










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.24 no.2 Bogotá June 2004
Paquimeningitis craneal hipertrófica idiopática
Jairo Lizarazo 1,2, Edgar Parra 3, Patricia Gutiérrez 1, Carlos Mora 1, Juan Vargas 1
1Hospital Universitario Erasmo Meoz, Cúcuta, Norte de Santander, Colombia.
2
Universidad de Pamplona, Pamplona, Norte de Santander, Colombia.3
Grupo de Patología, Instituto Nacional de Salud, Bogotá, D.C., Colombia.La paquimeningitis craneal hipertrófica idiopática es un raro proceso inflamatorio crónico de etiología desconocida que produce engrosamiento de la duramadre y alteraciones neurológicas progresivas debido a la compresión de estructuras vecinas. Se presenta el caso de una mujer adulta con un cuadro clínico crónico de cefalea, fiebre, pérdida progresiva de la visión y neuropatía óptica bilateral. El diagnóstico se basó en la visualización de la duramadre engrosada en las neuroimágenes y en la exclusión de causas conocidas mediante estudios histopatológicos. La terapia con corticoesteroides mejoró la cefalea pero no el déficit visual. El diagnóstico y el seguimiento de esta entidad se han facilitado con la utilización de la resonancia magnética cerebral con medio de contraste. La biopsia de duramadre sigue siendo la regla de oro para el diagnóstico definitivo de la enfermedad. La terapia con esteroides produce mejoría en un buen número de pacientes; sin embargo, son frecuentes las recaídas por lo que es necesario el uso concomitante de otro inmunosupresor como la ciclofosfamida o la azatioprina. La mortalidad es baja, pero son comunes las secuelas neurológicas definitivas.
Palabras clave: paquimeningitis, neuropatía óptica, cefalea, meningitis, esteroides.
Idiopathic hypertrophic cranial pachymeningitis
Idiopathic hypertrophic cranial pachymeningitis is an infrequent chronic inflammatory process of unknown etiology which causes thickening of the dura mater and progressive neurologic alterations due to the compression of adjacent structures. A case is presented of an adult woman with a clinical syndrome consisting of headache, progressive visual loss and bilateral optic neuropathy. The diagnosis was based upon visualization of the thickened dura mater in neuroimaging studies and the exclusion of known causes by histopathological examination. Diagnosis and follow-up of this condition are currently easier with the use of nuclear magnetic resonance with contrast medium. Biopsy of the dura mater continues to be the gold standard for the definitive diagnosis of this disease. Steroid therapy causes clinical improvement in most of the patients; however, relapses are frequent, making necessary the concomitant use of other immunosuppressive agents such as cyclophosphamide or azathioprine. Mortality is low but definitive neurologic sequelae are common.
Key words: pachymeningitis, optic neuropathy, headache, meningitis, steroids.
La paquimeningitis hipertrófica idiopática es un proceso inflamatorio crónico fibrosante de etiología desconocida infrecuente que causa engrosamiento de la duramadre y que produce déficit neurológico progresivo debido a la compresión de las estructuras adyacentes (1). De acuerdo con la localización anatómica y en orden de frecuencia, los casos de paquimeningitis hipertrófica se pueden subdividir en espinal, craneal y craneoespinal (2).
La primera descripción de paquimeningitis hipertrófica, en este caso de la médula espinal y probablemente luética, fue realizada por Charcot y Joffroy en 1869 (3). Naffziger y Stern describieron en 1949 el primer caso de paquimeningitis hipertrófica craneal idiopática (3). La paquimeningitis hipertrófica ha sido frecuentemente atribuida a etiologías específicas como la tuberculosis y la sífilis (3,4). Sin embargo, los casos más recientes parecen ser idiopáticos ya que una intensa búsqueda para definir la etiología fue negativa (1,5-7).
En este artículo presentamos los hallazgos clínicos, imaginológicos e histopatológicos de un caso de paquimeningitis craneal hipertrófica idiopática en una mujer adulta con un cuadro clínico crónico de cefalea, fiebre, pérdida progresiva de la visión y neuropatía óptica.
Caso clínico
Se trata de una paciente de sexo femenino de 51 años de edad, natural del área rural de Aguachica (Cesar) y procedente de El Carmen (Norte de Santander) quien consultó por primera vez al Hospital Universitario Erasmo Meoz de Cúcuta el 05/02/2004.
