










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.25 no.1 Bogotá Mar. 2005
El virus de la inmunodeficiencia humana tipo 1 y el sistema
nervioso central en desarrollo
Jorge Alejandro Henao, Nora Vanegas, Oscar David Cano, Juan Carlos Hiromi, María Teresa Rugeles
Grupo de Inmunovirología y Neurociencias, Universidad de Antioquia, Medelín, Colombia
.Actualmente, hay más de 42 millones de infectados con el virus de la inmunodeficiencia humana tipo 1 (VIH-1), de los cuales, 3,2 millones son niños y, de éstos, el 90% con transmisión vertical de la infección. Se considera que en Colombia más de 200.000 personas se han infectado desde el inicio de la pandemia y los estudios muestran una tendencia constante al aumento en las cifras de seroprevalencia en mujeres embarazadas y, por ende, en el número de recién nacidos infectados. El VIH-1 es primordialmente linfotrópico, pero su tropismo por el sistema nervioso central (SNC) es bien conocido, lo que genera múltiples alteraciones neurológicas, particularmente prominentes en niños, de las cuales la más prevalerte es la encefalopatía. Clásicamente, se reconocen dos tipos de encefalopatía en esta población: encefalopatía temprana y tardía, ambas con diferentes características clínicas e inmunológicas. La infección por el VIH-1 en el SNC está limitada a los macrófagos, la microglía y los astrocitos en menor escala. Las neuronas, células principalmente afectadas en los pacientes con sida, raras veces son infectadas, por lo que se postula que factores solubles, provenientes tanto del huésped como del virus, son los principales causantes del daño neuronal. Los hallazgos presentados en esta revisión sugieren la posibilidad de que el SNC, en etapas tempranas del desarrollo, sea especialmente susceptible a la infección por el VIH-1. Las cifras epidemiológicas sugieren que este tipo de alteraciones clínicas serán cada vez más frecuentes; de ahí, la importancia de conocer la neuropatogénesis de la infección por el VIH-1.
Palabras clave: encefalitis, VIH-1, neuropatogénesis, neurogénesis, citocinas, embriología.
The human immunodeficiency virus type 1 and the developing central nervous system
Currently, at least 42 million people are infected worldwide with the human immunodeficiency virus type 1 (HIV-1). Of these, 3.2 million are children infected, in 90% of the cases, through vertical transmission. In Colombia, approximately 200,000 persons have been infected since the beginning of the pandemic, with an increasing trend in the seroprevalence among pregnant women. Although HIV-1 is basically lymphotropic, its capacity to invade the central nervous system (CNS) is well known, generating multiple neurological alterations, especially prominent in children, with encephalopathy being the most prevalent. Classically, two types of neurological disorders are recognized in children, consisting of early and late encephalopathies, each with differing clinical and immunological characteristics. HIV-1 infection of the CNS is limited to macrophages, microglia and astrocytes in a restricted manner. In patients with acquired immunodeficiency virus (AIDS), neurons are rarely infected, suggesting that cellular and viral soluble factors, are responsible for the neuronal damage. The conclusion is that the CNS in earlier stages of development is especially susceptible to HIV-1 infection. The epidemiological trends predict that these types of clinical manifestations of HIV-1 will increase in frequency, and increases the necessity for an understanding of the underlying neuropathogenesis.
Keywords: encephalitis, HIV-1, neuropathogenesis, neurogenesis, cytokines, embriology.
Método
Se realizó una búsqueda sistemática en la red de la base de datos de Pubmed y en la página de los Centers for Disease Control and Prevention (CDC). Se restringió la búsqueda a las siguientes palabras clave: "neurogénesis + VIH-1", "neuropatogénesis + VIH-1", "encefalitis + VIH-1" y "VIH-1 + SNC + desarrollo", tanto en inglés como en español. Se seleccionaron trabajos originales y de actualización de las principales revistas médicas nacionales e internacionales, que hacían referencia al tema de la neuropatogénesis de la encefalopatía observada en niños con infección de transmisión vertical por el VIH-1. Lo anterior se restringió a lo publicado en los últimos 20 años. Para cumplir con el objetivo planteado, se hizo énfasis en la revisión de trabajos originales que evaluaran los principales hallazgos clínicos, inmunológicos, imaginológicos y moleculares encontrados en los pacientes pediátricos con encefalopatía asociada al VIH-1. Finalmente, se elaboró la revisión, el resumen y las conclusiones.
Introducción
Según datos del Programa de Naciones Unidas para el VIH/SIDA y la Organización Mundial de la Salud (OMS), para fines del 2002 había 42 millones de personas infectadas con el VIH-1, de los cuales, 3,2 millones eran niños (1). Se estima, además, que en el mundo se infectan anualmente 600.000 niños con el VIH-1 (2). Según las estadísticas del Ministerio de Protección Social, se calcula que en Colombia se han presentado en los últimos 16 años, aproximadamente, 267.000 casos de infección por el VIH-1. Además, un estudio reciente reporta que del 0,1% al 0,7% de las mujeres embarazadas en las grandes ciudades de Colombia son positivas para VIH-1 (1,3,4).
En Colombia, los porcentajes de seropositividad encontrados entre 1991 y 1999 en mujeres embarazadas mostraron una tendencia constante al aumento; si a esto se le suma la ausencia de políticas estatales claras en el campo de la prevención de la transmisión vertical, es de esperar que el número de individuos recién nacidos con infección de transmisión vertical continúe aumentado (2,5). Por esta razón, es de vital importancia que los trabajadores de la salud se familiaricen con la fisiopatología de las diferentes alteraciones clínicas encontradas en esta población infectada, ya que con mayor frecuencia se enfrentarán a este grupo de pacientes en su práctica clínica.
