of safe techniques such as total intravenous anesthesia (TIVA).32 Difficulties for intubation and for regional epidural and spinal anesthesia have been reported in these patients.
Merosin-deficient congenital muscular dystrophy
Merosin-deficient congenital muscular dystrophy belongs to the group of congenital muscular dystrophies and is the result of a merosin protein defect. Merosin protein surrounds the muscle fibers. The condition is characterized by progressive, severe muscle weakness with hypotony since birth, multiple contractures, deglutition disorders, scoliosis, severe restrictive lung disease, cardiac involvement ranging from branch blocks to dilated cardiomyopathy, in addition to elevated creatinkinase.2,33,34
The most salient clinical characteristics of muscular dystrophies are described in Table 3.35-40
Table 3 Clinical characteristics of muscular dystrophies
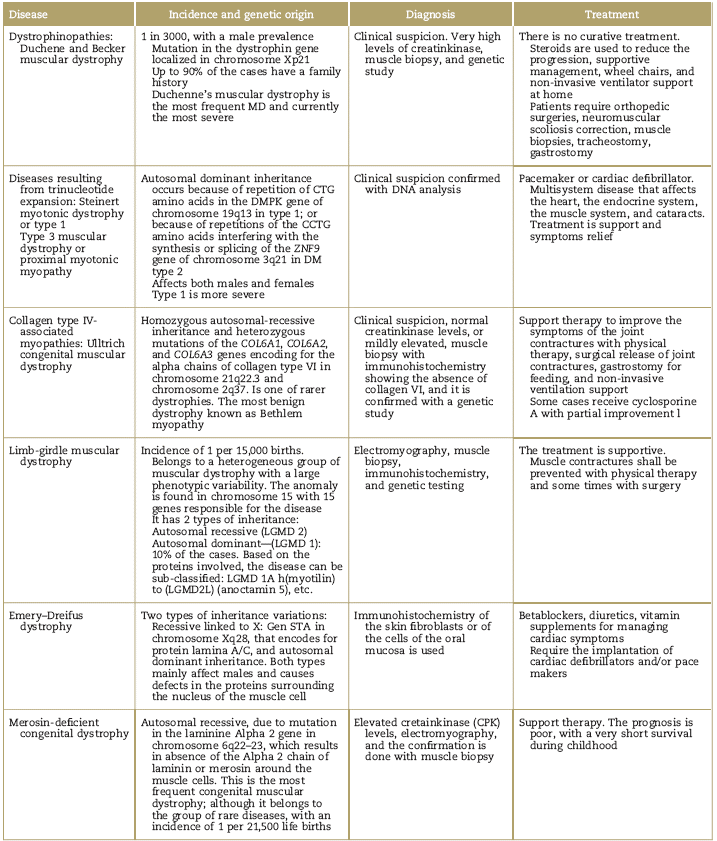
CBC=Complete Blood Count; CCTG=cytidine, cytidine, guanine, thymine; CPK=Creatin Phosphokinase; CTG=cytidine, thymine, guanine; DM=dystrophy myotonic; DMPK=dystrophia myotonia protein kinase; LGMD=Limb-Girdle Muscular Dystrophy; STA=Emerin was identified as a small gene: the STA gene in chromosome Xq28; VA=ventricular atrium; MD=merosin-deficient.
Source: Authors.
Anesthetic implications of muscular dystrophies
Pre-anesthetic evaluation
Patients with muscular dystrophy usually require orthopedic surgeries early in life, scoliosis correction, muscle biopsies, tendon release, or tendon transfers.3,41 In case of dystrophies affecting the heart, such as Steinert myotonic dystrophy, Emery-Dreifus, and limb-girdle, the patients will require anesthesia for implanting cardio deibrillators and pacemakers1,30,31,36,42 because of the high risk of sudden death caused by ventricular fibrillation, ventricular tachycardia, or complete heart block26; in advanced stages, patients require tracheostomies, gastrostomies, and therapeutic or diagnostic procedures such as computarized tomography-scans and magnetic resonance image.2
The recommendation is a comprehensive evaluation of the patient's functional condition, with emphasis on the most affected organs based on the type of dystrophy. The difficult airway criteria should be assessed as these are frequently present in this type of patients, and any pressure ulcers due to chronic positions should be ruled out.
