










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.25 n.2 Bogotá jun. 2005
Leishmania:
papel de la glicoproteína P en la mediación de resistencia a medicamentos y estrategias de reversiónEdison J. Osorio 1,2, Sara M. Robledo 2, Gabriel J. Arango 1, Carlos E. Muskus 2
1 Grupo de Investigación en Sustancias Bioactivas (GISB), Facultad de Química Farmacéutica, Corporación de Patologías Tropicales, Universidad de Antioquia, Medellín, Colombia.
2
Programa de Estudio y Control de Enfermedades Tropicales (PECET), Corporación de Patogías Tropicales, Universidad de Antioquia, Medellín, Colombia.Actualmente, los parásitos protozoarios son uno de los principales agentes causantes de morbilidad y mortalidad en el mundo, un problema complicado, además, por la aparición de resistencia a medicamentos en estos organismos. La resistencia a medicamentos observada en parásitos protozoarios se debe a diferentes mecanismos como la disminución de la entrada del medicamento a la célula por cambios en el transportador requerido, la pérdida de la activación del medicamento por parte del hospedero, las alteraciones en el blanco del medicamento y la expresión exagerada del transportador múltiple de medicamentos o glicoproteína P (Pgp). En esta revisión, nos centramos en: 1) el papel de las glicoproteínas P (Pgp) de la familia de proteínas ABC (ATP binding cassette) como los transportadores de múltiples medicamentos en la mediación de resistencia en protozoarios, especialmente en Leishmania, y en el desarrollo de resistencia cruzada para medicamentos estructural y funcionalmente no relacionados, y 2) en algunos conceptos relacionados con los mecanismos moduladores que podrían revertir la resistencia a medicamentos por fármacos y productos naturales. Numerosos moduladores o quimiosensibilizadores son conocidos por alterar la capacidad de las glicoproteínas P para mantener concentraciones intracelulares subtóxicas del medicamento; algunos ejemplos incluyen los bloqueadores de los canales de calcio como el verapamilo; sin embargo, se requieren altas concentraciones para una inhibición eficiente y duradera, las cuales producen efectos adversos indeseables. Por tanto, se necesitan más investigaciones relacionadas con los moduladores naturales para Pgp, los cuales podrían presentar menor toxicidad para el hospedero.
Palabras clave: Leishmania, protozoos, multirresistencia, glicoproteína P, productos naturales.
Leishmania: role of P glycoprotein in drug resistance and reversion strategies
Protozoan parasites are important causative agents of morbidity and mortality throughout the world -a problem further complicated by the emergence of drug resistance in these parasites. Mechanisms of drug resistance include the following: decreased uptake of the drug into the cell, loss of drug activation, alterations in the drug target, and over-expression of a well-known multiple drug transporter proteins. In this review, two critical components of resistance are stressed: (1) the role of ATP binding cassette proteins, such as P-glycoproteins, in mediating drug resistance in Leishmania and other protozoans, followed by development of cross-resistance to many structurally and functionally unrelated drugs, and (2) some concepts concerning the reversal mechanism of multidrug resistance by drugs and natural products. Several modulators or chemosensitizers alter the capacity of P-glycoproteins to maintain subtoxic intracellular drug concentrations. Calcium channel blockers such as verapamil act in this mode; however, high concentrations are required for an efficient and effective inhibition and, in addition, produce undesirable side effects. The discovery of new, natural product modulators of P-glycoproteins is stressed. This category of modulators offer potentially improved efficacy and lowered toxicity for the mammalian host.
Keywords: Protozoa, Leishmania, multidrug resistance, P-glycoprotein, natural products.
El género Leishmania comprende alrededor de 30 especies de parásitos protozoos y, al menos, 12 son patógenas para el humano (1,2) (cuadro 1).

El parásito es el agente causal de un grupo de enfermedades conocidas como leishmaniasis y que comprometen diferentes tejidos (piel, mucosas, medula ósea) y órganos (hígado, bazo). Dependiendo del tejido u órgano comprometido en la infección, la leishmaniasis se clasifica en cutánea (cuando hay compromiso exclusivo de la piel), mucosa (cuando hay compromiso de las mucosas, principalmente del tracto naso-oro-faríngeo) y visceral (cuando hay compromiso de la medula ósea, el hígado y el bazo).
La leishmaniasis es una de las enfermedades "olvidadas" que es endémica en numerosos países de Latinoamérica, Asia y África; se estima que, aproximadamente, 20 millones de personas están infectadas, 350 millones en riesgo de adquirir la infección y se presentan alrededor de 400.000 nuevos casos por año (3).
Entre las medidas de control más importantes en los sitios en los que el hombre puede estar participando en el ciclo de transmisión se incluyen el diagnóstico y el tratamiento oportuno de los casos con el fin de disminuir el riesgo de transmisión de la infección; sin embargo, se dispone de muy pocos medicamentos para el tratamiento adecuado de la enfermedad.
Entre los medicamentos potencialmente efectivos para el tratamiento de la leishmaniasis están los antimoniales pentavalentes (SbV) como el antimoniato de meglumina (Glucantime®) o el estibogluconato de sodio (Pentostam®), que han sido los medicamentos de primera elección desde hace más de 50 años para el tratamiento de cualquiera de las formas clínicas producidas por cualquiera de las diferentes especies de Leishmania (4); aunque el tratamiento con SbV resulta en tasas de curación superiores al 80%, la eficacia depende de la especie y cepa de Leishmania involucrada (5) y se asocia con efectos secundarios moderados o graves por la alta toxicidad de estos metales, por requerir una administración prologada (20 a 28 días según sea una forma cutánea o visceral, respectivamente) y por ser de administración parenteral (intravenosa para el estibogluconato de sodio e intramuscular para el antimoniato de meglumina).
Otros medicamentos potencialmente efectivos son: la pentamidina, la anfotericina B, el alopurinol, la mefloquina y la miltefosina (4,6-8); estos medicamentos aunque pueden resultar en eficacias similares a la obtenida con los SbV, varía en función de la forma clínica de la enfermedad y de la especie de Leishmania involucrada. Así, por ejemplo, el alopurinol y la mefloquina, que son medicamentos potencial-mente efectivos contra Leishmania mexicana (9), no lo son para el tratamiento de la leishmaniasis cutánea causada por Leishmania panamensis y Leishmania braziliensis en Colombia y Brasil (10-12). La pentamidina, aunque similar a los SbV en eficacia (13,14), su costo es mayor, por lo que no constituye una alternativa real para su uso en países pobres donde la enfermedad es endémica.
Actualmente, se tiene como candidato a la miltefosina, un medicamento oral que ha presentado tasas de curación superiores al 95% para casos de leishmaniaisis visceral producida por Leishmania donovani en India (8,15), al igual que para casos de leishmaniaisis cutánea por L. panamensis en Colombia donde se ha observado una tasa de curación mayor del 91% a la dosis de 2,5 mg/kg por día (16,17). Sin embargo, la efectividad de la miltefosina para el tratamiento de la leishmaniasis cutánea producida por L. braziliensis o L. mexicana en Guatemala ha sido alrededor del 50% (17).
El hecho de que la miltefosina haya sido efectiva contra L. panamensis pero no contra L. braziliensis o L. mexicana sugiere que la eficacia varía en función de la especie de Leishmania imvolucrada. La diponibilidad de la miltefosina como alternativa terapéutica permanece aún por definirse en países como Colombia, donde la leishmaniasis cutánea es producida por diferentes especies de Leishmania.
Mientras se define el papel de la miltefosina como alternativa para el tratamiento de la enfermedad, el medicamento aceptado hasta ahora continúa siendo el SbV. Sin embargo, el valor clínico de la terapia con los SbV se está viendo amenazado por la aparición cada vez más frecuente de fracasos terapéuticos, principalmente en India donde, aproximadamente, falla el 50% de los tratamientos para leishmaniasis visceral con las dosis estándar de SbV (18). Estas fallas terapéuticas pueden deberse a variaciones no sólo en el contenido de SbV en los lotes del medicamento, como se ha evidenciado previa-mente (19), sino también por la aparición de parásitos resistentes al SbV (8,20-22).