Se sabe que el VIH-1, además de ser un virus linfotrópico, también es neurotrópico, lo cual genera una amplia variedad de alteraciones neurológicas especialmente prominentes en niños (6). Entre las principales manifestaciones clínicas neurológicas observadas en los pacientes pediátricos positivos para VIH-1 están: la encefalopatía, caracterizada por un retardo importante de su desarrollo neurológico, que afecta la adquisición de funciones motoras y cognitivas; además de la neuropatía periférica, la mielopatía y la miopatía, entre otras (6). La encefalopatía es, sin lugar a dudas, la alteración neurológica más grave y prevalente en esta población. Además, la variabilidad en su presentación clínica nos permite inferir una interacción específica importante entre el VIH-1 y el SNC en desarrollo, la cual trae consigo profundas implicaciones en el normal funcionamiento de este sistema en formación.
Encefalopatía por el VIH-1
Manifestaciones clínicas y alteraciones inmunológicas
Según el CDC, la encefalopatía por VIH-1 se diagnostica por la presencia, por lo menos, de una de las siguientes alteraciones progresivas durante dos meses: 1) pérdida o retraso en la obtención de pautas de maduración cognitivas o pérdida de habilidad intelectual; 2) crecimiento cerebral anormal o microcefalia adquirida o atrofia cerebral demostrada por tomografía computarizada (TC) o por resonancia magnética (RM); 3) déficit motor simétrico adquirido, manifestado por paresia, ataxia o alteraciones en la marcha, (7,8). En la población pediátrica con sida, la encefalopatía alcanza cifras de prevalencia del 31% en Francia (9) y del 21% en Estados Unidos (10), países donde la terapia antirretroviral altamente efectiva está al alcance de la mayor parte de la población infectada.
Debido a la alta prevalencia de la encefalopatía, numerosos estudios han evaluado los efectos del VIH-1 en el desarrollo cognitivo y motor de infantes nacidos de madres positivas para VIH-1; se ha encontrado que la progresión es significativamente más lenta en los pacientes positivos para VIH-1, comparada con la de los niños negativos para VIH- 1 nacidos de madres positivas para VIH-1 o con individuos negativos para VIH-1 nacidos de madres no infectadas (9-16). Se ha establecido claramente que esta diferencia no se debe a otros factores de riesgo que hubieran podido influir en el desarrollo neurológico como el abuso de drogas (13), la exposición perinatal a fármacos (16), o el ambiente y cuidados inadecuados (14). De igual manera, se ha evaluado el desarrollo motor y cognitivo en pacientes tratados con diferentes esquemas antirretrovirales, y se encontró una menor prevalencia de alteraciones neurológicas en el grupo de pacientes en cuyo esquema terapéutico estaban incluidos fármacos con buena penetración de la barrera hematoencefálica (16).
Clásicamente, se han observado dos patrones diferentes de encefalopatía en infantes transmisión vertical de la infección por el VIH-1. La encefalopatía que se presenta precozmente -durante el primer año de vida- es un cuadro neurológico catastrófico típico, de cuadriparesia espástica, retardo mental y trastorno en la adquisición del lenguaje expresivo. La encefalopatía de inicio tardío que se presenta en niños mayores es semejante a la observada en la fase tardía de la infección por el VIH en adultos; se caracteriza por un cuadro demencial de pérdida de la memoria, inatención, apraxia, irritabilidad, empeoramiento o inicio de una parálisis espástica y alteraciones del lenguaje (17,18).
Tardieu et al. reportaron que mientras la incidencia de encefalopatía en pacientes transmisión vertical de la infección es del 9,9% durante el primer año de vida; para el segundo año, la incidencia disminuye dramáticamente a 4,2%, y en los años siguientes, al 1%. Estos mismos autores reportaron que la encefalopatía temprana es la primera manifestación del sida en el 56% de los infantes menores de un año, en el 32% de los niños mayores de un año y en el 9% de los adultos. Además, se observó que el nivel de inmunocompetencia, con base en el conteo de linfocitos T CD4+, era mayor en el momento del desarrollo de la encefalopatía si ésta ocurría durante el primer año de vida; sin embargo, la supervivencia luego del inicio de la misma fue idéntica en infantes y niños, aunque mucho menor en adultos (17). Otros autores han encontrado que el porcentaje de linfocitos T CD8+ es un marcador que predice el desarrollo de encefalitis, independiente del conteo de linfocitos T CD4+ (19), y que su porcentaje es significativamente menor en los niños que desarrollan encefalopatía, en especial, si se desarrolla tempranamente. Estos hallazgos sugieren que el SNC, en las etapas tempranas de su desarrollo, tiene mayor tendencia a sufrir alteraciones que un SNC más desarrollado o completamente maduro.
Alteraciones imaginológicas y electroencefalográficas
Tanto en la población adulta como en la pediátrica se han llevado a cabo diversos estudios con el objetivo de relacionar los hallazgos clínicos neurológicos con las alteraciones imaginológicas detectadas.
Las alteraciones más frecuentemente encontradas incluyen la atrofia cerebral, las alteraciones de la sustancia blanca y las calcificaciones de los ganglios basales (20-22). Se ha encontrado una asociación importante entre estos hallazgos imaginológicos con cargas virales altas y un estado inmunológico y clínico deficiente (23). La calcificación de los ganglios basales es el hallazgo por excelencia en niños con encefalopatía por VIH- 1, independiente del momento en que se presente la encefalopatía (20).
Se ha encontrado una mayor frecuencia y gravedad de atrofia cerebral en los pacientes pediátricos positivos para VIH-1 con encefalopatía de inicio temprano, quienes presentan simultáneamente un retardo significativo de la mielinización encefálica (21).
Además, se ha encontrado una correlación importante entre el déficit del lenguaje expresivo y las anormalidades mayores en la TC de niños positivos para VIH-1 (24-27).
Algunas pruebas de seguimiento menos costosas, como el electroencefalograma (EEG), han demostrado ser herramientas importantes para la evaluación de las alteraciones neurológicas en adultos infectados por el VIH-1 (28-30); sin embargo, hasta el momento se han realizado pocos estudios en niños. El estudio más importante en población pediátrica con VIH-1 es el realizado por Vigiliano et al., en el cual se encontró una relación importante entre la progresión de la enfermedad y la aparición de alteraciones en el EEG, y la prevalencia llegó a ser del 47,7% en los pacientes clasificados en la categoría C del sida (según la clasificación del CDC) y del 77% en infantes ya diagnosticados con encefalopatía (31). Aunque otros estudios han mostrado resultados semejantes (32-34), hasta el momento no se ha investigado la posible relación entre el desarrollo neurológico de los niños infectados con la aparición de patrones patológicos específicos en el EEG. Teniendo en cuenta que el EEG es una técnica poco invasiva, es fundamental que se adelanten estudios que permitan encontrar este tipo de asociaciones, y mejorar el pronóstico de los niños transmisión vertical de la infección o de los niños no infectados nacidos de madres positivas para el VIH-1.
