










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.24 suppl.1 Bogotá June 2004
Polimorfismo del TNF - a en autoinmunidad y tuberculosis
Paula A. Correa, Luis M. Gómez, Juan Manuel Anaya
Unidad de Biología Celular e Inmunogenética, Corporación para Investigaciones Biológicas,
Medellín, Colombia
El factor de necrosis tumoral alfa (TNF-
a ) está incriminado tanto en enfermedades autoinmunes como en infecciosas. En el presente estudio se examinó el polimorfismo de la región promotora -308 del gen del TNF- a en enfermedades autoinmunes; [ lupus eritematoso sistémico (LES), artritis reumatoidea (AR), síndrome de Sjögren primario (SSp)] ; y en tuberculosis.La genotipificación del polimorfismo -308 del TNF-
a se realizó en ADN de pacientes con AR (N=165), LES (N=118), SSp (N=67), tuberculosis (N=138) y controles sanos (N=419), mediante reacción en cadena de la polimerasa con polimorfismos en los tamaños de los fragmentos de restricción (PCR-RFLP).El alelo TNF2 se asoció con la AR (OR=1,6; IC95% 1,2-2,3, p=0,008), el LES (OR=2,3; IC95% 1,6-3,3, p<0,0001) y el SSp (OR=2,7; IC95% 1,7-4,1, p<0,0001). El alelo TNF1 se asoció con la tuberculosis (OR=1,9: IC95% 1,2-3,1, p=0,02). La heterocigosis TNF1/TNF2 fue factor de riesgo para AR (OR=1,7; IC95% 1,2-2,6, p=0,01), LES (OR=3; IC95% 2-4,7, p<0,0001) y SSp (OR=3,8; IC95% 2,2-6,5, p<0,0001), mientras que la homocigosis TNF1/TNF1 fue protectora para autoinmunidad (OR<0,6, p<0,01). Por el contrario, el genotipo TNF1/TNF2 fue protector para tuberculosis (OR=0,5; IC95% 0,3-0,9, p=0,02) y la homocigosis TNF1/TNF1 se asoció con susceptibilidad a la misma (OR=2; IC95% 1,2-3,4, p=0,02).
Los resultados indican que el alelo TNF2 es un factor común de riesgo para enfermedades autoinmunes reumatológicas pero protector para tuberculosis. Esto sugiere una selección genética en nuestra población.
Palabras clave: factor de necrosis tumoral, polimorfismo, artritis reumatoidea, lupus eritematoso sistémico, síndrome de Sjögren, autoinmunidad, tuberculosis.
Polymorphism of TNF -
a in autoimmunity and tuberculosisTumor necrosis factor alpha (TNF-
a ) has been incriminated in several autoimmune and infectious diseases. The influence of TNF-a -308 polymorphism was examined in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), primary Sjögren's syndrome (pSS) and tuberculosis. Genomic DNA from patients with RA (N=165), SLE (N=118), pSS (N=67), tuberculosis (N=138), as well as ethnic-matched controls (N=419) were characterized for the TNF-a -308 genetic polymorphism using the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) method. TNF2 allele was associated with RA (OR: 1.6, C.I.95% 1.2-2.3, p=0.008), SLE (OR: 2.3, 95%C.I. 1.6-3.3, p<0.0001), and pSS (OR: 2.7, 95%C.I. 1.7-4.1, p<0.0001). TNF1 was associated with tuberculosis (OR: 1.9, 95%C.I. 1.2-3.1, p=0.02). TNF1/TNF2 heterozygosity was associated with susceptibility for RA (OR: 1.7, 95%C.I. 1.2-2.6, p=0.01), SLE (OR: 3, 95%C.I. 2-4.7, p<0.0001), and pSS (OR: 3.8, 95%C.I. 2.2-6.5, p<0.0001). The homozygous state TNF1/TNF1 was protective for autoimmunity (OR<0.6, p<0.01). In contrast, the TNF1/TNF2 genotype was a protective factor for tuberculosis (OR 0.5, 95%C.I. 0.3-0.9, p= 0.02) whereas TNF1/TNF1 homozygosity was associated with susceptibility (OR: 2, 95%C.I. 1.2-3.4, p=0.02). These results indicate that TNF2 is a common susceptibility allele for autoimmune rheumatic diseases and a protective one for tuberculosis. In addition, the data point towards a genetic selection in our population that might be maintained through dominant selection (heterozygote advantage) to infection by M. tuberculosis but susceptible to autoimmunity.Key words: tumor necrosis factor, polymorphism, rheumatoid arthritis, systemic lupus erythematosus, Sjögren's syndrome, autoimmunity, tuberculosis.