Teniendo en cuenta que en el proceso de generación de resistencia a los medicamentos en los protozoos como Leishmania pueden intervenir diferentes mecanismos que involucran la acción de proteínas transportadoras de medicamentos conocidos como transportadores ABC (del inglés, ATP Binding Cassette), siendo los más conocidos la glicoproteína P (Pgp) y la proteína MRP (del inglés, Multidrug Resistance Associated Protein), la presente revisión tiene como objetivo discutir el posible mecanismo por medio del cual se puede presentar el fenómeno de resistencia conferido por estas proteínas transportadoras de medicamentos en las especies de Leishmania y las posibles alternativas para contrarrestar dicha resistencia.
Resistencia a medicamentos y fenotipo de multirresistencia
Existen dos grandes manifestaciones de resistencia a medicamentos en los micro-organismos: 1) la resistencia intrínseca, relacionada con la capacidad natural de los microorganismos para resistir la quimioterapia inicial, y 2) la resistencia adquirida que se presenta cuando un microorganismo inicialmente es sensible al medicamento pero luego se torna moderado o fuertemente resistente al tratamiento (23).
La resistencia a los medicamentos entendida como "la capacidad de un microorganismo para multiplicarse o para sobrevivir en presencia de concentraciones de un fármaco que normalmente destruye los microorganismos de la misma especie o, al menos, previene su multiplicación" (24) constituye un impedimento importante para el control de enfermedades consideradas como problemas de salud pública en el mundo.
Complicando el panorama del control de las enfermedades endémicas y prevalentes, la resistencia que muestran los agentes infecciosos a los medicamentos no se está limitando a un medicamento en particular sino a medicamentos diferentes, fenómeno que se conoce como resistencia múltiple a medicamentos. La manifestación de resistencia a varios medicamentos se conoce como fenotipo MDR (del inglés, multidrug resistance) descrito inicialmente en células tumorales de mamíferos (25) y, posteriormente, en bacterias (26-29), protozoos (29) y hongos (30).
El fenotipo MDR es un caso particular de resistencia adquirida a medicamentos, observada tanto in vitro como in vivo, que describe la aparición de resistencia cruzada a diversos medicamentos no relacionados estructuralmente (31).
Tanto en el cáncer como en las enfermedades infecciosas, el fenotipo MDR se asocia con la expresión exagerada de proteínas pertenecientes a la superfamilia ABC, una familia de moléculas transportadoras de nucleótidos de adenina, función que se conoce como tráfico de ATPasas (32,33).
Los transportadores ABC son polipéptidos grandes de 140-190 kd. Presentan como característica estructural el poseer dos unidades homólogas, cada una con seis segmentos transmembrana (TM) y un sitio de fijación a nucleótidos (NBD); las regiones TM fijan la proteína a la membrana y, probablemente, constituyen el sitio de interacción con el sustrato y, por tanto, son los responsables del transporte del medicamento.
Por su parte, la región NBD contiene secuencias de nucleótidos conocidas como motivos Walker A y B y una secuencia corta conocida como secuencia ABC que es típica de los miembros de la familia ABC (34) (
figura 1).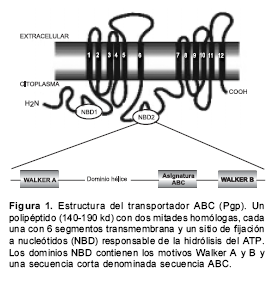
La topología general de los transportadores ABC es: TM2-NBD2 (31,32); sin embargo, en Plasmodium, Leishmania, Trypanosoma y Entamoeba se han informado topologías adicionales tales como TM-NBD, TM2-NBD y NBD2 (34).
Los transportadores ABC son responsables del transporte de compuestos (inclusive de medicamentos) desde el interior hacia el exterior de una célula. Al facilitar la entrada y la salida del medicamento, el transportador permite que las células o los microorganismos sean capaces de eliminar las sustancias tóxicas derivadas del medicamento evadiendo así los efectos terapéuticos del mismo.
En la familia de transportadores ABC se incluye la Pgp que es una glicoproteína de membrana producto del gen mdr1 también conocida como pg-170, PGP o P-1 (35), y la proteína MRP, una glicoproteína de 190 kd producto del gen mrp (36). Se ha propuesto a la Pgp como la proteína responsable del transporte de diferentes compuestos hacia el exterior de la célula, que actúa como una bomba de eflujo dependiente de ATP y que lleva a la disminución intracelular del medicamento que estaría en contacto con la molécula blanco (25).
La Pgp está constituida por 12 segmentos transmembrana que forman seis asas con estructura de alfa hélice en la membrana plasmática, las cuales constituyen el sitio de interacción con el sustrato y, por tanto, participan en el transporte del medicamento. Se postula que las Pgp actúan como bombas capaces de expulsar drogas en diferentes tipos celulares, ya que poseen dos sitios de unión a ATP en la cara citoplásmica. La hidrólisis del ATP provee la energía necesaria para la expulsión de las drogas.
Las proteínas homólogas a la Pgp y a la MRP de las células mamíferas también están presentes en hongos como Candida albicans, Saccharomyces cerevisiae y Schizosaccharomyces pombe (31), y en protozoos como Leishmania tarentolae (37), Trypanosoma cruzi (38) y Plasmodium falciparum (39). Además, en Leishmania tropica se describió una tercera proteína transportadora perteneciente a la superfamilia ABC que por presentar una alta homología con miembros de la subfamilia ABC A de mamíferos se conoce como transportador ABC tipo A (40).
La aparición de resistencia a los medicamentos en protozoos se atribuye principalmente a uno o varios de los siguientes mecanismos: 1) disminución de la entrada del medicamento a la célula hospedera como se observa en la
resistencia a arsenicales y análogos de purina en Leishmania major y otras especies (29,30,41) o a diamidinas en Trypanosoma brucei (29); 2) inactivación del medicamento por el hospedero, por ejemplo, resistencia al metronidazol en tricomonas (42) y Giardia spp. (43); 3) alteraciones en la molécula blanco del medicamento, por ejemplo, sustituciones de aminoácidos en la enzima dihidrofolato-reductasa-timidilato sintetasa (DHFR-TS) de P. falciparum lo que le confiere resistencia a la pirimetamina (44) y de L. major asociada a la resistencia al metotrexato (45) y, 4) expresión exagerada de la Pgp lo cual incrementa la salida del medicamento del interior del parásito disminuyendo así la cantidad de compuestos activos que estarían en contacto con el parásito (46), como ocurre en promastigotes de L. tarentolae resistentes al metotrexato (37,46) o en P. falciparum resistente a la mefloquina, la halofantrina y la quinina (47,48).
Papel de los genes pgp asociados con el fenotipo MDR en la mediación de resistencia a medicamentos en Leishmania
El mecanismo para adquirir resistencia en Leishmania, al igual que para otros protozoos y células mamíferas, es un proceso multifactorial que involucra múltiples genes y se asocia con la amplificación de regiones específicas de su genoma. Se ha propuesto que estas regiones cromosómicas actúan como un mecanismo de resistencia a medicamentos ya que las secuencias repetidas facilitan la amplificación de los genes de resistencia presentes en las regiones amplificadas (amplicones).
La amplificación de estas regiones específicas se demostró en promastigotes de L. tarentolae y Leishmania enriettii al observarse que los parásitos resistentes a metotrexato y vinblastina amplificaban regiones extracromosómicas conocidas como amplicones H y V, respectiva-mente (37,49), y que el amplicón H de L. tarentolae y L. major resistentes a la primaquina o la terbinafina y con resistencia cruzada a metotrexato contenía el gen para la Pgp (37,50). El papel fundamental de la Pgp quedó confirmado por estudios de mutación dirigida de los genes ltpgpA que da como resultado la hipersensibilidad de los parásitos a los medicamentos (51).
Desde entonces, numerosas investigaciones sugieren que la Pgp juega un papel importante en la mediación de resistencia a diferentes medicamentos, incluso el antimonio trivalente (SbIII) y el SbV.