El sistema nervioso central en desarrollo y el VIH-1
Durante las tres primeras semanas de vida embrionaria, la división neuroblástica, la multiplicación y la migración neuronal están en pleno desarrollo. Esta última se completa, en gran medida, hacia el final del quinto mes de vida fetal y continúa lentamente hasta la semana cuarenta de gestación. Del quinto al noveno mes tienen lugar cambios sutiles en la organización neuronal de la corteza cerebral, en la cual participan los procesos de sinaptogénesis y de establecimiento de trayectorias axónicas. Alrededor del sexto mes de vida fetal tiene lugar el inicio de la mielinización por parte de los oligodendrocitos; este proceso se termina casi completamente hacia el segundo año después del nacimiento y se puede controlar por medio de la RM (35).
Los monocitos infectados y la migración de viriones libres a través de las células endoteliales de la barrera hematoencefálica son la principal vía de ingreso del VIH-1 al SNC (36,37). Las células blanco de la infección en el cerebro son los macrófagos y la microglía, aunque los astrocitos se infectan de manera restringida (38,39). Las neuronas, células principalmente afectadas en los pacientes con sida, raras veces se infectan (18,40).
Por lo anterior, se ha postulado que factores solubles como las citocinas proinflamatorias, las quimiocinas, los metabolitos neurotóxicos y las proteínas virales provenientes de macrófagos infectados, de la microglía activada o de la periferia son las principales causantes del daño neuronal que da origen a las alteraciones neurológicas del individuo infectado (40,41).
Mediadores inmunológicos y proteínas virales solubles, actores esenciales del daño neuronal
La evidencia acumulada indica que las citocinas secretadas por las células inmunes activadas pueden modular el crecimiento y la función, prácticamente, de todas las células del SNC (42); es más, los estudios del tejido cerebral y de los cultivos neuronales han confirmado que estas citocinas y sus receptores están presentes en el momento adecuado del desarrollo para mediar una acción neurotrófica (43). Sin embargo, ciertos estados patológicos, entre ellos la infección por el VIH-1, generan una producción no regulada de estos factores, lo que produce alteraciones en el desarrollo neurológico. En este mismo sentido, varias investigaciones recientes señalan que mediadores solubles producidos por monocitos/ macrófagos de pacientes infectados con el VIH- 1, alteran el crecimiento y la supervivencia de las neuronas y de las células gliales inmaduras en cultivo (44-46). Se han documentado niveles alterados de IL-1, IL-6, TNF-a y MCP-1 en el líquido cefalorraquídeo (LCR) de pacientes con sida, tanto adultos como pediátricos (47-49). Estas citocinas demostraron ser altamente neurotóxicas en los estudios realizados in vitro, alterando la proliferación y la supervivencia de los oligodendrocitos (50), mediando el daño de la mielina (51) y dañando el tejido neuronal en desarrollo de ratones transgénicos (52).
Diversos estudios sugieren que algunas proteínas virales, como la gp120 y la proteína Tat, secretadas por las células infectadas, juegan un papel primordial en la alteración del desarrollo normal del SNC, probablemente por tres mecanismos diferentes. En primer lugar, por citotoxicidad directa; la gp120 afecta la supervivencia de las neuronas inmaduras y los astrocitos fetales por su interacción directa con el receptor para una aquimiocina, la molécula CXCR4, conocida como uno de los correceptores virales (53). La Tat, por su parte, altera directamente la viabilidad neuronal fetal (54) y las propiedades migratorias de las neuronas inmaduras (55). En segundo lugar, induce la alteración del equilibrio metabólico normal del SNC; la Tat interactúa con las neuronas fetales por medio de la proteína relacionada con el receptor de lipoproteína (PRRLP), lo que induce la acumulación de metabolitos neurotóxicos como la proteína precursora amiloide (PPA), la proteína b amiloide y la ApoE4, de una manera similar a lo observado en pacientes con la enfermedad de Alzheimer (38); además, nuestro laboratorio, en colaboración con la Universidad de Indiana, demostró que la interacción de las proteínas de la cápside viral con el ligando natural del virus en los astrocitos, el receptor de manosa humano (RMh), induce la producción de metaloproteinasa 2 (MMP2) (39), la cual junto con la MMP9 se encuentran aumentadas durante la infección por el VIH-1 (56,57). Esta alteración del equilibrio conduce, probablemente, a un aumento en la permeabilidad de la barrera hematoencefálica, rasgo característico de la infección por VIH-1 (58). Por último, el tercero de estos mecanismos neuropatogénicos probablemente se deba a que la secreción de estas proteínas virales es el paso inicial de un ciclo vicioso neurotóxico, ya que su presencia lleva a la producción incrementada de citocinas inflamatorias (59-75) que, a su vez, llevan a una mayor activación de la microglía (76) y a un aumento en la replicación viral (figura 1) (71,72).
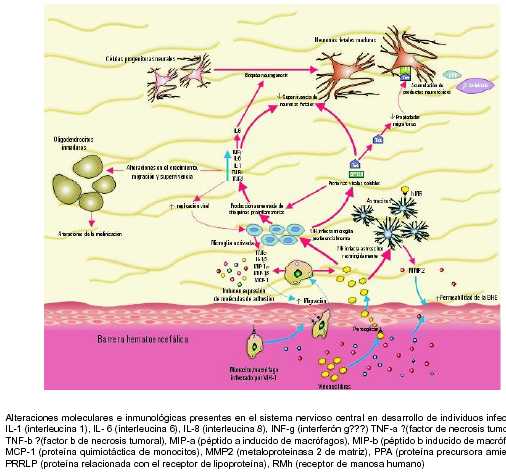
La apoptosis es un evento crucial en el sistema nervioso central en desarrollo; durante este periodo se producen neuronas de forma abundante, la mayoría de las cuales deben ser eliminadas para lograr una correcta sinaptogénesis. Este proceso es dependiente de un balance entre los elementos tróficos neurales, como las citocinas, las quimiocinas y sus receptores, cuya ausencia o exceso genera profundas repercusiones en la formación de las vías axonales (77,78).