Presentaba un cuadro clínico de 6 meses de evolución, caracterizado por fiebre casi diaria que cedía con la ingestión de antipiréticos y cefalea global no pulsátil acompañada de fotofobia. En el examen físico de ingreso se encontró una paciente pálida, con signos vitales normales y como único hallazgo positivo se anotó un moderado espasmo muscular de la nuca; la evaluación neurológica fue normal. Como antecedentes se informó una rinosinusitis alérgica crónica y una hospitalización 5 años atrás en el hospital de Ocaña por un proceso febril y poliartralgias de etiología sin aclarar. La paciente fue sometida a estudio y se encontró una anemia microcítica e hipocrómica; la glucemia y las pruebas de función renal fueron normales; hubo leve hipoalbuminemia con pruebas normales de la función hepática; el examen parcial de orina fue normal aunque en el urocultivo se aisló Klebsiella sp. (80.000 unidades formadoras de colonias), sensible al trimetropimsulfametoxasol. La prueba de VDRL no fue reactiva.
La radiografía del tórax y la ecografía abdominal fueron normales. La radiografía simple de senos paranasales reportó una pansinusitis mientras que la tomografía computarizada de cráneo fue informada como normal; sin embargo, un análisis posterior demostró marcada hiperdensidad y engrosamiento de la hoz del cerebro, especialmente en su parte posterior (
figura 1a).
Las pruebas de función tiroidea estaban dentro de los límites normales; los antígenos febriles fueron negativos al igual que los hemocultivos; se observaron quistes de Entamoeba coli en el examen coprológico y de las heces se cultivó Klebsiella sp. La biopsia de médula ósea fue informada como normal. El 14/02/2004 se le dio salida y se le formuló trimetropim-sulfametoxasol.
Durante la hospitalización presentó varios picos febriles. En las semanas siguientes, la fiebre desapareció totalmente pero persistió la cefalea, la cual se hizo más intensa y se acompañó de pérdida progresiva de la agudeza visual por ambos ojos, motivo por el cual consultó nuevamente al hospital de Cúcuta el 29/04/2004 y fue internada para estudio.
En el examen físico se encontró una paciente desnutrida (40 kg) con signos vitales normales, sin alteraciones en el examen general; en el examen neurológico se encontró una paciente alerta, diestra, orientada globalmente, sin alteraciones mentales y con lenguaje fluente normal.
No percibía la luz por el ojo derecho y por el ojo izquierdo solo percibía los movimientos de las manos a 1 m. En el fondo de ojo se detectó edema de los discos ópticos con áreas de atrofia en el ojo derecho. Los movimientos oculares eran completos. La sensibilidad facial y la audición se conservaban normales y los pares bajos no mostraban alteraciones. No tenía signos meníngeos. La exploración motora y sensitiva de los cuatro miembros fue normal; los reflejos miotáticos se valoraron como normales y simétricos (++); la marcha estaba limitada por el déficit visual y era de características normales. La paciente controlaba los esfínteres.
Los exámenes paraclínicos se consignan en el
cuadro 1; los hallazgos positivos fueron una anemia microcítica e hipocrómica, una sinusitis maxilar bilateral y aumento de las proteínas en el LCR.