La artritis reumatoidea (AR), el lupus eritematoso sistémico (LES) y el síndrome de Sjögren (SS), son enfermedades autoinmunes, inflamatorias y sistémicas de etiología desconocida y cuya fisiopatología es compleja. En ellas participan muchas proteínas de importancia inmunológica que incluyen las citocinas que son sintetizadas y reguladas a partir de genes individuales. El polimorfismo de estos últimos contribuye al carácter poligénico de estas enfermedades (1,2). Entre las citocinas incriminadas en la fisio-patología de estas enfermedades autoinmunes sobresalen aquéllas con propiedades pro-inflamatorias, una de las cuales es el factor de necrosis tumoral alfa (TNF-a ), proteína de 17 kd y una longitud de 157 aminoácidos (3). Es secretada por macrófagos, monocitos, neutrófilos, linfocitos T, principalmente CD4+, y también por células asesinas naturales (4). Su secreción es inhibida por la interleucina 6, el factor de crecimiento y transformación beta, la vitamina D3, la prostaglandina E2 y la dexametasona, entre otros. Actúa reclutando neutrófilos y monocitos en sitios de inflamación, a los que activa para que eliminen agentes microbianos. El TNF-a también estimula las células endoteliales para que expresen moléculas de adhesión que facilitan el tráfico leucocitario y, a su vez, estimula la secreción de quimiocinas e induce apoptosis en la célula blanco (5). Cuando esta citocina es sintetizada en grandes cantidades produce efectos sistémicos nocivos que incluyen inducción de fiebre, caquexia y síntesis de proteínas de fase aguda; también, puede causar trombosis intravascular y choque (3,4). En los modelos animales de algunas patologías, como la AR, se ha podido observar que la expresión aumentada o persistente del TNF-a puede, por sí sola, inducir artritis (6).
El gen que codifica para el TNF-
a está ubicado en el brazo corto del cromososma 6 (6p21.31), inmerso en el complejo mayor de histocompatibilidad (CMH). Este gen es regulado a nivel transcripcional y postranscripcional. Ciertas variaciones puntuales en la región promotora de este gen pueden favorecer un cambio en la síntesis de proteína, fenómeno que se ha asociado con patología autoinmune y enfermedades infecciosas (7-14).El polimorfismo en un solo nucleótido (single nucleotide polymorphism, SNP), en el que se sustituye una guanina por una adenina (G®A) en la posición _308, genera los alelos conocidos como TNF1 (G) y TNF2 (A). La síntesis del TNF-
a puede variar en función de este polimorfismo. El TNF2 se ha asociado con una transcripción significativamente aumentada de la citocina (13). Se ha encontrado, en diferentes estudios, que el alelo TNF2 puede estar asociado con varias patologías autoinmunes, incluso la AR y el LES (9,13).Las características moleculares y propiedades patogénicas del TNF-
a en enfermedades crónicas e inflamatorias como la AR promovieron el desarrollo de terapias biológicas anti- TNF- a que resultaron eficaces (15). No obstante, se ha observado que tienen también efectos adversos como son el riesgo de reactivación o el desarrollo de tuberculosis (16).La tuberculosis es una enfermedad granulomatosa crónica, causada por Mycobacterium tuberculosis. En su patogénesis intervienen factores tanto del medio ambiente, como sociales y genéticos. Aproximadamente, la tercera parte de la población mundial inmunocompetente está infectada con M. tuberculosis; se estima que el 10% de esta población desarrollará la enfermedad (17). La infección por M. tuberculosis ocurre por vía aérea y el primer contacto con el hospedero se lleva a cabo en el macrófago alveolar que, luego de fagocitar la micobacteria, sintetiza citocinas proinflamatorias como el TNF-
a (18), el cual juega un papel clave no sólo en la respuesta del hospedero contra M. tuberculosis sino también en las manifestaciones clínicas de la enfermedad. Esta citocina interviene en la formación, la organización y el control del granuloma y, en sinergismo con el interferón gamma, induce la expresión de la óxido nítrico sintasa inducible, lo cual genera la producción de óxido nítrico, uno de los principales agentes microbicidas para micobacterias (18,19). Además, el TNF- a puede afectar la migración celular, la localización tisular de la micobacteria y la expresión de moléculas de adhesión, de quimiocinas y sus respectivos receptores (19). La expresión del TNF- a en la tuberculosis, así como en otras patologías de origen infeccioso, puede variar de un individuo a otro y también entre poblaciones. Estas variaciones pueden ser explicadas, en parte, por polimorfismos en la región promotora del gen (13). La influencia de factores genéticos en el desarrollo de la tuberculosis es sustentada por el alto grado de concordancia de la enfermedad en gemelos univitelinos (20).Dado que la producción del TNF-
a es variable entre individuos y que esta variación puede ser explicada por factores genéticos encargados de regular su síntesis y que, además, el polimorfismo del gen correspondiente se puede asociar con algunas enfermedades autoinmunes e infecciosas, el presente trabajo evaluó simultáneamente la asociación del SNP _308 del TNF- a en pacientes con enfermedades autoinmunes reumatológicas y tuberculosis.Materiales y métodos
Pacientes y controles
Se realizó la genotipificación del SNP -308 del TNF-
a en 350 individuos con enfermedades autoinmunes reumatológicas, todos los cuales habían cumplido con los criterios internacionales de clasificación correspondientes a cada enfermedad y distribuidos así: 165 pacientes con AR (21), 118 con LES (22) y 67 pacientes con SS primario (23).También se incluyeron 138 pacientes con tuberculosis pulmonar, negativos para infección por el virus de inmunodeficiencia humano (VIH) y 419 individuos sanos (controles), pertenecientes al mismo grupo étnico y geográfico de los pacientes pero sin relación con ellos, exentos de enfermedad autoinmune o infecciosa. El diagnóstico de tuberculosis se confirmó por presencia de bacilos ácido alcohol resistentes en esputos o por aislamiento de M. tuberculosis en
cultivo. La serología para el VIH fue negativa en todos los casos.