El proceso de eliminación de neuronas inmaduras se lleva a cabo por apoptosis, por medio de una secuencia estrictamente ordenada de eventos moleculares, altamente dependiente del balance en la localización celular y mitocondrial de los miembros de la familia de proteínas BCL2 (B cell leukemia). Esta familia de proteínas comprende tres subgrupos, de acuerdo con su función proapoptótica o antiapoptótica, y a la presencia de motivos estructurales (79). De crucial relevancia en las neuronas inmaduras son la proteína antiapoptótica BCL2 y las proteínas proapoptóticas BAX y BIM.
La ausencia de factores tróficos neuronales, activa vías transduccionales que llevan al secuestro de la proteína BCL2, suprimiendo su habilidad de inhibir la translocación de la proteína proapoptótica BAX a la mitocondria. Esta alteración lleva a la formación de poros en la membrana mitocondrial, lo cual permite la salida excesiva de citocromo c al citoplasma y la posterior activación de las caspasas que llevan a la fragmentación de la cromatina (80).
De otra parte, el exceso de factores moduladores del desarrollo neuronal, como el TNFa, induce apoptosis neuronal pobremente controlada. Esta última se desencadena por medio de la activación de la vía extrínseca, en la cual la interacción entre el TNFa y el receptor TNFR1 activa la vía tansduccional de la familia Janus tirosina cinasas lo que, a su vez, activa las caspasas (81,82). El papel que tiene el VIH-1 en la alteración del delicado equilibrio de la apoptosis en el sistema nervioso central en desarrollo está por definirse.
La mielinización, un evento fundamental en el desarrollo del SNC
La mielinización, característica fundamental de los vertebrados, provee una ventaja selectiva, ya que los axones mielinizados pueden propagar los impulsos nerviosos más rápidamente a través del SNC. En los humanos, la mielinización se lleva a cabo durante la fase embrionaria tardía y el periodo posnatal temprano. Las células de Schwann proveen la mielina de los nervios periféricos, mientras que los oligodendrocitos proveen la mielina del SNC; el desarrollo de estas células, a partir de células madres neuronales, y su migración, están estrechamente regulados por factores solubles como las citocinas (83). Lo anterior sugiere que la alteración en el balance de estas últimas, en etapas tempranas del desarrollo, podría generar alteraciones en la mielinización del SNC. Las manifestaciones clínicas observadas en infantes con encefalopatía temprana sugieren una alteración importante del proceso de mielinización, Alteraciones moleculares e inmunológicas presentes en el sistema nervioso central en desarrollo de individuos infectados. IL-1 (interleucina 1), IL- 6 (interleucina 6), IL-8 (interleucina 8), INF-g (interferón g???) TNF-a ?(factor de necrosis tumoral a), TNF-b ?(factor b de necrosis tumoral), MIP-a (péptido a inducido de macrófagos), MIP-b (péptido b inducido de macrófagos), MCP-1 (proteína quimiotáctica de monocitos), MMP2 (metaloproteinasa 2 de matriz), PPA (proteína precursora amieloide), PRRLP (proteína relacionada con el receptor de lipoproteína), RMh (receptor de manosa humano) lo cual se ha comprobado imaginológicamente con la RM (21,25,31).
Los gangliósidos, componentes estructurales de la membrana citoplasmática, poseen igualmente funciones reguladoras del crecimiento y la migración celular (84); además, las alteraciones en su metabolismo generan una disminución importante de la mielinización axonal (85). Recientemente se reportaron niveles significativamente aumentados de productos metabólicos de los gangliósidos en el tejido cerebral y el LCR de individuos con demencia asociada con el VIH-1 (86). Existe, también, evidencia que sugiere la presencia de estados desmielinizantes en individuos infectados por el VIH-1; se ha reportado una asociación significativa entre los niveles elevados de la proteína mielínica básica (PMB) en el LCR y el desarrollo de demencia en individuos infectados (87). La PMB es una de las proteínas más abundantes en el SNC y se le reconoce como un indicador de desmielinización, lo que podría convertirla en un marcador importante en el seguimiento temprano de infantes nacidos de madres positivas para VIH-1.
Neurogénesis y un ambiente proinflamatorio
Hasta hace muy poco, el dogma central en el campo de la neurociencia era el hecho ampliamente aceptado de que la neurogénesis o el desarrollo de nuevas neuronas en el cerebro mamífero estaba limitado a las etapas iniciales del desarrollo y terminaba antes de la pubertad (88). A finales del siglo anterior, hubo evidencia contundente que desvirtuó dicha afirmación, demostrando que el desarrollo de neuronas en la región del hipocampo continúa durante toda la adultez y que este fenómeno no es un vestigio de las etapas tempranas del desarrollo (89,90).
Además, la magnitud de la neurogénesis se correlaciona estrechamente con las funciones del aprendizaje y la memoria del hipocampo (91,92).
La generación de nuevas neuronas a partir de células progenitoras neuronales está estrechamente regulada y es dependiente de un microambiente ideal; su alteración puede llevar a zonas ectópicas de neurogénesis o al bloqueo total de este fenómeno (93,94), lo que lleva a déficit en el aprendizaje y la memoria. La gran activación de la microglía, caracterizada por una amplia secreción de citocinas proinflamatorias, probablemente juega el papel más importante en la alteración de dicho microambiente. La evidencia experimental que soporta este hecho fue reportada por Monje et al.; en su estudio se demostró que las cantidades excesivas de citocinas proinflamatorias bloquean la neurogénesis en la región del hipocampo, pero más importante aún es el hecho de que el bloqueo inflamatorio con indometacina, un antiinflamatorio no esteroideo (AINE), restaura de manera importante la neurogénesis en esta región (94).