La tomografía computarizada de cráneo simple, al igual que la practicada anteriormente, evidenció engrosamiento e hiperdensidad de la parte posterior de la hoz del cerebro (
figura 1b). En las imágenes ponderadas, la resonancia magnética de cerebro mostró en el T1 un engrosamiento lineal de la duramadre, especialmente de la hoz del cerebro y de la tienda del cerebelo con una señal ligeramente hipointensa en relación con el parénquima cerebral (figura 2a). En el T2, la señal era hipointensa y se acompañaba de un delgado borde de hiperintensidad (figura 2b). Luego de la aplicación del medio de contraste paramagnético, se apreció una captación acentuada y uniforme del medio de contraste por la parte posterior de la duramadre. Representativas de esta alteración fueron la parte posterior de la hoz del cerebro y la tienda del cerebelo (figura 2c, 2d y 2e). Los nervios ópticos se notaron engrosados (no se muestran en las figuras).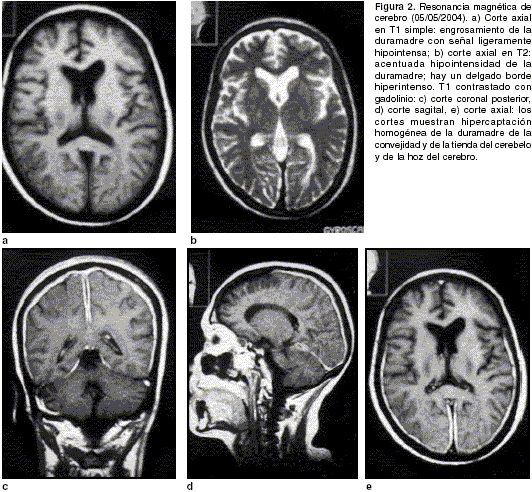
La paciente fue llevada a cirugía el 10/05/2004 y se le practicó craneotomía parietal derecha para la obtención de una biopsia de la duramadre. En el acto operatorio se encontró una duramadre muy engrosada (8 mm) de color amarillento y ricamente vascularizada. Las leptomeninges y el cerebro subyacentes parecían normales. El estudio histopatológico correspondiente mostró una meninges con importante proliferación fibroconjuntival, difusa, con algunas áreas semejantes a queloide y se observaron, además, focos de infiltrado inflamatorio linfocitario. Con las tinciones de PAS, Ziehl-Nielsen y Wartin-Starry no se observaron microorganismos(
figuras 3a,3b 3c). Tampoco se identificó tumor alguno. Estos hallazgos morfológicos corresponden a una paquimeningitis hipertrófica.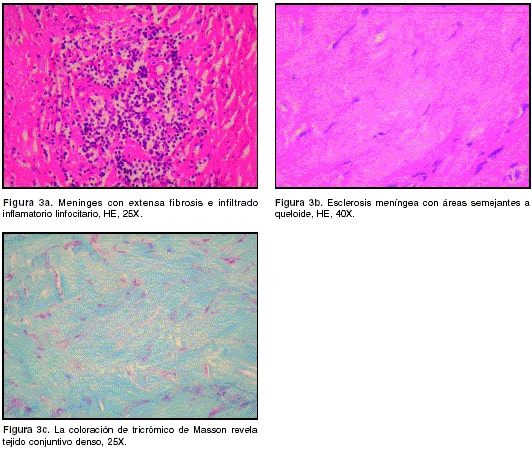
Se inició tratamiento con esteroides, inicialmente dexametasona, 8 mg intravenosos cada 6 horas y, luego, prednisona oral, 40 mg diarios (1 mg/kg de peso por día). La cefalea desapareció pero el déficit visual no cambió. La paciente fue dada de alta y en el último control realizado (26/05/2004) refirió estar sin cefalea; en el fondo de ojo se notó disminución del edema de la papila con evolución hacia la atrofia óptica en el ojo derecho y sin cambios en la agudeza visual. Se decidió continuar con el mismo tratamiento y hacer control de resonancia magnética contrastada de cerebro en 3 meses.
Discusión
La paquimeningitis craneal hipertrófica idiopática es una entidad de rara ocurrencia. Parney et al. encontraron en la literatura informes sobre 33 casos hasta 1996 en Medline (3). Al utilizar esta misma base de datos, identificamos 38 casos adicionales hasta abril de 2004 (1,5-9). La entidad afecta especialmente a adultos mayores, generalmente después de los 50 años, con un ligero predominio del sexo masculino (1,5,7). La evolución del cuadro clínico es usualmente crónica, en algunos casos de años (8). El síntoma de presentación más frecuente es la cefalea, observada en casi todos los pacientes y que puede ser la única manifestación clínica; no obstante, el patrón es indefinido (orbital, frontotemporal o global) y se describe como no pulsátil (8). Siguen, en orden de frecuencia, la pérdida de la visión debida a neuropatía óptica y la parálisis de los nervios oculomotores (III, IV y VI) (5). El déficit visual puede ir es desde leve alteración de la agudeza visual hasta ceguera total; algunos pacientes tienen papiledema asociado con hipertensión intracraneana o con aumento de las proteínas en el LCR (5). Otros cumplen los criterios del síndrome de Tolosa-Hunt debido a que presentan dolor ocular y parálisis de los nervios oculomotores (1,5,7).