Todos los organismos que presentan el fenotipo MDR expresan una o más Pgp, codificadas por varios genes que tienen secuencias homólogas y mantienen las principales características de estas proteínas, pero difieren entre sí. Es decir, son genes diferentes y no varias copias de un mismo gen, aunque es probable que su origen provenga de un mismo gen. Los genes que codifican por la Pgp pertenecen a la familia de genes mdr los cuales se han identificado, clonado y secuenciado en diferentes protozoos que incluyen varias especies de Leishmania (37,49,50,52-58), P. falciparum (39,53,59,60), T. cruzi y T. brucei spp. (38,59,61,62) y E. histolytica (59,63-65).
El
cuadro 2 resume los diferentes genes mdr encontrados en estos protozoos. En el caso de Leishmania, el primer gen mdr, denominado ltpgpA se detectó en el amplicón H de promastigotes de L. tarentolae resistente al metotrexato (37); la amplificación de este gen se define como un fenotipo MDR no convencional, es decir, no asociado al fenotipo MDR de los mamíferos (66) ya que se asocia con resistencia a agentes hidrofílicos tipo arsenicales y SbIII (67) los cuales no son substratos para el mecanismo de eflujo de la Pgp en mamíferos (52).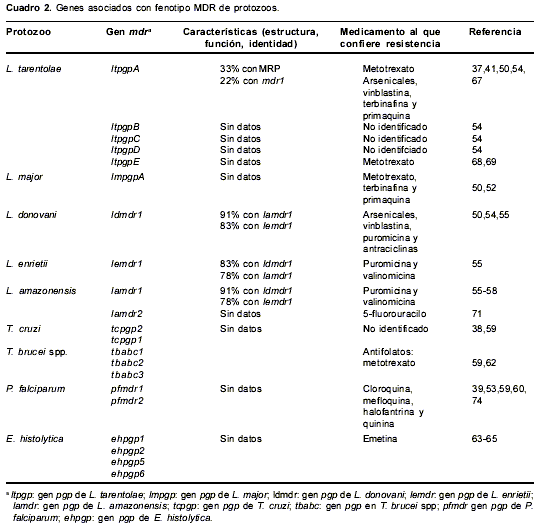
Los experimentos de hibridación indican que el gen ltpgpA pertenece a una familia de genes en donde existen otros cuatro genes: ltpgpB, ltpgpC, ltpgpD y ltpgpE, pero sólo el gen ltpgpA es el que se encuentra más frecuentemente asociado a resistencia a medicamentos en las diferentes especies de Leishmania hasta ahora estudiadas (54).
Un gen homólogo al gen ltpgpE se amplificó en promastigotes de L. tropica resistentes a metotrexato; la amplificación de este gen se asoció con una actividad ATPasa aumentada (68,69). A su vez, el gen mdr en L. major corresponde al gen lmpgpA localizado en el amplicón H (52) y cuyo perfil de resistencia es similar al del gen ltpgpA; este gen también se amplifica en parásitos resistentes a terbinafina y primaquina (50) que son medicamentos estructuralmente no relacionados, mientras que el gen mdr de L. donovani corresponde al gen ldmdr1 que amplifica en parásitos resistentes a vinblastina que también muestran resistencia cruzada a los compuestos hidrofóbicos puromicina y antraciclinas (55).
En L. enriettii, el gen mdr corresponde a lemdr1 y en L. amazonensis a lamdr1 y lamdr2; tanto el gen lemdr1 como el gen lamdr1 se describieron en parásitos con resistencia cruzada a puromicina y valinomicina pero no a SbV (57,70), mientras que el gen lamdr2 se describió en una cepa resistente a 5-fluorouracilo (71).
La secuencia de ltpgpA predice una estructura similar a la de otros transportadores ABC como la proteína MRP con cuyo gen presenta mayor homología que con el gen mdr1 (33% versus 22%, respectivamente) (37,53). Las secuencias de los genes ldmdr1 (L. donovani) y lemdr1 (L. enriettii) son homólogas en 83% aunque también predicen una estructura similar a otros transportadores ABC como la proteína MRP (57), mientras que la secuencia de lamdr1 (L. amazonensis) es 91% y 78% homóloga a los genes ldmdr1 y lemdr1, respectivamente (72).
Por otro lado, el fenotipo de resistencia mostrado por L. donovani es similar al de células mamíferas con fenotipo MDR en las cuales la resistencia es exclusivamente para moléculas de naturaleza hidrofóbicas y difiere del fenotipo observado en L. tarentolae y L. major en donde se produce resistencia para compuestos hidrofílicos (56).
En promastigotes de L. braziliensis, L. guyanensis y L. mexicana, aunque no hay informes de genes mdr asociados con resistencia, si se sugiere la existencia de otros sistemas transportadores de medicamentos, todos dependientes de energía y que comparten similitud con la MRP1. Estos sistemas incluyen: 1) una bomba de eflujo de pirarrubicina cuya actividad se inhibe por la acción del verapamilo y por algunos derivados de la fenotiazina como la tiodirazina, la proclorperazina, la trifluoperazina, la clorpromazina y la trifluoropromazina; 2) una bomba de eflujo de acetoximetiléster de calceína que se puede inhibir por la acción de los derivados de la fenotiazina pero no por el verapamilo; y 3) una bomba de eflujo de calceína, la cual se demostró en L. braziliensis y L. guyanensis y que sólo es inhibida por la proclorperazina y la trifluoperazina (73). Tanto la calceína como el acetoximetiléster de calceína y el derivado antracíclico pirarrubicina son sustratos conocidos de las bombas de eflujo MDR de las células mamíferas.
En P. falciparum los genes mdr se denominan pfmdr1 y, pfmdr2 (39,59,60,74) y se asocian con resistencia a cloroquina, mefloquina, halofantrina y quinina (47,48,75-79). Los genes mdr de T. cruzi se conocen con los nombres tcpgp1, tcpgp2 (38,59) y en T. brucei spp. como tbabc1, tbabc2 y tbabc3 (59,62) los cuales están asociados al gen ptr1 que confiere resistencia a los compuestos antifolatos (80). En E. histolytica, los genes mdr se denominan ehpgp1, ehpgp2, ehpgp5 y ehpgp6 y se relacionan con resistencia a la emetina (63-65).
A pesar de la existencia comprobada de genes pgp en especies de Leishmania tanto patógenas
para el humano (por ejemplo, L. donovani, L. major, L. tropica, L. amazonensis y L. guyanensis) como no patógenas (por ejemplo, L. tarentolae), es poco probable que niveles altos de resistencia necesiten únicamente de la amplificación de un gen como el pgp.
Los diferentes estudios concuerdan en sugerir que la resistencia es multifactorial por lo que factores adicionales a la expresión exagerada de la Pgp son necesarios para la generación de resistencia, lo que se demuestra por los siguientes hallazgos: 1) la transfección de los genes ltpgpA, lemdr1 y lmpgpA muestra ausencia o sólo niveles parciales de resistencia (52,57,66); 2) la transfección del amplicón V demuestra la presencia de otros genes, además del gen lemdr1, los cuales podrían estar asociados también con resistencia (49), y 3) la caracterización de un fenómeno que conduce a la permeabilidad de la membrana a medicamentos citotóxicos como un componente de difusión pasiva a través de la misma (81).
En el caso de resistencia al SbV que, como se mencionó previamente, es prácticamente el único medicamento disponible para el tratamiento de cualquiera de las formas clínicas de leishmaniasis producidas por cualquiera de las especies de Leishmania, el conocimiento acerca del mecanismo o mecanismos que participan en la generación de resistencia al SbV no es completamente claro.
Probablemente, el escaso conocimiento al respecto se debe a que el mecanismo de acción del SbV todavía no está bien definido. Hasta ahora se acepta que el parásito es susceptible a los antimoniales porque el amastigote es capaz de reducir la forma pentavalente a una forma trivalente capaz de matar el parásito por la acción de una enzima reductasa denominada TDR1 (del inglés, Thiol dependent reductase), un trímero cuyos monómeros conservan similitud con la enzima glutatión S transferasa y que utiliza el glutatión como agente reductor (82).