Los pacientes se atendieron en la Clínica Universitaria Bolivariana y en el Hospital La María de Medellín. Esta investigación se llevó a cabo de acuerdo con la Resolución No 008430 de 1993 del Ministerio de Salud de la República de Colombia y se consideró como de riesgo mínimo. El estudio contó con la aprobación del comité de ética de la Corporación para Investigaciones Biológicas.
Extracción de ADN
Se extrajo de cada uno de los individuos, pacientes y controles, el ADN genómico de células mononucleares de sangre periférica obtenida por punción venosa, según el método de precipitación con sales (salting out), previamente descrito (24).
Polimorfismo del TNF-
aSe realizó la genotipificación del SNP _308 por el método de reacción en cadena de la polimerasa (PCR). La reacción se llevó a cabo en un volumen final de 50 ul, los cuales contenían 100 ng de ADN genómico, 10 pmol de cada uno de los iniciadores específicos para la posición _308, tanto sentido (5'AGGCAATAGGTTTTGAGGGCC AT3') como antisentido (5'TCCTCCCTGCTCCGATTCCG 3' ) (25), 1,5 mM de Mg2Cl y 200 uM de dNTPs (Promega, Madison, WI, EU); se utilizó 1 unidad de Taq ADN polimerasa (Promega, Madison, WI, EU) con amplificación de anidaje a 60°C, durante 35 ciclos, en un termociclador MJ PTC 200 (Promega, Madison, WI, EU). El producto esperado de la PCR correspondía a 107 pb y se analizó en un gel de agarosa al 2% (SeaKem LE, BMA, Rockland, ME, EU), teñido con bromuro de etidio (Sigma, St. Louis, MI, EU).
El análisis final del SNP _308 se evaluó por la técnica de polimorfismo en el tamaño de fragmentos de restricción (RFLP). Para este análisis se utilizó la endonucleasa NcoI (Promega, Madison, WI, EU), la cual genera cortes específicos en el producto de la amplificación de la PCR, los que, luego de la restricción, permiten discriminar los alelos según el patrón de bandas que fuera originado: 87 pb y 20 pb para el TNF1 (alelo nativo o TNFG) y 107 pb para el alelo TNF2 (alelo mutado o TNFA) (25), el cual no presenta la secuencia específica para que la endonucleasa pueda fragmentar el ADN.
Análisis estadístico
El diseño del presente estudio asumió la presencia de una probabilidad de error del 0,05, con un poder de detección del 90%. Se asumió, igualmente, una correlación del 10% o menos entre el caso y su control. La relación caso:control fue superior a 1:2,5, y el mínimo riesgo relativo indirecto previsto para marcadores genéticos en los pacientes fue de 3. Por lo tanto, el tamaño de la muestra fue suficiente para observar diferencias significativas. El presente trabajo cumplió con las guías para el desarrollo de estudios de asociación genética (26,27).
Tanto en pacientes como en controles se determinaron las frecuencias alélicas y génicas. Las comparaciones entre frecuencias fueron realizadas utilizando la prueba de c2. Se calcularon la razón de disparidad (OR) y los intervalos de confianza (IC) de 95% (28). En todos los casos, el grado de significación fue inferior al 5%. Dado que no se hicieron comparaciones múltiples, no fue necesario corregir el valor de la significancia.
Resultados
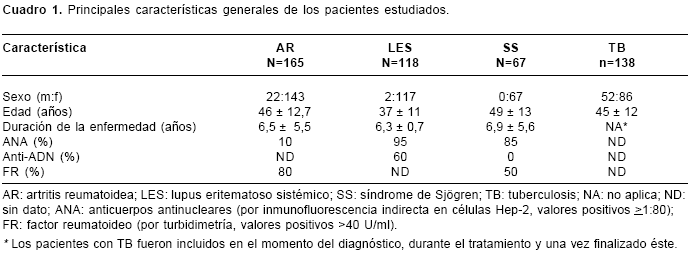
Las características generales de los pacientes se resumen en el cuadro 1.