La asociación entre la activación de la microglía y la neuroinflamación en diversas entidades neurodegenerativas, entre las cuales se encuentra la demencia asociada al sida, y el hecho de que estos fenómenos tengan una repercusión tan profunda en la neurogénesis señala la importancia de dicho proceso. Por lo anterior, es importante cuestionar el papel que juega este microambiente proinflamatorio, tan común en los recién nacidos de madres positivas para VIH-1, en las etapas tempranas del desarrollo, en las cuales la neurogénesis está en su máxima expresión; y más aún, qué alternativas terapéuticas se podrían ofrecer para recién nacidos positivos para VIH-1, teniendo en cuenta los hallazgos anteriores y el hecho de que la farmacoterapia con AINE reduzca el riesgo o la progresión de la pérdida de memoria (95). Ambos cuestionamientos deben ser objeto de amplia investigación en los próximos años.
¿Podrían los recién nacidos no infectados de madres positivas para VIH-1 desarrollar algún tipo de alteración neurológica?
Es importante resaltar que muchos de los bebés negativos para VIH-1, nacidos de madres positivas para VIH-1, estuvieron en contacto directo con el virus, lo cual es evidente por la presencia de células CD4 y CD8 positivas específicas para el VIH-1 presentes en la gran mayoría de bebés no infectados que fueron expuestos in útero al VIH-1 (96). Sin embargo, las implicaciones de esta exposición intrauterina al VIH-1 en el desarrollo del SNC no han sido suficientemente estudiadas. Por el contrario, las alteraciones en los distintos componentes del sistema inmune han sido claramente identificadas en niños no infectados nacidos de madres positivas para VIH-1. En un estudio realizado en neonatos negativos para VIH- 1, nacidos de madres infectadas y no infectadas con este virus, se encontró que los niños nacidos de madres positivas para VIH-1 tenían un menor porcentaje de células vírgenes CD4+ y CD8+ y el porcentaje de células CD8+ activadas era mayor. Así mismo, se encontró un número elevado de células inmaduras, doblemente negativas, CD25- y CD44-, lo cual sugiere que la maduración fisiológica del timo está alterada en estos bebés. Algunas de estas anormalidades persistieron por varios años (97). En una cohorte similar (neonatos negativos para VIH-1 nacidos de madres infectadas y no infectadas), en la cual se estudió la capacidad de producción de citocinas por parte de los monocitos aislados de sangre de cordón umbilical, se encontró que la capacidad de producir IL-12 por parte de las células presentadoras de antígeno (CPA) de todos los neonatos era menor a la exhibida por monocitos aislados de sangre periférica de adultos sanos. Sin embargo, la producción de IL-12 fue significativamente menor en el grupo de bebés nacidos de madres positivas para VIH-1 (98).
El hecho de que las citocinas puedan modular el crecimiento y la función de prácticamente todas las células del SNC en conjunto con los hallazgos de alteraciones inmunológicas en niños negativos para VIH-1, nacidos de madres positivas para VIH- 1, particularmente de la red de citocinas, hace posible pensar que el ambiente intrauterino de una madre positiva para VIH-1, independiente de que el bebé adquiera o no la infección por transmisión vertical, tiene un efecto negativo en el desarrollo integral del SNC y podría llevar a alteraciones neurológicas importantes en etapas posteriores de la vida.
Reflexiones finales
Las observaciones anteriores sugieren que la forma de encefalopatía que se desarrolla, temprana versus tardía, se puede deber al momento en que se adquiere la infección y a la etapa del desarrollo en que se encuentra el SNC en el feto/infante recién infectado. Esto implicaría que, aunque existen mecanismos fisiopatológicos comunes entre las dos formas de manifestaciones neurológicas, el momento en que se establece la infección es crítico para el desarrollo neurológico y, por ende, para determinar el tipo de alteraciones neurológicas que va a desarrollar el paciente pediátrico. Es lógico pensar que los bebés que se infectan temprano in útero, cuando el SNC se encuentra en su principal etapa de crecimiento y desarrollo, van a presentar más frecuentemente encefalopatía temprana con daños y manifestaciones más graves del SNC que los bebés que se infectan en el último trimestre del embarazo o en el período perinatal. Las alteraciones neurológicas, muy probablemente, sean debidas a una interacción directa entre citocinas, metabolitos neurotóxicos, proteínas virales y las neuronas del SNC en formación, lo que altera el balance metabólico normal de dichas células.
Conflicto de intereses
Los autores manifiestan que no existe ningún conflicto de interés.
Financiación
Esta revisión hace parte del proyecto financiado por Colciencias y la Universidad de Antioquia, código 11150416401.
Correspondencia:
María Teresa Rugeles, Carrera 53 No. 61-30, Torre, piso 2,
Laboratorio 532, Medellín, Colombia.
Teléfono: (574) 210 6481
Recibido: 09/07/04; aceptado: 08/11/04
Referencias
1.UNAIDS/WHO. Epidemiological fact Sheet on HIV/AIDS and Sexually Transmitted Infections.
http://www.who.int/emc-hiv/fact_sheets/All_countries.html2.Watts DH. Management of human immunodeficiency virus infection in pregnancy. N Engl J Med 2002;346:1879-91. [ Links ]
3. UNAIDS/WHO. Colombia: Epidemiological fact Sheet on HIV/AIDS and Sexually Transmitted Infections. 2000.
4. UNAIDS/WHO. HIV and AIDS in the Américas: and epidemic with many faces. 2000:1-56.
5.Garcia R. SIDA: Situación en el mundo y en Colombia veinte años después. Biomedica 2003;23:247-51.
6.Orejon de Luna G, Mateos F, Simon de las Heras R, Martinez Menendez B, Ramos Amador JT, Munoz Gonzalez A. Neurological impairment in children with HIV infection. Rev Neurol 1996;24:278-84. [ Links ]
7.Center Disease Control and Prevention. Revised classification system for Human Immunodeficiency Virus infection in children less than 13 years of age. MMWR 1994;43(RR-12):1-14.