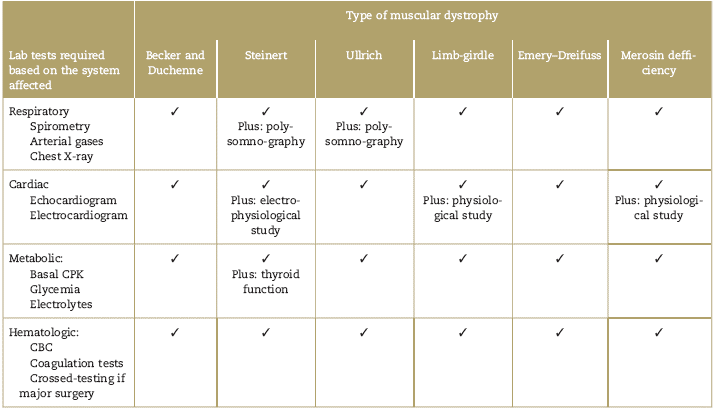
Table 4 describes the pre-surgical laboratory examinations and complementary tests that should be ordered to patients with muscular dystrophy undergoing surgery. The most frequent recommended tests are creatinkinase levels, Complete Blood Count (CBC) coagulation tests, echocardiogram, EKG, and lung function tests.
Table 4 Laboratory examinations and complementary tests recommended for the pre-anesthetic evaluation of patients with muscular dystrophies programed for surgery
Source: Authors.
The forced vital capacity (FVC) and the forced expiratory volume in both supine decubitus and seating position should be evaluated. If FVC is below 50% of the expected level, the patient is at high risk of requiring postoperative non-invasive ventilation.17,19,20,38,39 Occasionally, there may be a need to do additional tests to assess the diaphragmatic function; a dysfunctional diaphragmatic function requires non-invasive mechanical ventilation support before surgery.
The severity and high perioperative risk criteria are as follows:
PEP: peak expiratory pressure, LVEF: left ventricular ejection fraction.39
Due to the incidence of malignant arrhythmia and sudden death in these patients, it is recommended to conduct electrophysiological studies, in particular for limb-girdle dystrophies, Steinert disease and Emery Dreifuss.12
Anesthetic management
The anesthetic management of patients with muscular dystrophies is particularly difficult because of the risk of rhabdomyolysis leading to hyperpotassemia and cardiac arrest, malignant arrhythmias, increased muscle weakness, airway management problems, and exacerbation of the respiratory failure. It is important to stress that the anesthetic risk depends on the type of surgery, the clinical condition of the individual patient, and the progression of the disease; the following recommendations are general but sound clinical judgment should prevail for each particular patient.
Complete monitoring of these patients will require EKG, non-invasive blood pressure, capnography, pulse oxime-try, temperature, diuresis, ventilation parameters, and neuromuscular relaxation.
The administration of glucose solutions is recommended to lower the risk of rhabdomyolysis and acute hyperpotassemia43; extended fasting shall be avoided and consideration of bronchoaspiration prophylaxis with H2 antagonists, metoclopramide, and sodium citrate shall be considered.44-46
TIVA is beneficial for these patients.3 Propofol is the most commonly used hypnotic for the induction and maintenance of anesthesia in patients with Duchenne muscular dystrophy.47 If the heart is affected, other options may be used, including etomidate and thiopental which have been successfully used in patients with myotonic dystrophies.46 Volatile anesthetic agents are not used in most case reports, as these may trigger myotony and crisis of rhabdomyolysis; nitrous oxide is the only inhaled agent used in around 34% to 78% of these reports.3,48 However, the use of sevofluorane has been reported for anesthetic induction in patients with muscular dystrophy with no associated complications. The reported events of rhabdomyolysis associated with exposure to inhaled agents have 2 situations in common: first, the onset of the adverse event presents in the recovery area when patients are moved, and second, it is more frequent in children less than 8 years old, in whom the disease is less severe, but the muscular cell membrane is more instable and vulnerable to the development of rhabdomyolysis, even with normal muscular activity.45,48,49 For this reason, some authors conclude that the evidence is not strong enough to associate inhaled agents with adverse events such as rhabdomyolysis and that association is multifactorial, but there are safer anesthetic techniques.48,49 In myotonic dystrophy, halogenated agents are not contraindicated, but may increase the muscular weakness.1,12,50 Based on the author's experience, the recommendation is to use inhaled induction to facilitate catheterization of an intravenous access and immediately change to TIVA or regional anesthesia-associated sedation. This strategy shortens the time of exposure to anesthetics which theoretically lowers the probability of muscular contractures and rhabdomyolysis.