En el
cuadro 2 se muestran los resultados de las frecuencias alélicas y genotípicas. El alelo TNF2 se asoció con la AR (OR=1,6; IC95% 1,2-2,3, p=0,008), el LES (OR=2,3; IC95% 1,6-3,3, p<0,0001) y el SS (OR=2,7, IC95% 1,7-4,1, p<0,0001). El alelo TNF1 se asoció con la tuberculosis (OR=1,9; IC95% 1,2-3,1, p=0,02). La heterocigosis TNF1/TNF2 (GA) fue factor de riesgo para AR (OR=1,7; IC95% 1,2-2,6, p=0,01), para LES (OR=3; IC95% 2-4,7, p<0,0001) y para el SS (OR=3,8; IC95% 2,2-6,5, p<0,0001), mientras que la homocigosis TNF1/TNF1 (GG) fue protectora para AR (OR=0,6; IC95% 0,4-0,8, p=0,007), LES (OR=0,33; IC95% 0,2-0,5, p<0,0001) y SS (OR=0,3; IC95% 0,2-0,4, p<0,0001). Por el contrario, el genotipo TNF1/TNF2 (GA) fue protector para tuberculosis (OR=0,5; IC95% 0,3-0,9, p=0,02), mientras que la homocigosis TNF1/TNF1 (GG) se asoció con susceptibilidad a la misma enfermedad (OR=2; IC95% 1,2-3,4, p=0,02).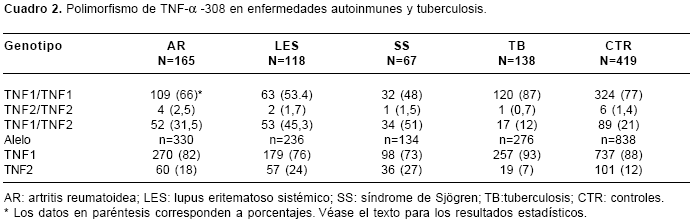
La comparación entre el grupo de enfermedades autoinmunes y la tuberculosis puso en evidencia que la asociación entre TNF2 u autoinmunidad era más fuerte que cuando se comparaban los grupos de autoinmunidadcon el grupo control (OR>0,3, p<0,0001). Así mismo, el papel protector de TNF1 para autoinmunidad fue más fuerte cuando se comparó con el grupo de tuberculosis (OR<0,3, p<0,0001).
En el grupo control se observó una heterocigosis del 21% (
cuadro 2), muy similar a aquella observada en población caucásica (29), sin que se detectaran, en función del sexo, diferencias significativas en alelos y genotipos (p=0,7).Discusión
En el presente estudio se examinó simultánea-mente el polimorfismo _308 del TNF-
a en un número importante de pacientes con enfermedades autoinmunes, tuberculosis e individuos clínicamente sanos, provenientes de un mismo grupo de población. Tres aspectos importantes pueden ser discutidos a la luz de estos resultados. En primer lugar, muestran una asociación opuesta del polimorfismo _308 del TNF- a con autoinmunidad y tuberculosis, pues mientras el TNF2 resultó ser un alelo común de susceptibilidad para autoinmunidad, y protector para tuberculosis, el alelo TNF1 mostró ser un factor de riesgo para tuberculosis. De la misma manera, la heterocigosis TNF1/TNF2 (GA) fue factor de riesgo para autoinmunidad pero protector para tuberculosis. En segundo lugar, los datos sugieren la presentación de una selección genética en nuestra población. Es posible que los altos niveles de polimorfismo se mantengan por selección de equilibrio entre enfermedades, en la cual los alelos que predisponen a una enfermedad, resultan ser protectores para otra. Podría, también, suceder que tal selección se mantenga por dominancia (ventaja de heterocigosis), en la cual los heterocigotos para un determinado alelo son resistentes a una enfermedad (tuberculosis) pero susceptibles a otras (autoinmunidad). En tercer lugar, este estudio, realizado sobre el particular en población colombiana, servirá para futuras comparaciones de frecuencias alélicas y genotípicas, así como para el estudio de la genética de poblaciones (29).La gran diversidad genética, así como la distribución de frecuencias alélicas observadas en la mayoría de los genes polimórficos, están influenciadas y generadas por fuerzas selectivas propias del entorno en el que se desarrollan las especies, tales como agentes infecciosos, supervivencia y dominancia genética, entre otros. Basados en esta última, se ha postulado la teoría de la 'ventaja de heterocigosis', en humanos o el 'vigor híbrido' en plantas y algunas especies animales (30). Esta teoría propone que los individuos heterocigotos son capaces de responder de manera más eficiente a las exigencias del medio, ya que poseen una mayor variedad genética que les permite adaptarse a las condiciones adversas, en particular, al ataque de diversos microorganismos (20,31). Esta teoría parece estar sustentada por la coevolución entre patógenos y hospederos, en la cual existe selección positiva para aquellos genes involucrados en la interacción hospedero-parásito (30,32).