Luego, el SbIII inhibe en forma reversible la enzima tripanotión reductasa (TR) (83). La TR es un enzima responsable de conservar el tripanotión (N,N-(bis)glutationilespermidina) en estado ditiol denominado T(SH)2. El sistema T(SH)2/TR, conocido como metabolismo tiol, participa en varias funciones metabólicas importantes en el parásito como son: síntesis de deoxirribo-nucleótidos (84), conjugación, secuestro y transporte de metales y medicamentos (85-87), homeostasis del ácido ascórbico (88) y reducción de radicales oxidativos tales como H2O2, O2-, OH- (89-93).
Dado que la TR es una enzima clave en el metabolismo redox de Leishmania (y otros parásitos tripanosomátidos como T. cruzi y T. brucei spp.) y, por ende, necesaria para proteger al parásito de la acción de los radicales oxidativos generados por el hospedero durante la respuesta inmune y de los efectos tóxicos de los metales pesados, permitiendo la supervivencia del parásito al interior de la célula hospedera y que esta vía de detoxificación de compuestos nocivos no la comparten el parásito y el hospedero mamífero, es posible pensar que un mecanismo de resistencia a los antimoniales involucre las enzimas del metabolismo Tiol. Se ha observado, por ejemplo, que los niveles de T(SH)2 están incrementados en algunas especies de L. mexicana, L. tropica y L. tarentolae resistentes a antimoniales y arsenicales (85,86,94).
Las hipótesis en estudio implican al T(SH)2 y al glutatión (g-L-glutamil-L-cisteinilglicina o GSH) en la detoxificación del SbIII y otros metales pesados mediada por bombas de eflujo dependientes de ATP (85,95,96) contradiciendo un estudio inicial en el cual se evidenció que la amplificación del gen lmpgpA estaba asociada con resistencia a SbIII al disminuir el influjo y sin aumentar el eflujo del medicamento (41). Actualmente, se sugiere que la resistencia al SbIII se asocia con la expresión exagerada de g-glutamilcisteína sintetasa (87) y ornitina descarboxilasa (97) que son enzimas necesarias para la biosíntesis de GSH y T(SH)2, respectivamente, y con la expresión exagerada de PgpA (87,94).
Los diferentes estudios concuerdan en sugerir que la generación de resistencia a un medicamento implica la participación de varios genes y que dependiendo del perfil de los genes amplificados ocurriría resistencia a un tipo de SbV pero no a otro. El hecho de que un parásito sea resistente a un tipo de SbV pero no al otro como ocurre con promastigotes de L. guyanensis resistentes a antimoniato de meglumina pero sensibles al estibogluconato de sodio, paromicina y vinblastina, y en los cuales la resistencia se asocia con la amplificación de un gen homólogo al gen ltpgpA (98), sugiere también que la resistencia puede deberse a mecanismos diferentes, tema que aún no se ha estudiado.
Actualmente estamos ejecutando un proyecto de investigación que consiste en la detección y caracterización de genes que estén asociados con la resistencia al antimoniato de meglumina y que, además, se expresen en forma diferencial en promastigotes y amastigotes de L. panamensis a fin de dilucidar si la resistencia se debe a la expresión exagerada de genes, entre ellos, los genes mdr.
Posibles estrategias para revertir
el fenotipo MDR
Se han diseñado diferentes estrategias bioquímicas, farmacológicas y clínicas para contrarrestar el fenotipo MDR en diferentes líneas celulares. Una de estas estrategias consiste en utilizar altas concentraciones del medicamento para compensar la pérdida por el eflujo de la célula y mantener, por lo tanto, los niveles terapéuticos; sin embargo, el incremento en las dosis del medicamento podría favorecer la aparición o el aumento de los efectos colaterales.
Otra estrategia consiste en la utilización de medicamentos que no sean afines a las Pgp, tales como la ciclofosfamida y el cis-platino aunque son pocos los medicamentos disponibles con estas propiedades y no se lograría inhibir el transporte de algunos compuestos análogos (35).
La efectividad de los agentes moduladores de las Pgp en la quimiosensibilización de células cancerosas resistentes a medicamentos estimuló la búsqueda de agentes capaces de contrarrestar el fenotipo MDR en parásitos protozoos (25,29).
Dado que, al parecer, el fenotipo MDR se asocia con altos niveles de proteína cinasa C (PKC) y, en especial, con la isoforma a (99,100), se sugiere que la Pgp constituye un blanco de fosforilación lo que, a su vez, sustenta la hipótesis que la PKC puede servir como un importante modulador en el desarrollo de resistencia a medicamentos y sugiere que puede ser otra estrategia para regular la actividad de la Pgp.
Se ha demostrado que la exposición de células con fenotipo MDR a activadores de PKC aumenta la fosforilación de la Pgp, disminuye la acumulación del medicamento e incrementa la resistencia a los fármacos (101,102); al contrario, la presencia de inhibidores de PKC disminuye la fosforilación e incrementa la acumulación del medicamento (103).
Sin embargo, muchos de los activadores e inhibidores de PKC utilizados para alterar el estado de fosforilación de la Pgp no son muy específicos por lo que podrían causar múltiples efectos en las células. En este sentido, se han descrito tres tipos de complicaciones producidas por la utilización de agonistas o antagonistas de PKC que dificultan el análisis de los datos: 1) varios activadores e inhibidores de PKC pueden afectar la expresión de Pgp por activación o desactivación de la transcripción, lo cual sugiere que las señales transmitidas por PKC pueden regular la expresión del gen mdr (100); 2) algunos moduladores de PKC son moléculas amfipáticas que se pueden unir a la Pgp e inhibir la actividad de transporte de medicamentos si se tiene en cuenta que los trabajos recientes en L. tropica y otros tipos de células muestran que los moduladores de la PKC se unen directamente con la Pgp e inhiben la fosforilación en la Pgp y de su actividad (104), y 3) el tiempo de exposición de las células a un modulador particular de PKC puede afectar el mecanismo de acción como ocurre con la exposición de células al activador TPA (12-O-tetradecanoilforbol-13-acetato) que puede activar la PKC. Sin embargo, el tiempo necesario para que ocurra esta unión puede disminuir los niveles de PKC al aumentar la tasa de proteólisis (105,106). Además, algunas investigaciones señalan que la fosforilación no juega un papel importante en la regulación de la actividad de las Pgp en líneas de células tumorales de mama y fibroblastos (107,108) lo cual sugiere que esta estrategia puede ser poco eficaz.
Como la actividad de la Pgp se reconoce como un factor clave en la dirección del fenotipo MDR, la reversión de la resistencia mediante el bloqueo del eflujo de los medicamentos por inhibición de las funciones de la Pgp se ha convertido en la estrategia más ampliamente utilizada (32). Actualmente, los medicamentos que actúan bloqueando la Pgp reciben el nombre de quimiosensibilizadores (109) o moduladores (35,110).
El
cuadro 3 muestra algunos de los moduladores en células cancerígenas que alteran la capacidad de las Pgp para mantener concentraciones intracelulares alteradas o bajas de los medicamentos. Entre ellos, tenemos productos naturales hidrofóbicos (derivados de plantas o microorganismos), análogos semisintéticos y compuestos orgánicos sintéticos. Aunque la química no es compartida por estos diversos moduladores, todos son compuestos amfipáticos preferiblemente solubles en lípidos (31,111), lo cual facilita las interacciones hidrofóbicas entre el medicamento y la Pgp debido a la gran cantidad de aminoácidos aromáticos presentes en estas proteínas (112,113).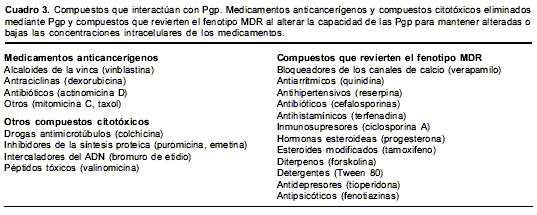
Entre los primeros moduladores de Pgp que se evaluaron están algunos bloqueadores de los canales de calcio como el verapamilo, la nifedipina, la imipramina y la azidopina (114-117); se ha encontrado que los inhibidores de los canales de calcio como el verapamilo (118,119), la eritromicina (120) y los derivados de las fenotiazinas (121) son capaces de revertir el fenotipo MDR en P. falciparum.