Opioids such as remifentanil and fentanyl have proven to be safe in these patients.1
Low-dose ketamine and midazolam have been reported as safe options for sedation-analgesia associated with regional techniques (spinal or epidural) in these patients.47,51,52 It should be highlighted that the onset of action of non-depolarizing muscle relaxants is faster, their behavior is erratic, with longer duration and a high risk of residual paralysis; hence, monitoring of the neuromuscular function is critical.9,53
The most frequently used non-depolarizing muscle relaxant is rocuronium,47,48 which is able to neutralize its pharmacological action using a specific reversal agent. Although the use of sugamadex in patients with neuro-muscular disorders has not been fully established and its role in patients with dystrophies and dilated cardiomyopathy is not totally clear; sugamadex use has been reported in patients with dystrophies and dilated cardiomyopathy,10 at a dose of 4mg/kg (dose described for adults) with 100% recovery of the neuromuscular function, and minimal adverse cardiovascular events.54 In contrast, neostigmine may trigger acute myotony, rhabdomyolysis, malignant arrhythmias, and heart failure.10,54 The risk of malignant hyperthermia is similar to that of the general population.55
Regional caudal or epidural spinal anesthesia may be reliably and successfully used in these patients, although cases of myotonic contractures have been described in incomplete neuraxial block, and technically, it can be more challenging because of the spinal abnormalities.3,42,55-58
Patients programed for major surgery should receive anti-fibrinolytic therapy with tranexamic acid and blood-sparing techniques, as well as warming strategies with hot fluids and electric blankets to prevent hypothermia that may lead to myoclonic contractures and increased bleeding.47,58 These patients require complete monitoring, including non-invasive blood pressure monitoring, capnography, cardioscope, pulse oximeter, airway pressure, temperature, urinary catheter, central venous catheter, and arterial line.3,59 It should be highlighted that fluid therapy in these patients should be done with potassium-free crystalloids, and there are no absolute contraindications to the use of colloids.60
Special warnings for the anesthetic management of patients with muscular dystrophies
Patients with muscular dystrophy have an absolute contraindication to receive depolarizing muscle relaxants such as succinylcholine and care should be exercised with the use of inhaled anesthetics that may trigger a rhabdomyolysis crisis.8,47,55 The Food and Drug Administration in 1992 issued an alert to avoid succinylcholine in patients with a history of myopathies or unclear congenital hypotony. Inhaled anesthetics may be used with care in children with dystrophies and the pre-surgical potassium and creatine kinase levels must be monitored, in addition to cardiac function.9
Postoperative management and perioperative complications
Patients with muscular dystrophies must have an anesthetic recovery in intensive monitoring units, particularly in symptomatic patients following major abdominal surgery or when the muscle dysfunction is severe.57 Good analgesia is a must as it reduces any respiratory complications. However, keep in mind that these patients are more sensitive to the effects of opiates (systemic and neuraxial) and therefore present a higher risk of respiratory depression, exacerbated gastrointestinal paresis, and increased risk of reflux, aspiration, and ventilation dysfunction. In case of respiratory depression following reversal of the neuromuscular relaxation, consider deferring the extubation 24 to 48 hours, or consider the use of non-invasive mechanical ventilation.55
Non-steroidal anti-inflammatory drugs should be used with care as these may also trigger crises of rhabdomyolysis.43 The use of intravenous or rectal paracetamol has been reported,61,62 regional epidural anesthesia, and ultrasound-guided plexus nerve block, as well as para-vertebral blocks for thoracic procedures.61,63,64