El gen del TNF-a es altamente polimórfico (33). Se han identificado 5 microsátelites denominados TNF a, b, c, d, e, cada uno de los cuales presenta diferentes variables, así: TNFa AC/GT, que origina 13 alelos diferentes; TNFb TC/GA con 7 alelos diferentes; TNFc TC/GA con 2 alelos diferentes; TNFd TC/GA con dos alelos diferentes, y TNFe TC/GA con 4 alelos. Igualmente, se han descrito varias mutaciones puntuales dentro del gen, ubicadas tanto en regiones promotoras como en sitios de transcripción y en regiones no transcritas. De las mutaciones en la región reguladora, el cambio de G®A en el sitio _308 del promotor es considerado uno de los más importantes, dado su efecto en la síntesis de la citocina (13,25).
Un análisis del TNF-
a y las enfermedades autoinmunes reumatológicas revela que existe suficiente evidencia para sustentar que esta citocina es un elemento clave en la patogénesis de la AR (35). La membrana sinovial normal no expresa TNF- a , pero sí la de pacientes con AR. Se ha encontrado expresado en los macrófagos, las células endoteliales y en la interfase pannus-cartílago. Es el principal factor de crecimiento de los fibroblastos, liberado espontáneamente por las células mononucleares sinoviales encontradas en la sinovial reumatoidea. En las articulaciones inflamadas, el TNF-a tiene una variedad de efectos que incluyen la activación de los osteoclastos y la estimulación de la adherencia de los neutrófilos a las células endoteliales (34). Los pacientes con AR que presentan destrucción ósea tienen niveles mayores de TNF- a en el líquido sinovial que los pacientes sin destrucción ósea (35).En nuestra población, observamos que el alelo TNF2 es un factor de predisposición a la AR (OR=1,6). En otras poblaciones, este alelo se ha asociado con la AR tanto como factor de susceptibilidad como de gravedad (36-38). Para evitar asociaciones espurias y múltiples, y tal como ha sido sugerido para este tipo de estudios (26), no se analizaron en el presente estudio las relaciones entre los alelos del TNF-
a y las características clínicas o de laboratorio de los pacientes.En el LES, el papel del TNF-
a es controvertido en la medida que hay discordancia entre los estudios en humanos y los modelos murinos (39,40). No obstante, en pacientes con LES esta citocina pudiera tener más un papel protector que deletéreo (41,42). Cerca de 20% de los pacientes con AR tratados con bloqueadores del TNF- a desarrollan anticuerpos anti-ADN específicos de LES (39,43) y algunos presentan el cuadro clínico de LES (44).El alelo TNF2 se ha asociado con susceptibilidad al LES en poblaciones caucásicas, tanto en desequilibrio de ligamiento con HLA-DR3 (45,46) como independientemente de este locus (47,48). Por el contrario, en población mexicana con alto porcentaje amerindio (49) o en africanos (46) no se ha observado tal asociación. Un estudio de ligamiento en familias de California (Estados Unidos) reveló que el TNF2 no confería susceptibilidad para el LES (50). Nuestros resultados, en pacientes con LES de Medellín, corroboran la asociación del TNF2 con esta enfermedad (OR=2,3). De otra parte, los hallazgos en el grupo control muestran resultados similares a los obtenidos en población caucásica americana (29).
El SS es una exocrinopatía autoinmune caracterizada por la resequedad de las mucosas, principalmente oral y lagrimal (51). El análisis de los infiltrados linfocitarios provenientes de las glándulas salivares de pacientes con SS ha mostrado un predominio de linfocitos T CD4+, seguido de linfocitos T CD8+, linfocitos B y macrófagos (52). El TNF-
a , tanto su ARNm como la proteína, son expresados en forma significativa en canalículos y en células mononucleares del infiltrado glandular de pacientes con SS e induce proteólisis de los acinos glandulares (53-57). Nuestros resultados muestran, por primera vez, que tal como sucede para la AR y el LES, el TNF2 es un alelo de susceptibilidad para el SS primario (OR=2,7).Las enfermedades autoinmunes son entidades frecuentes que se desarrollan en individuos genéticamente susceptibles y cuya expresión clínica es modificada por ambientes permisivos y protectores. Desde el punto de vista genético, estas enfermedades son complejas, es decir, su herencia no es mendeliana y son poligénicas. Nuestros resultados indican que el TNF2 es un alelo común de susceptibilidad para enfermedades autoinmunes como la AR, el LES y el SS, y sustentan la hipótesis de mecanismos inmuno-genéticos similares para estas enfermedades (51,58-60). La herencia de la patología autoinmune, con fenotipos diferentes (enfermedades), puede tener un ancestro genético común para varias de ellas.
En cuanto al TNF-
a y la tuberculosis, se considera que esta citocina es necesaria para el control de la infección aguda por M. tuberculosis (19). En particular, el TNF-a es de mucha importancia en la formación del granuloma en ésta y en otras enfermedades por micobacterias (19). Esto explica, en parte, la reactivación de la tuberculosis en pacientes con AR que han sido tratados con agentes biológicos inhibidores del TNF- a (15,16). Si bien el papel del TNF- a en la tuberculosis es considerado importante, no hay reportes sobre su polimorfismo en poblaciones caucásicas ni en mestizas. En un estudio previo, realizado en India, no se observó asociación entre el SNP -308 y tuberculosis pulmonar (61). En este sentido, nuestros resultados aportan un conocimiento nuevo en cuanto a la asociación del alelo TNF1 y esta enfermedad en nuestra población.Los estudios en curso evalúan el polimorfismo de otros genes inmersos en la región del CMH, con el fin de examinar si la asociación diferencial del SNP _308 del TNF con autoinmunidad y tuberculosis es primaria o secundaria, es decir, si está o no en desequilibrio de ligamiento con otros loci del CMH incriminados en la respuesta inmune. Es también importante considerar en estudios futuros, los niveles de expresión de la citocina con el fin de determinar la relación funcional con su polimorfismo.