Otros moduladores de la Pgp que se han evaluado incluyen compuestos tipo antisicóticos y antidepresivos como las fenotiazinas y los tioxantenos (122-124), inmunosupresores como la ciclosporina A (125) y algunos esteroides y hormonas análogas (126). Sin embargo, las dosis óptimas de estos moduladores producen serios efectos tóxicos inherentes a su actividad farmacológica que incluyen complicaciones cardiacas, inmunosupresión y nefrotoxicidad (127), lo cual hace urgente la búsqueda de nuevos moduladores que sean altamente selectivos.
Moduladores de Pgp en Leishmania spp.
Similar a lo observado en P. falciparum, el verapamilo modula la resistencia de los promastigotes de L. donovani resistentes a los arseniatos al producir una disminución de la expresión de Pgp (128). No obstante, y a pesar de lo anterior, estos moduladores no son utilizados por su alta toxicidad y baja eficacia (129).
Varios compuestos de origen natural se han utilizado para revertir el fenotipo MDR. Hasta ahora, se han establecido varios grupos químicos que, al parecer, inhiben la función de la Pgp al competir con algunos medicamentos por la unión a dicha glicoproteína en el dominio TM que participa directamente en el transporte del medicamento o por la unión al sitio de fijación del nucleótido conocida como región NBD y, de esta forma, evitan que los medicamentos sean finalmente expulsados mediante la proteína de eflujo Pgp.
Entre los compuestos que poseen capacidad de unión a la Pgp y, por ende, capacidad para revertir el fenotipo MDR, se incluyen terpenoides y flavonoides. A continuación se discuten algunas de las principales características y propiedades de estos compuestos al igual que las observaciones encontradas hasta ahora en especies de Leishmania.
Terpenoides: son carbohidratos de origen biológico derivados del isopreno [CH2=C(CH3)CH=CH2]. Algunos se caracterizan por poseer átomos de oxígeno en diferentes grupos funcionales y se subdividen según el número de átomos de carbono en hemiterpenos (C5), monoterpenos (C10), sesquiterpenos (C15), diterpenos (C20), sesterterpenos (C25), triterpenos (C30) y tetraterpenos o carotenoides (C40). Varios trabajos recientes sugieren que estos compuestos tienen la capacidad de revertir el fenotipo MDR en células cancerígenas (130-135).
Así mismo, algunos sesquiterpenos hallados en la familia Celastraceae son compuestos naturales conocidos por su actividad moduladora del fenotipo MDR en varias líneas celulares cancerígenas humanas y en cepas de Leishmania (132,136,137) (
figura 2). Entre estos tenemos los sesquiterpenos del tipo dihidro-b-agarofuranos obtenidos de Maytenus magellanica y Maytenus chubutensis como promisorios moduladores del fenotipo MDR en L. tropica resistente a daunomicina (138).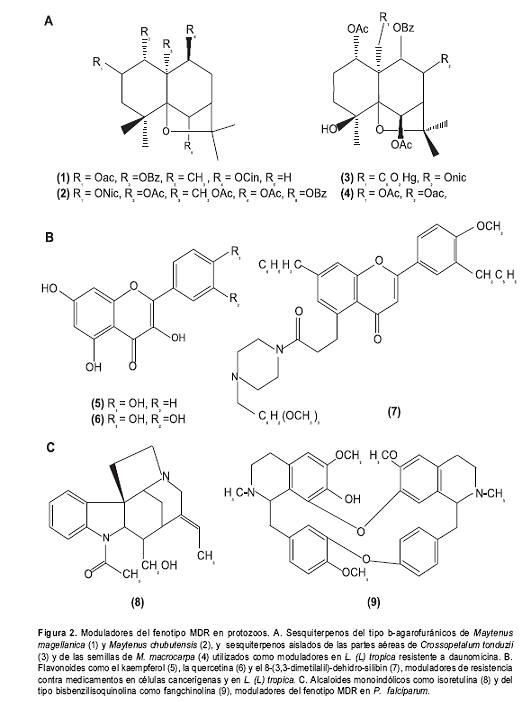
Otro ejemplo lo constituyen los sesquiterpenos aislados de las partes aéreas de Crossopetalum tonduzii y de las semillas de Maytenus macrocarpa, los cuales inhiben el crecimiento de parásitos resistentes a daunomicina en el 75% (139), mientras que los sesquiterpenos obtenidos de M. magellanica y M. chubutensis inhiben el crecimiento de estos mismos parásitos en el 95% (138).
Hasta hace poco, el mecanismo molecular por el cual los sesquiterpenos revertían la resistencia de L. (L) tropica al medicamento no había sido caracterizado, se especulaba que posiblemente ocurría por la fijación de los compuestos al dominio TM de la Pgp y no a la región NBD, al menos, en el bloqueo del eflujo de daunomicina (139).
En un trabajo reciente, se estudiaron 28 sesquiterpenos capaces de revertir el fenotipo MDR dependiente de Pgp para elucidar su mecanismo molecular de acción (137). La investigación sugiere que los sesquiterpenos interactúan, principalmente, con los dominios transmembrana de Pgp y a altas concentraciones pueden actuar como inhibidores no competitivos de la actividad ATPasa (137).
Flavonoides: constituyen una clase de compuestos polifenólicos con 15 átomos de carbono; dos anillos de benceno unidos por una cadena de tres carbonos conocido como sistema C6-C3-C6 (
figura 2). Son moduladores naturales de diferentes proteínas transportadoras al unirse a ellas (140-143); en el caso de la Pgp, al parecer, los flavonoides interactúan al contrario de los sesquiterpenos, con el sitio de fijación del ATP o dominio NBD y con una región hidrofóbica adyacente a este dominio (144-146). Varios de estos compuestos son capaces de inhibir el eflujo de medicamentos y revierten el fenotipo MDR en una línea de L. tropica resistente a la daunomicina (147,148).Entre los flavonoides naturales y sintéticos promisorios por presentar afinidad de fijación a la Pgp en diferentes líneas celulares, se encuentran algunos flavonoides como el kaempferol y la quercetina, isoflavones como la genisteína, chalconas halogenadas, flavonoides que contienen una cadena de N-benzilpiperazina y el flavanolignano derivado del silibin denominado 8-(3,3-dimetilalil)-dehidro-silibin (144,145,148).
Los diferentes compuestos naturales evaluados hasta ahora son capaces de interactuar con las Pgp y ejercen sus efectos en varias posiciones de la proteína como son los sitios de transporte o dominios TM y con los sitios de regulación o sitios NBD (149). Sin embargo, futuras investigaciones en el campo de los productos naturales como posibles moduladores del fenotipo MDR en Leishmania y en otros tipos celulares ayudaría al entendimiento de la interacción entre dichos compuestos y la Pgp del parásito.
Un grupo de compuestos naturales promisorio para futuras evaluaciones contra la Pgp de especies de Leishmania incluye los alcaloides que son compuestos orgánicos nitrogenados de carácter básico, con potentes acciones fisiológicas. Aunque los alcaloides no se han implicado en la modulación de la resistencia a los medicamentos en Leishmania, sí se sabe que los alcaloides de tipo indólico como la kopsoflorina presenta un incremento en la citotoxicidad de las drogas contra las células tumorales resistentes (150).
Lo mismo sucede con los alcaloides del tipo monoindólico, como la isorretulina, y del tipo bisbenzilisoquinolina, como la fangchinolina (
figura 2); se han utilizado para revertir la resistencia a la cloroquina y la mefloquina en P. falciparum (151,152).Los estudios de relación estructura-actividad de la interacción entre los moduladores con la Ppg han permitido identificar farmacóforos y propiedades fisicoquímicas para dichos moduladores que incluyen estructuras con anillos aromáticos, alta lipofilicidad y la presencia de átomos de nitrógeno básico (153,154). Esto sugiere que los alcaloides aromáticos son promisorios para ser modulares activos del fenotipo MDR en parásitos potozoos, incluida Leishmania.