Prácticamente todos los pares craneanos pueden verse lesionados pero existen dos patrones de parálisis de los nervios craneanos que reflejan los dos principales sitios de afección: por un lado, la fisura orbitaria superior y la región del seno carvernoso (pares craneanos III, IV, VI y VI) y por el otro, las regiones petrosa y tentorial (pares craneanos V, VII, VIII, IX y X) (7). Con cierta frecuencia los enfermos presentan fiebre y son raros los signos meníngeos. Con menor frecuencia, se presenta disfunción del sistema nervioso central manifestada como convulsiones, ataxia, temblor, psicosis, alteración de la conciencia, ceguera cortical o déficit motor (1,5). Otra manifestación clínica que se ha descrito como asociada es la diabetes insípida por afección del eje hipotálamo-hipófisis posterior (7). Por el compromiso de las estructuras adyacentes, también se puede observar encefalitis, hidrocefalia, trombosis de senos venosos e hipopituitarismo (5). La paquimeningitis espinal hipertrófica idiopática compromete especialmente las regiones cervical y torácica y se manifiesta como mielorradiculopatía (2).
En la mayoría de los pacientes el estudio del LCR muestra una presión de apertura normal, elevación de las proteínas y, en algunos casos, aumento moderado de las células a expensas de los linfocitos y que no se correlaciona con la gravedad de la enfermedad (1,5). En una minoría de casos el LCR es normal. En los exámenes de laboratorio es frecuente observar el aumento de la velocidad de la sedimentación globular y de la proteína C reactiva (6). Recientemente, y especialmente en pacientes japoneses, se han descrito títulos positivos para anticuerpos anticitoplasma del neutrófilo tipo perinuclear (p-ANCA, por su sigla en inglés) (1,9,10). Estos p-ANCA se pueden detectar en pacientes que presentan una variedad de enfermedades como: glomerulonefritis idiopática de medias lunas, síndrome de Churg-Strauss, poliarteritis nodosa, colitis ulcerativa, hepatitis activa crónica, colangitis esclerosante primaria y otras enfermedades autoinmunes; también, con el uso del propiltiouracilo, un medicamento usado en el tratamiento del hipertiroidismo. Existe otro tipo de ANCA, el citoplásmico (c-ANCA) que es un marcador muy específico para la granulomatosis de Wegener (9).
Las imágenes diagnósticas son pilares en el diagnóstico de esta entidad. La tomografía computarizada revela lesiones durales hiperdensas con un patrón de realce homogéneo con el medio de contraste (1). Sin embargo, la resonancia magnética es el método más importante para identificar las lesiones y para excluir tumores u otro tipo de patología. Las paquimeninges hipertróficas se muestran iso o hipointensas con relación al parénquima nervioso en la imágenes ponderadas en T1 y acentuadamente hipointensas en las ponderadas en T2. Luego de la administración del gadolinio, la duramadre se realza intensamente con un patrón lineal, nodular o mixto. A veces, como en nuestro caso, se nota un delgado borde de señal alta en las imágenes ponderadas en el T2 (
figura 2b). En algunos, se aprecia un patrón de realce intenso de las márgenes periféricas de la dura engrosada, sin realce de las lesiones más internas; esta última característica se correlaciona con la mayor actividad inflamatoria documentada en la periferia de la lesión.En la mayoría de las series, las regiones posteriores son las más afectadas y se destacan la mitad posterior de la hoz del cerebro y la tienda del cerebelo (1,5). La resonancia magnética también es de ayuda en el seguimiento de los pacientes; con la disminución de la inflamación como consecuencia del tratamiento, el realce anormal dural disminuye o desaparece. De igual manera, permite detectar la reactivación de la enfermedad al presentarse, nuevamente, los hallazgos característicos (1,5-7). Recientemente, se ha llamado la atención acerca de la utilidad de la tomografía computarizada por emisión de fotón único (SPECT, por su sigla en inglés) con talio 201; este trazador se acumula significativamente en la duramadre de los pacientes con paquimeningitis hipertrófica (11). De forma similar, la tomografía por emisión de positrones (PET) es útil en el seguimiento de los pacientes (12).
El estudio histopatológico muestra una dura engrosada que, al microscopio, se observa compuesta por tejido fibroso denso y por infiltración celular de linfocitos y, en menor cantidad, por células plasmáticas y macrófagos. Los estudios inmunohistoquímicos identifican a los linfocitos como células B o T y no son monoclonales. Lo característico de esta entidad son los cambios inflamatorios inespecíficos de la duramadre. Ocasionalmente, se ha reportado vasculitis necrosante de pequeñas arterias o cambios granulomatosos. En los casos de autopsias no se ha encontrado vasculitis en ningún otro órgano (1,5).