Agradecimientos
Los autores expresan su gratitud a Alexandra Flórez, Rosa Hinojosa, Ángela M. Tobón, Lizeth Paniagua, José Cadena, Mauricio Luján y Jaime Robledo por sus aportes en el inicio del presente trabajo y por haber permitido estudiar algunos de sus pacientes, así como a Ángela Restrepo y William Rojas por sus pertinentes comentarios y corrección del manuscrito. También agradecemos la colaboración de los médicos del Hospital La María y la Clínica Universitaria Bolivariana, en donde fueron atendidos la mayoría de los pacientes estudiados. Este trabajo fue financiado por Colciencias (22130412672), la Asociación Colombiana de Reumatología y la Corporación para Investigaciones Biológicas.
Correspondencia:
Juan Manuel Anaya, Carrera 72-A N°78-B-141, Medellín, Colombia.
Teléfono: (4) 441 0855; fax: (4) 441 5514
Recibido: 08/25/03; aceptado: 05/02/04
Referencias
1. Kuchroo VK, Sarvetnick N, Hafler DA, Nicholson LB. Cytokines and autoimmune diseases. En: Kuchroo VK, Sarvetnick N, Hafler DA, Nicholson LB, editors. Cytokines and autoimmune diseases. New Jersey, NJ, Humana Press; 2001. p. 3-422. [ Links ]
2. Jirholt J, Lindqvist AKB, Holmdahl R. The genetics of rheumatoid arthritis and the need for animal models to find and understand the underlying genes. Arthritis Res 2001; 3: 87-97. [ Links ]
3. Aggarwal BB, Kohr WJ, Hass PE, Moffat B, Spencer SA, Henzel WJ, et al. Human tumor necrosis factor. Production, purification, and characterization. J Biol Chem 1985; 260: 2345-54. [ Links ]
4. Oppenheim JJ, Francis W, Ruscetti, Faltynek C. Cytokines. En: Stites DP, Terr AI, Parslow TG, editors. Immunology. Basic and clinical. 8th edition. Norwalk: Appleton & Lance; 1994. p. 105-23. [ Links ]
5. Gupta S. A decision between life and death during TNF-alpha-induced signalling. J Clin Immunol 2002; 22: 185-94. [ Links ]
6. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumor necrosis factor: a predictive genetic model of rheumatoid arthritis. EMBO J 1991; 10: 4025-31. [ Links ]
7. Stuber F, Peterson M, Bokelmann, Schade U. A genomic polymorphism within the TNF locus influences TNF-plasma levels and outcome of patients with severe sepsis. Crit Care Med 1996; 24: 381-4. [ Links ]
8. Roy S, McGuire W, Mascie-Taylor CN, Saha B, Hazra SK, Hill AV, et al. Tumor necrosis factor promoter polymorphism and susceptibility to lepromatous leprosy. J Infect Dis 1997; 17: 530-2. [ Links ]
9. Cornelis LV. Tumor necrosis factor gene polymorphisms as severity markers in rheumatoid arthritis. Ann Rheum Dis 1999; 58 (Suppl. I): 120-6. [ Links ]
10. Kolliaas G, Douni E, Kassiootis G, Kontoyiannis D. The function of tumor necrosis factor and receptors in models of multi-organs inflammation, rheumatoid arthritis, multiple sclerosis and immflamatory bowel disease. Ann Rheum Dis 1999; 58:(Suppl. 1): 132-9. [ Links ]
11. Shapira L, Stabholz A, Rieckmann P, Kruse N. Genetic polymorphism of the tumor necrosis factor (TNF)-alpha promoter region in families with localized early-onset periodontitis. J Periodontal Res 2001; 36: 183-6. [ Links ]
12. Wilson AG, Symons JA, McDowell TL, Duff GW. Effects of polymorphism in the human tumor necrosis alpha promoter with secretion of TNF-alpha and TNF-beta by human mononuclear cells: a possible link to insulin-dependent diabetes mellitus. Eur J Immunol 1993; 23: 224-31. [ Links ]
13. Hajeer AH, Hutchinson IV. Influence of TNFa gene polymorphisms on TNFa production and disease. Hum Immunol 2001; 62: 1191-9. [ Links ]
14. McGuire W, Hill AV, Allospp CE, Greenwood BM, Kwiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 1994; 371: 508-11. [ Links ]
15. Criscione LG, St Clair EW. Tumor necrosis factor-alpha antagonists for the treatment of rheumatic diseases. Curr Opin Rheumatol 2002; 14: 204-11. [ Links ]
16. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with Infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345: 1098-104. [ Links ]
17. World Health Organization. Global tuberculosis Control. WHO