Dado que los alcaloides son compuestos con potentes efectos toxicológicos y farmacológicos, derivados exclusivamente de plantas, y a que, en la actualidad, la búsqueda de nuevos medicamentos se concentra en gran parte en la evaluación de productos derivados de plantas, es importante incluir en las evaluciones no sólo una actividad antimicrobiana o citotóxica determinada, sino también la capacidad de dichos compuestos para modular la resistencia a otros medicamentos.
Conclusiones
Aunque el mecanismo que induce la aparición de resistencia a diferentes medicamentos en Leishmania y otros protozoos, al parecer, es multifactorial con la participación de múltiples genes, es claro que la Pgp juega un papel importante en la generación de resistencia, toda vez que las mutaciones en los genes de Pgp resultan en una hipersensibilidad de los parásitos a los medicamentos.
El patrón de resistencia o de sensibilidad y las características fisicoquímicas de los medicamentos para los cuales los protozoos tienen amplificado los genes pgp dependen de la especie del parásito involucrado. Por un lado, la expresión exagerada de los genes ltpgpA de L. tarentolae y lmpgpA de L. major provocan resistencia a los agentes hidrófilos como los arseniatos y a agentes antimoniales los cuales no son substratos para la Pgp de las células de mamíferos y de P. falciparum que actúan preferiblemente sobre moléculas hidrofóbicas.
Por otro lado, los genes ldmdr1 de L. donovani, lemdr1 de L. enriettii y el gen lamdr1 de L. amazonensis se asocian con la resistencia a medicamentos como la vinblastina, la puromicina, la valinomicina y las antraciclinas, todos ellos hidrofóbicos por lo que el patrón de resistencia es similar al de las células mamíferas con fenotipo MDR.
Sin embargo, se requiere más investigación en estos sistemas transportadores en protozoos a fin de obtener un mayor conocimiento sobre la forma en que los diferentes medicamentos interactúan con la Pgp para poder ser expulsados del interior del parásito.
La aparición aumentada de fallas terapéuticas con los SbV pone en evidencia la necesidad de identificar nuevos blancos terapéuticos, o desarrollar nuevos compuestos con propiedades anti-Leishmania, o ambas. Sin embargo, es importante evaluar dichos compuestos promisorios con el fin de asegurar que no sean substratos para la Pgp y otros transportadores ABC y garantizar que no se desarrolle un fenotipo de resistencia al medicamento.
Aunque el uso de compuestos naturales como moduladores del fenotipo MDR en Leishmania y en otros tipos celulares ayuda a la compresión del papel ejercido por la Pgp en la resistencia a los medicamentos, se necesitan trabajos adicionales para caracterizar las moléculas efectivas en la regresión de la resistencia o en la generación de nuevos compuestos que actúen sobre las vías metabólicas en las cuales no se realice extrusión del medicamento por parte de estas proteínas.
Una observación interesante son los mecanismos análogos de expulsión de medicamentos llevados a cabo por los microorganismos, lo cual hace más evidente que los sistemas de salida de múltiples medicamentos encontrados en células procarióticas son muy similares a los observados en las células eucarióticas. Por esta razón se puede establecer que algunas sustancias químicas relacionadas con los productos naturales o derivados sintéticos utilizados para revertir el fenotipo MDR en las líneas tumorales tienen, posiblemente, igual o mayor eficacia para revertir el fenotipo MDR en Leishmania y otros protozoos.
Conflicto de intereses
Los autores declaramos que no existe ningún tipo de interés que pudiere influir en los resultados de esta revisión.
Financiación
Los autores agradecen al Instituto para el Desarrollo de la Ciencia y la Tecnología en Colombia Colciencias (contrato 1115-0535396) y al Programa ECOS-Nord/ICFES/COLCIENCIAS/ICETEX (contrato 99S03) por la financiación de los proyectos de investigación que han permitido desarrollar la línea de investigación en blancos parasitarios para medicamentos. Edison Osorio recibió apoyo del Programa Jóvenes Investigadores de la Universidad de Antioquia.
Correspondencia:
Carlos Muskus, Calle 62 No. 52-59, SIU Laboratorio 632: apartado aéreo 1226, Medellín, Colombia.
Teléfono: (574) 210 6502/07; fax: (574) 210 6511
Recibido: 30/07/04; aceptado: 28/03/05
Referencias
1. Shaw JJ. Taxonomy of the genus Leishmania: present and future trends and their implications. Mem Inst Oswaldo Cruz 1994;89:471-8. [ Links ]
2. Noyes H. Implications of a neotropical origin of the genus Leishmania. Mem Inst Oswaldo Cruz 1998;93: 657-61. [ Links ]
3. World Health Organization. Leishmaniasis. 2004. Disponible en
http://www.who.int/health-topics/leishmaniasis.htm Revisada el 9 de marzo de 2005. [ Links ]4. Davies CR, Kaye P, Croft SL, Sundar S. Leishmaniasis: new approaches to disease control. BMJ 2003;326:377-82. [ Links ]
5. Robledo S, Valencia A, Saravia N. Sensitivity to glucantime of Leishmania Viannia isolates from patients prior to treatment. J Parasitol 1999;85:360-6. [ Links ]
6. Berman J. Leishmaniasis. Curr Treat Opt Infect Dis 2001;3:333-6. [ Links ]
7. Batista P, Arribas A, Ferreira E. Leishmaniasis. What do we know about its chemotherapy. Brazilian J Phar Sci 2000;36:69-96. [ Links ]
8. Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, Fischer C et al. Oral miltefosine for Indian visceral leishmaniasis. N Eng J Med 2002;347:1739-46. [ Links ]
9. Baum KF, Berens RL. Successful treatment of cutaneous leishmaniasis with allopurinol after failure of treatment with ketoconazole. Clin Infect Dis 1994;18: 813-5. [ Links ]
10. Vélez I, Agudelo S, Hendrickx E, Puerta J, Grogl M, Modabber F et al. Inefficacy of allopurinol as monotherapy for Colombian cutaneous leishmaniasis. A randomized controlled trial. Ann Intern Med 1997;126: 232-6. [ Links ]
11. Hendrikcx E, Agudelo S, Muñoz D, Puerta J, Vélez I. Lack of efficacy of mefloquine in the treatment of new world cutaneous leishmaniasis in Colombia. Am J Trop Med Hyg 1998;59:889-92. [ Links ]
12. Laguna-Torres VA, Silva CA, Correia D, Carvalho EM, Magalhaes AE, Macedo V de O. Efficacy of mefloquine in the treatment of cutaneous leishmaniasis in an endemic area of Leishmania (Viannia) braziliensis. Rev Soc Bras Med Trop 1999;32:529-32. [ Links ]
13. Soto J, Buffet P, Grogl M, Berman J. Successful treatment of Colombian cutaneous leishmaniasis with four injections of pentamidine. Am J Trop Med Hyg 1994;50:107-11. [ Links ]
14. Ribeiro de Paula CD, Duarte Sampaio JH, Rizzo Cardoso D, Ribeiro Sampaio RN. Estudo comparativo da eficacia de isotianato de pentamidina administrada em tres doses durante uma semana e de N-metil-glucamina 20 mgSbV/kg/ dia durante 20 dias para o tratamento da forma cutanea da leishmaniose tegumentar americana. Rev Soc Bra Med Trop 2003;36: 365-71. [ Links ]