El diagnóstico diferencial es amplio con todas las demás patologías que producen engrosamiento de la duramadre (
cuadro 2) (1,3-7,13-22). Por ello, el diagnóstico definitivo de paquimeningitis hipertrófica idiopática es por exclusión pero se deben hacer cultivos, tinciones especiales y demás estudios paraclínicos que permitan descartar razonablemente patologías infecciosas, inflamatorias o neoplásicas. En los países en desarrollo, la tuberculosis y la sífilis merecen especial consideración. En Colombia, Toro et al. describieron 3 casos de paquimeningitis hipertrófica luética en una revisión de 8.500 autopsias realizadas en el Hospital San Juan de Dios de Bogotá durante casi dos décadas (4). Se ha sugerido que una prueba terapéutica antituberculosa pudiera estar indicada en aquellos pacientes con paquimeningitis hipertrófica idiopática con una prueba de PPD positiva o con una historia de contacto tuberculoso (3).
La patogénesis de la paquimeningitis hipertrófica idiopática no está clara. Su estrecha relación con enfermedades del tejido conjuntivo como la granulomatosis de Wegener, la artritis reumatoidea o la enfermedad mixta del tejido conjuntivo y su reciente asociación con los p-ANCA en Japón, sugieren que pudiera tratarse de un síndrome vasculítico o de una enfermedad del tejido conjuntivo. Algunos han postulado que se trata de una inflamación sistémica autoinmune que se localiza en la duramadre (1).
Los esteroides han sido la terapia más ampliamente usada; algunos responden dramáticamente a dosis altas administradas en formas de pulsos de metilprednisolona y obtienen remisiones duraderas; otros requieren tratamientos prolongados con prednisona a dosis de 1 mg/kg al día debido a recaídas por suspensión del medicamento. Otra opción terapéutica ha sido la combinación con otros inmunosupresores como la ciclofosfamida en forma de ciclos o la administración continua de azatioprina o de metotrexato. La cirugía descompresiva se reserva para aquellas formas localizadas que afectan, por ejemplo, la fisura orbitaria superior. Algún paciente ocasional puede requerir una derivación del LCR para manejo de la hidrocefalia asociada. La radioterapia no ha mostrado ninguna utilidad (1,5,7). Por supuesto que todos los tratamientos descritos han sido anecdóticos y no existe ningún estudio controlado que avale su eficacia. La mortalidad por esta patología es baja pero las secuelas son frecuentes, especialmente las de tipo neurooftalmológico (1,5,7).
n conclusión, aunque la paquimeningitis hipertrófica idiopática es una entidad rara, sus hallazgos clínicos e imaginológicos son muy característicos. La cefalea, la neuropatía craneal múltiple y el realce de la duramadre en la resonancia magnética contrastada de cerebro llevan fácilmente al diagnóstico. La biopsia de duramadre es necesaria para su confirmación y para la exclusión de otras causas de paquimeningitis hipertrófica. Los esteroides a altas dosis son el tratamiento de elección y cuando la respuesta es pobre se puede adicionar otro inmunosupresor como la ciclofosfamida, la azatioprina o el metotrexato. Sin embargo, la patogénesis de este trastorno sigue siendo oscura y la respuesta final al tratamiento, impredecible.
Agradecimientos
A Gabriel Toro por el análisis patológico de la biopsia. A Elizabeth Castañeda y a Yeni Peña por su gran colaboración.
Correspondencia:
Jairo Lizarazo, Calle 13 Nº 1E-44 Consultorio 404B, Cúcuta, Norte de Santander, Colombia.