Report 2001, WHO/CDS/TB. Geneva, Switzerland; World Health Organization; 2001. [ Links ]18. van Crevel R, Ottenhoff T, van der Mer J. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev 2002; 15: 294-309. [ Links ]
19. Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol 2001; 19: 93-129. [ Links ]
20. Cooke GS, Hill AV. Genetics of susceptibility to human infectious disease. Nature Rev 2001; 2: 967-77. [ Links ]
21. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988; 31: 315-24. [ Links ]
22. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25: 1271-7. [ Links ]
23. Vitali C, Moutsopoulos HM, Bombardieri S. The European community study group on diagnostic criteria for Sjögren's syndrome. Sensitivity and specificity of test for ocular and oral involvement in Sjögren's syndrome. Ann Rheum Dis 1994; 53: 637-47. [ Links ]
24. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215. [ Links ]
25. Wilson AG, de Vries N, Pociot F, di Giovine FS, Van der Putte LB, Duff GW. An allelic polymorphism within the human tumor necrosis factor alpha promoter region is strongly associated with HLA-A1, B8 and DR3 alleles. J Exp Med 1993; 177: 557-60. [ Links ]
26. Cardon LR, Bell J. Association study designs for complex diseases. Nature Rev 2001; 2: 91-9. [ Links ]
27. Cooper DN, Nussbaum RL, Krawezak M. Proposed guidelines for papers describing DNA polymorphism-disease associations. Hum Genet 2002; 110: 207-8. [ Links ]
28. SPSS, Inc. SPSS 9.05 for Windows Reference Guide. Chicago, IL: SPSS Inc.; 1999. [ Links ]
29. Baena A, Leung JY, Sullivan AD, Landires I, Vasquez-Luna N, Quinones-Berrocal J, et al. TNF-alpha promoter single nucleotide polymorphisms are markers of human ancestry. Genes Immun 2002; 3: 482-7. [ Links ]
30. Hedrick PW. Evolution at HLA: possible explanations for the deficiency of homozygotes in two populations. Hum Hered 1990; 40: 213-30. [ Links ]
31. Carrington M, Nelson GW, Martín MP, Kissner T, Vlahov D, Goedert JJ, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 1999; 283: 1748-52. [ Links ]
32. Schliekelman P, Garner C, Slatkin M. Natural selection and resistance to HIV. Nature 2001; 411: 545-6. [ Links ]
33. Correa P, Anaya JM. Bases moleculares para el estudio del TNFa en la artritis reumatoidea. Rev Colomb Reumatol 2001; 8: 236-50. [ Links ]
34. Choy EHS, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 2001; 344: 907-16. [ Links ]
35. Neidel J, Schulze M, Lindschau J. Association between degree of bone-erosion and synovial fluid-levels of tumor necrosis factor a in the knee-joints of patients with rheumatoid arthritis. Inflamm Res 1995; 44: 217-21. [ Links ]
36. Brikman BM, Huizinga TW, Kurban SS, van der Velde EA, Schreuder GM, Hazes JM, et al. Tumor necrosis factor a gene polymorphisms in rheumatoid arthritis: association with susceptibility to, or severity of, disease? Br J Rheumatol 1997; 36: 516-21. [ Links ]
37. Cvetkovic JT, Wallberg-Jonsson S, Stegmayr B, Rantapaa-Dahlqvist S, Lefvert AK. Susceptibility for and clinical manifestations of rheumatoid arthritis are associated with polymorphisms of the TNF-alpha, IL-1 beta, and IL-1Ra genes. J Rheumatol 2002; 29: 212-9. [ Links ]
38. Waldron-Lynch F, Adams C, Amos C, Zhu DK, McDermott MF, Shanahan F, et al. Tumor necrosis factor 5' promoter single nucleotide polymorphisms influence susceptibility to rheumatoid arthritis (RA) in immunogenetically defined multiplex RA families. Genes Immun 2001; 2: 82-7. [ Links ]
39. Mageed RA, Isenberg DA. Tumour necrosis factor alpha in systemic lupus erythematosus and anti-DNA autoantibody production. Lupus 2002; 11: 850-5. [ Links ]
40. Aringer M, Smolen JS. Complex cytokine effects in a complex autoimmune disease: tumor necrosis factor in systemic lupus erythematosus. Arthritis Res Ther 2003; 5: 172-7. [ Links ]
41. Jones BM, Liu T, Wong RW. Reduced in vitro production of interferon-gamma, interleukin-4 and interleukin-12 and increased production of interleukin-6, interleukin- 10 and tumour necrosis factor-alpha in systemic lupus erythematosus. Weak correlations of cytokine production with disease activity. Autoimmunity 1999; 31: 117-24. [ Links ]