8.The European Collaborative Study. Neurologic signs in young children with human immunodeficiency virus infection. Pediatr Infect Dis J 1990; 9: 402-6. [ Links ]
9.Lobato MN, Caldwell MB, Ng P, Oxtoby MJ. Encephalopathy in children with perinatally acquired human immunodeficiency virus infection. Pediatric Spectrum of Disease Clinical Consortium. J Pediatr 1995;126:710-5. [ Links ]
10.Chase C, Ware J, Hittelman J, Blasini I, Smith R, Llorente A, et al. Early cognitive and motor development among infants born to women infected with human immunodeficiency virus. Women and Infants Transmission Study Group. Pediatrics 2000;106:E25. [ Links ]
11.Bruck I, Tahan TT, Cruz CR, Martins LT, Antoniuk SA, Rodrigues M, et al. Developmental milestones of vertically HIV infected and seroreverters children: follow up of 83 children. Arq Neuropsiquiatr 2001;59:691-5. [ Links ]
12.Macmillan C, Magder LS, Brouwers P, Chase C, Hittelman J, Lasky T, et al. Head growth and neurodevelopment of infants born to HIV-1-infected drug-using women. Neurology 2001;57:1402-11. [ Links ]
13.Gay CL, Armstrong FD, Cohen D, Lai S, Hardy MD, Swales TP, et al. The effects of HIV on cognitive and motor development in children born to HIV-seropositive women with no reported drug use: birth to 24 months. Pediatrics 1995;96:1078-82. [ Links ]
14.Drotar D, Olness K, Wiznitzer M, Guay L, Marum L, Svilar G, et al. Neurodevelopmental outcomes of Ugandan infants with human immunodeficiency virus type 1 infection. Pediatrics 1997;100:E5. [ Links ]
15.Mellins CA, Levenson RL, Jr., Zawadzki R, Kairam R, Weston M. Effects of pediatric HIV infection and prenatal drug exposure on mental and psychomotor development. J Pediatr Psychol 1994;19:617-27. [ Links ]
16.Raskino C, Pearson DA, Baker CJ, Lifschitz MH, O'Donnell K, Mintz M, et al. Neurologic, neurocognitive, and brain growth outcomes in human immunodeficiency virus-infected children receiving different nucleoside antiretroviral regimens. Pediatric AIDS Clinical Trials Group 152 Study Team. Pediatrics 1999;104:e32. [ Links ]
17.Tardieu M, Le Chenadec J, Persoz A, Meyer L, Blanche S, Mayaux MJ. HIV-1-related encephalopathy in infants compared with children and adults. French Pediatric HIV Infection Study and the SEROCO Group. Neurology 2000;54:1089-95. [ Links ]
18.Gendelman HE, Lipton SA, Tardieu M, Bukrinsky MI, Nottet HS. The neuropathogenesis of HIV-1 infection. J Leukoc Biol 1994;56:389-98. [ Links ]
19.Sanchez-Ramon S, Bellon JM, Resino S, Canto-Nogues C, Gurbindo D, Ramos JT, et al. Low blood CD8+ T-lymphocytes and high circulating monocytes are predictors of HIV-1-associated progressive encephalopathy in children. Pediatrics 2003;111:E168-75. [ Links ]
20.Angelini L, Zibordi F, Triulzi F, Cinque P, Giudici B, Pinzani R, et al. Age-dependent neurologic manifestations of HIV infection in childhood. Neurol Sci 2000;21:135-42. [ Links ]
21.Johann-Liang R, Lin K, Cervia J, Stavola J, Noel G. Neuroimaging findings in children perinatally infected with the human immunodeficiency virus. Pediatr Infect Dis J 1998;17:753-4. [ Links ]
22.Davenport C, Dillon WP, Sze G. Neuroradiology of the immunosuppressed state. Radiol Clin North Am 1992;30:611-37. [ Links ]
23.Wolters PL, Brouwers P, Moss HA, Pizzo PA. Differential receptive and expressive language functioning of children with symptomatic HIV disease and relation to CT scan brain abnormalities. Pediatrics 1995;95:112-9. [ Links ]
24.Salvan AM, Lamoureux S, Michel G, Confort-Gouny S, Cozzone PJ, Vion-Dury J. Localized proton magnetic resonance spectroscopy of the brain in children infected with human immunodeficiency virus with and without encephalopathy. Pediatr Res 1998;44:755-62. [ Links ]
25.Depas G, Chiron C, Tardieu M, Nuttin C, Blanche S, Raynaud C, et al. Functional brain imaging in HIV-1-infected children born to seropositive mothers. J Nucl Med 1995;36:2169-74. [ Links ]
26.Tepper VJ, Farley JJ, Rothman MI, Houck DL, Davis KF, Collins-Jones TL, et al. Neurodevelopmental/neuroradiologic recovery of a child infected with HIV after treatment with combination antiretroviral therapy using the HIV-specific protease inhibitor ritonavir. Pediatrics 1998;101:E7. [ Links ]
27.Mitchell W. Neurological and developmental effects of HIV and AIDS in children and adolescents. Ment Retard Dev Disabil Res Rev 2001;7:211-6. [ Links ]
28.Harrison MJ, Newman SP, Hall-Craggs MA, Fowler CJ, Miller R, Kendall BE, et al. Evidence of CNS impairment in HIV infection: clinical, neuropsychological, EEG, and MRI/MRS study. J Neurol Neurosurg Psychiatry 1998;65:301-7. [ Links ]
29.Diehl B, Evers S, Sylvester E, Sprinz A, Husstedt IW. Routine electroencephalogram in follow-up of patients with HIV infections of different stages. A long-term study. Nervenarzt 1998;69:485-9. [ Links ]
30.Gruzelier J, Burgess A, Baldeweg T, Riccio M, Hawkins D, Stygall J, et al. Prospective associations between lateralised brain function and immune status in HIV infection: analysis of EEG, cognition and mood over 30 months. Int J Psychophysiol 1996;23:215-24. [ Links ]
31.Vigliano P, Boffi P, Bonassi E, Gandione M, Marotta C, Raino E, et al. Neurophysiologic exploration: a reliable tool in HIV-1 encephalopathy diagnosis in children. Panminerva Med 2000;42:267-72. [ Links ]
32.Vigliano P, Rigardetto R, Capizzi G, Arfelli P, Barbicinti I, Boffi P, et al. EEG diagnostic and predictive value on HIV infection in childhood. Neurophysiol Clin 1994;24:367-79. [ Links ]
33.Roy S, Geoffroy G, Lapointe N, Michaud J. Neurological findings in HIV-infected children: a review of 49 cases. Can J Neurol Sci 1992;19:453-7. [ Links ]
34.do Prado GF, da Silva AB, Lima JG. Electroencephalogram base rhythm in AIDS patients. Arq Neuropsiquiatr 1993;51:169-74. [ Links ]
35.Victor M, Ropper A. Adam´s Principios de Neurología. México: Mc Graw Hill Interamericana; 2002. [ Links ]
36.Bobardt MD, Salmon P, Wang L, Esko JD, Gabuzda D, Fiala M, et al. Contribution of proteoglycans to human immunodeficiency virus type 1 brain invasion. J Virol 2004;78:6567-84. [ Links ]
37.Annunziata P. Blood-brain barrier changes during invasion of the central nervous system by HIV-1. Old and new insights into the mechanism. J Neurol 2003;250:901-6. [ Links ]
38.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, et al. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 2000;6:1380-7. [ Links ]
39.Lopez-Herrera A, Liu Y, Rugeles M, He J. HIV-1 interaction with hMR induces production of matrix metalloproteinase 2 through hMR-mediated intracellular signaling in astrocytes. Submitted to journal of neurochemistry.