15. Jha TK, Sundar S, Thakur CP, Bachmann P, Karbwang J, Fisher C et al. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N Engl J Med 1999;341:1795-800. [ Links ]
16. Soto J, Toledo J, Gutiérrez P, Nicholls RS, Padilla J, Fischer C et al. Treatment of American cutaneous leishmaniasis with miltefosine, an oral agent. Clin Infect Dis 2001;33:E57-61. [ Links ]
17. Soto J, Arana BA, Toledo J, Rizzo N, Vega JC, Díaz A et al. Miltefosine for new world cutaneous leishmaniasis. Clin Infect Dis 2004;38:1266-72. [ Links ]
18. Berhe N, Ali A, Hailu A, Yeneneh H. Relapse in Ethiopian visceral leishmaniasis (VL) patients after therapy with pentavalent antimonials: a ten year observation. Acta Trop 1994;57:83-90. [ Links ]
19. Jackson JE, Tally JD, Ellis WY. Quantitative in vitro drug potency and drug susceptibility evaluation of Leishmania sp. from patients unresponsive to pentavalent antimony therapy. Am J Trop Med Hyg 1990;90:464-80. [ Links ]
20. Farault-Gambarelli F, Piarroux R, Deniau M, Giusiano B, Marty P, Michel G et al. In vitro and in vivo resistance of Leishmania infantum to meglumine antimoniate: a study of 37 strains collected from patients with visceral leishmaniasis. Antimicrob Agents Chemother 1997;41:827-30. [ Links ]
21. Grogl M, Thomason T, Franke E. Drug resistance in leishmaniasis: its implication in systemic chemotherapy of cutaneous and mucocutaneous disease. Am J Trop Med Hyg 1992;47:117-26. [ Links ]
22. Bhattacharyya A, Mukherjee M, Duttagupta S. Studies on stibanate unresponsive isolates of Leishmania donovani. J Biosci 2002;27:503-8. [ Links ]
23. Kerbel RS, Kobayashi H, Graham CH. Intrinsic or acquired drug resistance and metastasis: are they linked phenotypes? J Cell Biochem 1994;56:37-47. [ Links ]
24. Hayes J, Wolff R. Molecular mechanisms of drug resistance. Biochem J 1990;272:281-95. [ Links ]
25. Gottesman M, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 1993;62:385-427. [ Links ]
26. Grkovic S, Brown M, Skurray R. Regulation of bacterial drug export systems. Microbial Mol Biol Rev 2002;66: 671-701. [ Links ]
27. Zgurskaya H. Molecular analysis of efflux pump based antibiotic resistance. Int J Med Microbiol 2002;292:95-105. [ Links ]
28. Veen H, Konings W. Drug efflux proteins in multidrug resistance bacteria. Biol Chem 1997; 378:769-77. [ Links ]
29. Borst P, Ouellette M. New mechanisms of drug resistance in parasitic protozoa. Annu Rev Microbiol 1995;49:427-60. [ Links ]
30. Wolfger H, Mamnun Y, Kuchler K. Fungal ABC proteins: pleiotropic drug resistance, stress response and cellular detoxification. Res Microbiol 2001; 152:375-89. [ Links ]
31. Ambudkar S, Dey S, Hrycyna C, Ramachandra M, Pastan I, Gottesman M. Biochemical, cellular and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 1999;39:361-98. [ Links ]
32. Higgins C. ABC transporters: from microorganisms to man. Annu Rev Cell Biol 1992;8:67-113. [ Links ]
33. Doige C, Ames G. ATP-dependent transport systems in bacteria and humans. Relevance to cystic fibrosis and multidrug resistance. Annu Rev Microbiol 1993;47: 291-319. [ Links ]
34. Klokouzas A, Shahi S, Hladky SB, Barrand MA, Veen HW. ABC transporters and drug resistance in parasitic protozoa. Int J Antimicro Agents 2003;22:301-17. [ Links ]
35. Borst P, Evers R, Kool M, Wijnholds J. The multidrug resistance protein family. Biochim Biophys Acta 1999; 1461:347-57. [ Links ]
36. Kruh GD, Chan A, Myers K, Gaughan K, Miki T, Aaronson SA. Expression complementary DNA library transfer establishes mrp as a multidrug resistance gene. Cancer Res 1994;54:1649-52. [ Links ]
37. Ouellette M, Fase FF, Borst P. The amplified H circle of methotrexate resistant Leishmania tarentolae contains a novel P-glycoprotein gene. EMBO J 1990;9: 1027-33. [ Links ]
38. Dallagiovanna B, Gamarro F, Castanys S. Molecular characterization of a P-glycoprotein related tcpgp2 gene in Trypanosoma cruzi. Mol Biochem Parasitol 1996;75: 145-57. [ Links ]
39. Wilson C, Serrano A, Wasley A, Bogenschutz M, Shankar A, Wirth D. Amplification of a gene related to mammalian mdr genes in drug resistant Plasmodium falciparum. Science 1989;244:1184-6. [ Links ]
40. Parodi-Talice A, Araujo JM, Torres C, Perez-Victoria JM, Gamarro F, Castanys S. The overexpression of a new ABC transporter in Leishmania is related to phospholipid trafficking and reduced infectivity. Biochim Biophys Acta 2003;161:195-207. [ Links ]
41. Callahan H, Roberts W, Rainey P, Beverley S. The pgpA gene of Leishmania major mediates antimony (SbIII) resistance by decreasing influx and not by increasing efflux. Mol Biochem Pasasitol 1994;68:145-9. [ Links ]
42. Jonson P. Metronidazole and drug resistance. Parasitol Today 1993;9:183-6. [ Links ]
43. Upcroft J, Upcroft P. Drug resistance and Giardia. Parasitol Today 1993;9:187-90. [ Links ]
44. Peterson D, Walliker D, Wellems T. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in P. falciparum malaria. Proc Natl Acad Sci USA 1988;85: 9114-8. [ Links ]
45. Arrebola R, Olmo A, Reche P, Garvey EP, Santi DV, Ruiz-Perez LM et al. Isolation and characterization of a mutant dihydrofolate reductase-thymidylate synthase from methotrexate resistant Leishmania cells. J Biol Chem 1994;269:10590-6. [ Links ]
46. Ouellette M, Hettema E, Wust D, Fase FF, Borst P. Direct and inverted DNA repeats associated with P-glycoprotein gene amplification in drug resistant Leishmania. EMBO J 1991;10:1009-16. [ Links ]
47. Cowman A, Galatis D, Thompson J. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc Natl Acad Sci USA 1994;91:1143-7. [ Links ]
48. Foote SJ, Kyle DE, Martin RK, Oduola AM, Forsyth K, Kemp DJ et al. Several alleles of the multidrug resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature 1990;345: 255-8. [ Links ]
49. Wong A, Chow L, Wirth D. A homologous recombination strategy to analyze the vinblastine resistance property of the V-circle in Leishmania. Mol Biochem Parasitol 1994;64:75-86. [ Links ]
50. Ellenberger T, Beverley M. Multiple drug resistance and conservative amplification of the H region in Leishmania major. J Biol Chem 1989;264:15094-103. [ Links ]
51. Papadopoulou B, Roy G, Dey S, Rosen B, Olivier M, Ouellette M. Gene disruption of the P-glycoprotein related gene pgpA of Leishmania tarentolae. Biochem Biophys Res Commun 1996;224:772-8. [ Links ]
52. Callahan H, Beverley S. Heavy metal resistance: a new role for P-glycoproteins in Leishmania. J Biol Chem 1991;266:18427-30. [ Links ]
53. Chow L, Volkman S. Plasmodium and Leishmania: the role of mdr genes in mediating drug resistance. Exp Parasitol 1998;90:135-41. [ Links ]
54. Legare D, Hettema E, Ouellette M. The P-glycoprotein related gene family in Leishmania. Mol Biochem Parasitol 1994;68:81-91. [ Links ]
55. Henderson D, Sifri D, Rodgers M, Wirth D, Hendrickson N, Ullman B. Multidrug resistance in Leishmania donovani is conferred by amplification of a gene homologous to the mammalian mdr1 gene. Mol Cell Biol 1992;12:2855-65. [ Links ]