42. Gómez D, Correa PA, Cadena J, Gómez LM, Molina JF, Anaya JM. Th1/Th2 cytokines in systemic lupus erythematosus. Protecting role of tumor necrosis factor alfa. Semin Arthritis Rheum 2004; 33: 404-13. [ Links ]
43. Charles PJ, Smeenk RJ, De Jong J, Feldmann M, Maini RN. Assessment of antibodies to double-stranded DNA induced in rheumatoid arthritis patients following treatment with Infliximab, a monoclonal antibody to tumor necrosis factor alpha: findings in open-label and randomized placebo-controlled trials. Arthritis Rheum 2000; 43: 2383-90. [ Links ]
44. Debant M, Vittecoq O, Descamps V, Le Loet X, Meyer O. Anti-TNF-a-induced systemic lupus syndrome. Clin Rheumatol 2003; 22: 56-61. [ Links ]
45. Wilson AG, Gordon C, di Giovine FS, de Vries N, van de Putte LB, Emery P, et al. A genetic association between systemic lupus erythematosus and tumor necrosis factor alpha. Eur J Immunol 1994; 24: 191-5. [ Links ]
46. Rudwaleit M, Tikly M, Khamashta M, Gibson K, Klinke J, Hughes G, et al. Interethnic differences in the association of tumor necrosis factor promoter polymorphisms with systemic lupus erythematosus. J Rheumatol 1996; 23: 1725-8. [ Links ]
47. Rood MJ, van Krugten MV, Zanelli E, van der Linden MW, Keijsers V, Schreuder GM, et al. TNF-308A and HLA-DR3 alleles contribute independently to susceptibility to systemic lupus erythematosus. Arthritis Rheum 2000; 43: 129-34. [ Links ]
48. van der Linden MW, van der Slik AR, Zanelli E, Giphart MJ, Pieterman E, Schreuder GM, et al. Six microsatellite markers on the short arm of chromosome 6 in relation to HLA-DR3 and TNF-308A in systemic lupus erythematosus. Genes Immun 2001; 2: 373-80. [ Links ]
49. Zuniga J, Vargas-Alarcón G, Hernandez-Pacheco G, Portal-Celhay C, Yamamoto-Furusho JK, Granados J. Tumor necrosis factor-alpha promoter polymorphisms in Mexican patients with systemic lupus erythematosus. Genes Immun 2001; 2: 363-6. [ Links ]
50. Tsuchiya N, Kawasaki A, Tsao BP, Komata T, Grossman JM, Tokunaga K. Analysis of the association of HLA-DRB1, TNF alpha promoter and TNFR2 (TNFRSF1B) polymorphisms with SLE using transmission disequilibrium test. Genes Immun 2001; 2: 317-22. [ Links ]
51. Anaya JM, Talal N. Sjögren's syndrome comes of age. Semin Arthritis Rheum 1999; 28: 355-9. [ Links ]
52. Skopouli FN, Fox PC, Galanopoulou V, Atkinson JC, Jaffe ES, Moutsopoulos HM. T cell subpopulations in the labial minor salivary gland histopathologic lesion of Sjögren's syndrome. J Rheumatol 1991; 18: 210-4. [ Links ]
53. Boumba D, Skopouli FN, Moutsopoulos HM. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjögren's syndrome. Br J Rheumatol 1995; 34: 326-33. [ Links ]
54. Fox RI, Kang HI, Ando D, Abrams J, Pisa E. Cytokine mRNA expression in salivary gland biopsies of Sjogren's syndrome. J Immunol 1994; 152: 5532-9. [ Links ]
55. Oxholm P, Daniels TE, Bendtzen K. Cytokine expression in labial salivary glands from patients with primary Sjogren's syndrome. Autoimmunity 1992; 12: 185-91. [ Links ]
56. Sun D, Emmert-Buck MR, Fox PC. Differential cytokine mRNA expression in human labial minor salivary glands in primary Sjögren's syndrome. Autoimmunity 1998; 28: 125-37. [ Links ]
57. Azuma M, Motegi K, Aota K, Hayashi Y, Sato M. Role of cytokines in the destruction of acinar structure in Sjögren's syndrome salivary glands. Lab Invest 1997; 77: 269-80. [ Links ]
58. Heward J, Gough SCL. Genetic susceptibility to the development of autoimmune disease. Clin Science 1997; 93: 479-9 1. [ Links ]
59. Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, et al. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci USA 1998; 95: 9979-84. [ Links ]
60. Myerscough A, John S, Barrett JH, Ollier WER, Worthington J. Linkage of rheumatoid arthritis to insulin-dependent diabetes mellitus loci. Arthritis Rheum 2000; 43: 2771-5. [ Links ]
61. Selvaraj P, Sriram U, Mathan Kurian S, Reetha AM, Narayanan PR. Tumor necrosis factor alpha (-238 and -308) and beta gene polymorphisms in pulmonary tuberculosis: haplotype analysis with HLA-A, B and DR genes. Tuberculosis 2001; 81: 335-41
. [ Links ]