40.Garden GA. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 2002;40:240-51. [ Links ]
41.Ensoli F, Fiorelli V, DeCristofaro M, Santini Muratori D, Novi A, Vannelli B, et al. Inflammatory cytokines and HIV-1-associated neurodegeneration: oncostatin-M produced by mononuclear cells from HIV-1-infected individuals induces apoptosis of primary neurons. J Immunol 1999;162:6268-77. [ Links ]
42.Patterson PH, Nawa H. Neuronal differentiation factors/cytokines and synaptic plasticity. Cell 1993;72 Suppl:123-37. [ Links ]
43.Burns TM, Clough JA, Klein RM, Wood GW, Berman NE. Developmental regulation of cytokine expression in the mouse brain. Growth Factors 1993;9:253-8. [ Links ]
44.Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990;250:1593-6. [ Links ]
45.Pulliam L, Clarke JA, McGrath MS, Moore D, McGuire D. Monokine products as predictors of AIDS dementia. Aids 1996;10:1495-500. [ Links ]
46.Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem 1999;274:17098-102. [ Links ]
47.Hober D, Haque A, Wattre P, Beaucaire G, Mouton Y, Capron A. Production of tumour necrosis factor-alpha (TNF-alpha) and interleukin-1 (IL-1) in patients with AIDS. Enhanced level of TNF-alpha is related to a higher cytotoxic activity. Clin Exp Immunol 1989;78:329-33. [ Links ]
48.Mintz M, Rapaport R, Oleske JM, Connor EM, Koenigsberger MR, Denny T, et al. Elevated serum levels of tumor necrosis factor are associated with progressive encephalopathy in children with acquired immunodeficiency syndrome. Am J Dis Child 1989;143:771-4. [ Links ]
49.McCoig C, Castrejon MM, Saavedra-Lozano J, Castano E, Baez C, Lanier ER, et al. Cerebrospinal fluid and plasma concentrations of proinflammatory mediators in human immunodeficiency virus-infected children. Pediatr Infect Dis J 2004;23:114-8. [ Links ]
50.Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, et al. Cell death and control of cell survival in the oligodendrocyte lineage. Cell 1992;70:31-46. [ Links ]
51.Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol 1991;147:1522-9. [ Links ]
52.Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, et al. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A 1993;90:10061-5. [ Links ]
53.Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol 1999;98:185-200. [ Links ]
54.Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D. Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol 1998;4:281-90. [ Links ]
55.Kolson DL, Buchhalter J, Collman R, Hellmig B, Farrell CF, Debouck C, et al. HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: RGD sequence dependence. AIDS Res Hum Retroviruses 1993;9:677-85. [ Links ]
56.Marshall DC, Wyss-Coray T, Abraham CR. Induction of matrix metalloproteinase-2 in human immunodeficiency virus-1 glycoprotein 120 transgenic mouse brains. Neurosci Lett 1998;254:97-100. [ Links ]
57.Conant K, McArthur JC, Griffin DE, Sjulson L, Wahl LM, Irani DN. Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann Neurol 1999;46:391-8.