Patients with muscular dystrophy have a high risk of apnea and death following extubation, over the next 24 hours after surgery.55
In the initial stages of the disease with mild involvement and preserved gait, sedation and analgesia with low-dose benzodiazepines and opioids may be used; however, in patients in advanced stages and with compromised respiratory function, the use of opioids and sedatives during the postoperative period should be avoided.3,45 If required, use them with care and keep the patient under strict surveillance in the intensive care unit.11,64
For extubation, the recommendation is to do it with the patient awake, and if non-depolarizing muscle relaxants need to be used, complete reversal must be ensured to avoid residual relaxation.4,10,44,64
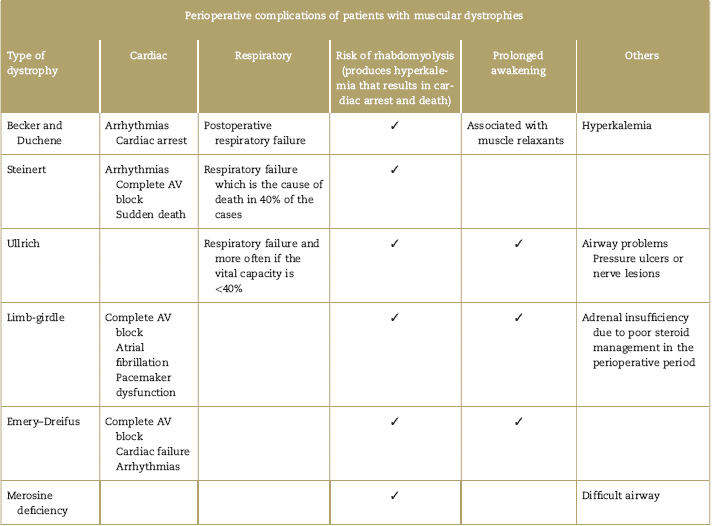
Regardless of the anesthetic agent used, patients with dystrophies have a high risk of complications including respiratory failure, rhabdomyolysis, arrhythmias, cardiac arrest, and reactions similar to malignant hyperthermia that need acute symptomatic treatment, but fail to resolve with dantrolene.44,46,55,63,64 Patients with dystrophies may be classified based on the perioperative risk, in accordance with MIRS (muscular impairment rating scale); the risk is intermediate in grade MIRS 3, and very high in grades 4 and 5 (Table 5).65 The surgical risk also increases in major surgery and in patients who were administered muscle relaxant. Table 6 describes the most frequent perioperative complications for each muscular dystrophy.
Table 5 MIRS (muscular impairment rating scale) to classify muscular involvement in muscular dystrophy65

Source: Authors.
Limitations
The purpose of this article is to do a review of the anesthetic implications in the most frequent dystrophies, based on a systematic literature review. However, due to the qualityof the evidence-which is based on case reports, case series, and subject reviews-the information is presented as a descriptive review. Further studies are needed to contribute with a higher level of evidence, although probably this may be difficult due to the low frequency of this type of pathology, to show the safest anesthetic strategies in patients with muscular dystrophies. Up to now, we just have the shared experience of the authors and of the institutions devoted to care for patients with rare diseases; it must be highlighted that most of the available studies are case series in pediatric patients.
Conclusion
Muscular dystrophies are a group of congenital, heterogeneous diseases, with a different genetic origin and with widely variable clinical manifestations. All of them have significant anesthetic implications that represent a very high perioperative risk for patients, and require accurate knowledge to anticipate and provide specific management in each case. The main complications of muscular dystrophies are ventilator failure, malignant cardiac arrhythmias, hyperkalemia, rhabdomyolisis, cardiovascular instability, and sudden death. Although these diseases are apparently unrelated to malignant hyperthermia, they do share the need to avoid the use of depolarizing muscle relaxants and prefer the use of intravenous anesthetic techniques or regional anesthesia to reduce the risk of these complications, that could be associated with the use inhaled anesthetics. A careful pre-surgical evaluation is recommended to identify the general condition of each patient, the surgical risk, and to adopt a multidisciplinary approach that ensures the best perioperative care and reduces the complications associated with the disease and the management of anesthesia. It is mandatory to assess the risk/ benefit of any surgical and anesthetic procedure in patients with muscular dystrophy, before an intervention.