Teléfonos: 5722635 - 5722652 Telefax: 5835161
Correo electrónico:
jflizar@telecom.com.coRecibido: 28/05/04; aceptado: 31/05/04
Referencias
1. Riku S, Kato S. Idiopathic hypertrophic pachymeningitis. Neuropathology 2003;23:335-44. [ Links ]
2. Dumont AS, Clark AW, Sevick RJ, Myles ST. Idiopathic hypertrophic pachymeningitis: a report of two patients and review of the literature. Can J Neurol Sci 2000 ;27:333-40. [ Links ]
3. Parney IF, Johnson ES, Allen PBR. "Idiopathic" cranial hypertrophic pachymeningitis responsive to antituberculous therapy: case report. Neurosurgery 1997; 41:965-71. [ Links ]
4. Toro G, Vergara I, Moya C. Paquimeningitis hipertrófica luética. Rev Latinoam Patol 1971;10:113-28. [ Links ]
5. Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ. Idiopathic hypertrophic pachymeningitis. Neurology 2004;62:686-94. [ Links ]
6. Sylaja PN, Cherian PJ, Das CK, Radhakrishnan VV, Radhakrishnan K. Idiopathic hypertrophic cranial pachymeningitis. Neurol India 2002;50:53-9. [ Links ]
7. Hatano N, Behari S, Nagatani T, Kimura M, Ooka K,Saito K, et al. Idiopathic hypetrophic cranial pachymeningitis: clinicoradiological spectrum and therapeutic options. Neurosurgery 1999;45:1336-44. [ Links ]
8. Wouda EJ, Vanneste JAL. A specific headache during 13 years as the only symptom of idiopathic hypertrophic pachymeningitis. J Neurol Neurosurg Psychiatry 1998; 64:408-9. [ Links ]
9. Takuma H, Shimada H, Inoue Y, Ishimura E, Himuto K, Miki T, et al. Hypertrophic pachymeningitis with antineutrophil cytoplasmic antibody (p-ANCA), and diabetes insipidus. Acta Neurol Scand 2001;104:397-401. [ Links ]
10. Takahashi K, Kobayashi S, Okada K, Yamaguchi S. Pachymeningitis with a perinuclear antineutrophil cytoplasmic antibody: response to pulse steroid. Neurology 1998;50:1190-1. [ Links ]
11. Ohta H, Takeuchi H, Suzuki T, Shintaku M. Tl-201 uptake in chronic hypertrophic pachymeningitis. Clin Nuclear Medicine 2003:28:252-3. [ Links ]
12. Voller B, Vass K, Wanschitz J, Machold K, Asenbaum S, Hoberstorfer M, et al. Hypertrophic chronic pachymeningitis as a localized immune process in the craniocervical region. Neurology 2001;56:107-9. [ Links ]
13. Bosch J, Ortega-Aznar A, Tintoré M, Río J, Ferreira R, Rubio E, et al. Paquimeningitis hipertrófica. Revisión histórica a propósito de dos casos y relación patogénica con el síndrome de Tolosa-Hunt y el pseudotumor orbitario. Rev Neurol 2000;31:946-51. [ Links ]
14. Kawano Y, Kira J. Chronic hypertrophic cranial pachymeningitis associated with HTLV-1 infection. J Neurol Neurosurg Psychiatry 1995;59:435-7. [ Links ]
15. Matsumoto K, Natori Y, Hirokawa E, Iwaki T. Hypetrophic pachymeningitis as a result of a retropharyngeal inflammatory pseudotumor: case report. Neurosurgery 2002;51:1061-5. [ Links ]
16. de Deus-Silva L, Queiroz L de S, Zanardi V de A, Ghizoni E, Pereira H da C, Malveira GLS, et al. Hypertrophic pachymeningitis. Arq Neuropsiquiatr 2003; 61:107-11. [ Links ]
17. Li J, Lai P, Lam H, Lu L, Cheng H, Lee J, et al. Hypertrophic cranial pachymeningitis and lymphocytic hypophysitis in Sjögren's syndrome. Neurology 1999; 52:420-3. [ Links ]
18. Fam AG, Lavine E, Lee L, Perez-Ordoñez B, Goyal M. Cranial pachymeningitis: an unusual manifestation of Wegener's granulomatosis. J Rheumatol 2003;30: 2070-4. [ Links ]
19. Case 2-1998. N Eng J Med 1998;338:180-8. [ Links ]
20. Case 9-1999. N Eng J Med 1999:340:945-53. [ Links ]
21. Caso 8-2001. N Eng J Med 2001;344:832-9. [ Links ]
22. Holodny AI, Kirsch CF, Hameed M, Sclar G. Tumefactive fibroinflammatory lesion of the neck with progressive invasion of the meninges, skull base, orbit and brain. Am J Neuroradiol 2001;22:876-9. [ Links ]