58.Weis S, Haug H, Budka H. Vascular changes in the cerebral cortex in HIV-1 infection: I. A morphometric investigation by light and electron microscopy. Clin Neuropathol 1996;15:361-6. [ Links ]
59.Butera ST, Roberts BD, Folks TM. Regulation of HIV-1 expression by cytokine networks in a CD4+ model of chronic infection. J Immunol 1993;150:625-34. [ Links ]
60.Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987;238:800-2. [ Links ]
61.Matsuyama T, Kobayashi N, Yamamoto N. Cytokines and HIV infection: is AIDS a tumor necrosis factor disease? Aids 1991;5:1405-17. [ Links ]
62.Poli G, Fauci AS. The effect of cytokines and pharmacologic agents on chronic HIV infection. AIDS Res Hum Retroviruses 1992;8:191-7. [ Links ]
63.Ameglio F, Capobianchi MR, Castilletti C, Cordiali Fei P, Fais S, Trento E, et al. Recombinant gp120 induces IL-10 in resting peripheral blood mononuclear cells; correlation with the induction of other cytokines. Clin Exp Immunol 1994;95:455-8. [ Links ]
64.Capobianchi MR, Barresi C, Borghi P, Gessani S, Fantuzzi L, Ameglio F, et al. Human immunodeficiency virus type 1 gp120 stimulates cytomegalovirus replication in monocytes: possible role of endogenous interleukin-8. J Virol 1997;71:1591-7. [ Links ]
65.De SK, Venkateshan CN, Seth P, Gajdusek DC, Gibbs CJ. Adenovirus-mediated human immunodeficiency virus-1 Nef expression in human monocytes/macrophages and effect of Nef on downmodulation of Fcgamma receptors and expression of monokines. Blood 1998;91:2108-17. [ Links ]
66.Chen P, Mayne M, Power C, Nath A. The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. Implications for HIV-1-associated neurological diseases. J Biol Chem 1997;272:22385-8. [ Links ]
67.Roux P, Alfieri C, Hrimech M, Cohen EA, Tanner JE. Activation of transcription factors NF-kappaB and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces interleukin-8 expression. J Virol 2000;74:4658-65. [ Links ]
68.Patella V, Florio G, Petraroli A, Marone G. HIV-1 gp120 induces IL-4 and IL-13 release from human Fc epsilon RI+ cells through interaction with the VH3 region of IgE. J Immunol 2000;164:589-95. [ Links ]
69.Brigino E, Haraguchi S, Koutsonikolis A, Cianciolo GJ, Owens U, Good RA, et al. Interleukin 10 is induced by recombinant HIV-1 Nef protein involving the calcium/calmodulin-dependent phosphodiesterase signal transduction pathway. Proc Natl Acad Sci U S A 1997;94:3178-82. [ Links ]
70.Hofman FM, Chen P, Incardona F, Zidovetzki R, Hinton DR. HIV-1 tat protein induces the production of interleukin-8 by human brain-derived endothelial cells. J Neuroimmunol 1999;94:28-39. [ Links ]
71.Ensoli F, Fiorelli V, De Cristofaro M, Muratori DS, Novi A, Isgro A, et al. Role of immune-derived diffusible mediators in AIDS-associated neurological disorders. Arch Immunol Ther Exp 2000;48:259-66. [ Links ]
72.Janabi N, Di Stefano M, Wallon C, Hery C, Chiodi F, Tardieu M. Induction of human immunodeficiency virus type 1 replication in human glial cells after proinflammatory cytokines stimulation: effect of IFNgamma, IL1beta, and TNFalpha on differentiation and chemokine production in glial cells. Glia 1998;23:304-15. [ Links ]
73.Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med 1992;176:1703-18. [ Links ]
74.Nuovo GJ, Gallery F, MacConnell P, Braun A. In situ detection of polymerase chain reaction-amplified HIV-1 nucleic acids and tumor necrosis factor-alpha RNA in the central nervous system. Am J Pathol 1994;144:659-66. [ Links ]
75.Wahl SM, Allen JB, McCartney-Francis N, Morganti-Kossmann MC, Kossmann T, Ellingsworth L, et al. Macrophage- and astrocyte-derived transforming growth factor beta as a mediator of central nervous system dysfunction in acquired immune deficiency syndrome. J Exp Med 1991;173:981-91. [ Links ]
76.Benveniste EN. Inflammatory cytokines within the central nervous system: sources, function, and mechanism of action. Am J Physiol 1992;263:C1-16. [ Links ]
77.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci 1991;14:453-501. [ Links ]
78.Calabrese V, Scapagnini G, Ravagna A, Giuffrida Stella AM, Butterfield DA. Molecular chaperones and their roles in neural cell differentiation. Dev Neurosci 2002;24:1-13. [ Links ]
79.Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res 1999;59:1693s-1700s. [ Links ]
80.Putcha GV, Johnson EM, Jr. Men are but worms: neuronal cell death in C elegans and vertebrates. Cell Death Differ 2004;11:38-48. [ Links ]
81.Saha RN, Pahan K. Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. J Neurochem 2003;86:1057-71. [ Links ]
82.Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem 2001;276:46639-46. [ Links ]
83.Rogister B, Ben-Hur T, Dubois-Dalcq M. From neural stem cells to myelinating oligodendrocytes. Mol Cell Neurosci 1999;14:287-300. [ Links ]
84.Perry DK, Hannun YA. The role of ceramide in cell signaling. Biochim Biophys Acta 1998;1436:233-43. [ Links ]
85.Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, et al. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A 1999;96:7532-7. [ Links ]
86.Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, et al. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol 2004;55:257-67. [ Links ]
87.Liuzzi GM, Mastroianni CM, Vullo V, Jirillo E, Delia S, Riccio P. Cerebrospinal fluid myelin basic protein as predictive marker of demyelination in AIDS dementia complex. J Neuroimmunol 1992;36:251-4. [ Links ]
88.Rakic P. DNA synthesis and cell division in the adult primate brain. Ann N Y Acad Sci 1985;457:193-211. [ Links ]
89.Gould E, Reeves AJ, Graziano MS, Gross CG. Neurogenesis in the neocortex of adult primates. Science 1999;286:548-52. [ Links ]
90.Gould E, Tanapat P, Rydel T, Hastings N. Regulation of hippocampal neurogenesis in adulthood. Biol Psychiatry 2000;48:715-20. [ Links ]
91.Gould E, Tanapat P, Hastings NB, Shors TJ. Neurogenesis in adulthood: a possible role in learning. Trends Cogn Sci 1999;3:186-192. [ Links ]
92.Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001;410:372-6. [ Links ]
93.Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature 2000;405:951-5. [ Links ]
94Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med 2002;8:955-62. [ Links ]
95.in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 2001;345:1515-21. [ Links ]
96.Clerici M, Sison AV, Berzofsky JA, Rakusan TA, Brandt CD, Ellaurie M, et al. Cellular immune factors associated with mother-to-infant transmission of HIV. Aids 1993;7:1427-33. [ Links ]
97.Clerici M, Saresella M, Colombo F, Fossati S, Sala N, Bricalli D, et al. T-lymphocyte maturation abnormalities in uninfected newborns and children with vertical exposure to HIV. Blood 2000;96:3866-71. [ Links ]
98.Goriely S, Vincart B, Stordeur P, Vekemans J, Willems F, Goldman M, et al. Deficient IL-12(p35) gene expression by dendritic cells derived from neonatal monocytes. J Immunol 2001;166:2141-6
. [ Links ]