56. Hendrickson N, Sifri D, Henderson D, Allen T, Wirth D, Ullman B. Molecular characterization of the ldmdr1 multidrug resistance gene from Leishmania donovani. Mol Biochem Parasitol 1993;60:53-64. [ Links ]
57. Chow L, Wong A, Ullman B, Wirth D. Cloning and functional analysis of an extrachromosomally amplified multidrug resistance-like gene in Leishmania enriettii. Mol Biochem Parasitol 1993;60:195-208. [ Links ]
58. Urbina J. Lipid biosynthesis pathways as chemo-therapeutic targets in kinetoplastid parasites. Parasitology 1997;114:91-9. [ Links ]
59. Ullman B. Multidrug resistance and P-glycoproteins in parasitic protozoa. J Bioenerg Biomembr 1995;27:77-84. [ Links ]
60. Zalis M, Wilson C, Zhang Y, Wirth D. Characterization of the pfmdr2 gene for Plasmodium falciparum. Mol Biochem Parasitol 1993;62:83-92. [ Links ]
61. Torres C, Barreiro L, Dallagiovanna B, Gamarro F, Castanys S. Characterization of a new ATP-binding cassette transporter in Trypanosoma cruzi associated to a L1Tc retrotransposon. Biochim Biophys Acta 1999; 1489:428-32. [ Links ]
62. Maser P, Kaminsky R. Identification of three ABC transporter genes in Trypanosoma brucei spp. Parasitol Res1998;84:106-11. [ Links ]
63. Descoteaux S, Shen P, Ayala P, Orozco E, Samuelson J. P-glycoprotein genes of Entamoeba histolytica. Arch Med Res 1992;23:23-5. [ Links ]
64. Descoteaux S, Ayala P, Orozco E, Samuelson J. Primary sequences of two P-glycoprotein genes of Entamoeba histolytica. Mol Biochem Parasitol 1992;54: 201-11. [ Links ]
65. Gómez M, Pérez D, Ayala P, Samuelson J, Orozco E. Physiology and molecular biology of multidrug resistance in Entamoeba histolytica. Arch Med Res 1996;27:421-5. [ Links ]
66. Papadopoulou B, Roy G, Dey S, Rosen B, Ouellete M. Contribution of the Leishmania P-glycoprotein-related gene ltpgpA to oxyanion resistance. J Biol Chem 1994;269:11980-6. [ Links ]
67. Grondin K, Papadopoulou B, Ouellette M. Homologous recombination between direct repeat sequences yields P-glycoprotein containing amplicons in arsenite resistant Leishmania. Nucleic Acids Res 1993;21:1895-901. [ Links ]
68. Sánchez A, Castanys S, Gamarro F. Increased P-type ATPase activity in Leishmania tropica resistant to methotrexate. Biochem Biophys Res Commun 1994; 199:855-61. [ Links ]
69. Gamarro F, Chiquero M, Amador M, Legare D, Ouellette M, Castanys S. P-glycoprotein over-expression in methotrexate-resistant Leishmania tropica. Biochem Pharmacol1994;47:1939-47. [ Links ]
70. Gueiros-Filho FJ, Viola JP, Gomes FC, Farina M, Lins U, Bertho AL et al. Leishmania amazonensis: multidrug resistance in vinblastine-resistant promastigotes is associated with rhodamine 123 efflux, DNA amplification, and RNA overexpression of a Leishmania mdr1 gene. Exp Parasitol 1995;81:480-90. [ Links ]
71. Katakura K, Fujise H, Takeda K, Kaneko O, Torii M, Suzuki M et al. Overexpression of LaMDR2, a novel multidrug resistance ATP-binding cassette transporter, causes 5-fluorouracil resistance in Leishmania amazonensis. FEBS Letters 2004;561:207-12. [ Links ]
72. Katakura K, Iwanami M, Ohtomo H, Fujise H, Hashiguchi Y. Structural and functional analysis of the LaMDR1 multidrug resistance gene in Leishmania amazonensis. Biochem Biophys Res Commun 1999; 255:289-94. [ Links ]
73. Essodaigui M, Frezard F, Moreira E, Dagger F, Suillerot A. Energy dependent efflux from Leishmania promastigotes of substrates of the mammalian multidrug resistance pumps. Mol Biochem Parasitol 1999;100:73-84. [ Links ]
74. Ruetz S, Delling U, Brault M, Schurr E, Gros P. The pfmdr1 gene of Plasmodium falciparum confers cellular resistance to antimalarial drugs in yeast cells. Proc Natl Acad Sci USA 1996;93:9942-7.
75. Volkman S, Wilson C, Wirth D. Stage-specific transcripts of the Plasmodium falciparum pfmdr1 gene. Mol Biochem Parasitol 1993;57:203-11. [ Links ]
76. Lim A, Galatis D, Cowman A. Plasmodium falciparum: Amplification and overexpression of pfmdr1 is not necessary for increased mefloquine resistance. Exp Parasitol 1996;83:295-303. [ Links ]
77. Wilson CM, Volkman SK, Thaithong S, Martin R, Kyle D, Milhous W et al. Amplification of pfmdr1 associated with mefloquine and halofantrine resistance in Plasmodium falciparum from Thailand. Mol Biochem Parasitol 1993;57:151-60. [ Links ]
78. Grogl M, Martin R, Oduola A, Milhous W, Kyle D. Characteristics of multidrug resistance in Plasmodium and Leishmania: detection of P-glycoprotein-like components. Am J Trop Med Hyg 1991;45:98-111. [ Links ]
79. Ekong R, Robson K, Baker D, Warhurst D. Transcripts of the multidrug resistance genes in chloroquine sensitive and chloroquine resistant Plasmodium falciparum. Parasitology 1993;106:107-15. [ Links ]
80. Robello C, Navarro P, Castanys S, Gamarro F. A pteridine reductase gene ptr1 contiguous to a P-glycoprotein confers resistance to antifolates in Trypanosoma cruzi. Mol Biochem Parasitol 1997;90: 525-35. [ Links ]
81. Chiquero MJ, Perez JM, O'Valle F, Gonzalez JM, del Moral RG, Ferragut JA et al. Altered drug membrane permeability in a multidrug resistance Leishmania tropica line. Biochem Pharmacol 1998;55:131-9. [ Links ]
82. Denton H, McGregor C, Cooms GH. Reduction of anti-leishmanial pentavalent antimonial drugs by a parasite-specific thiol-dependent reductase, TDR1. Biochem J 2004;381:405-12. [ Links ]
83. Cunningham ML, Fairlamb AH. Trypanothione reductase from Leishmania donovani. Purification, characterisation and inhibition by trivalent antimonials. Eur J Biochem 1995;230:460-8. [ Links ]
84. Ludemann H, Dormeyer M, Sticherling C, Stallmann D, Follmann H, Krauth-Siegel RL. Trypanosoma brucei tryparedoxin, a thioredoxin-like protein in African trypanosomes. FEBS Lett 1998;431:381-5. [ Links ]
85. Mukhopadhyay R, Dey S, Xu N, Gage D, Lightbody J, Ouellette M et al. Trypanothione overproduction and resistance to antimonials and arsenicals in Leishmania. Proc Natl Acad Sci USA 1996;93:10383-7. [ Links ]
86. Legare D, Papadopoulou B, Roy G, Mukhopadhyay R, Haimeur A, Dey S et al. Efflux systems and increased trypanothione levels in arsenite-resistant Leishmania. Exp Parasitol 1997;87:275-82. [ Links ]
87. Grondin K, Haimeur A, Mukhopadhyay R, Rosen B, Ouellette M. Co-amplification of the glutamyl-cysteine synthetase gene gsh1 and of the ABC transporter gene pgpA in arsenite resistant Leishmania tarentolae. EMBO J 1997;16:3057-65. [ Links ]
88. Krauth-Siegel RL, Ludemann H. Reduction of dehydroascorbate by trypanothione. Mol Biochem Parasitol 1996;80:203-8. [ Links ]
89. Nogoceke E, Gommel DU, Kiess M, Kalisz HM, Flohe L. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism in Crithidia fasciculata. Biol Chem 1997;378:827-36. [ Links ]
90. Gommel DU, Nogoceke E, Morr M, Kiess M, Kalisz HM, Flohe L. Catalytic characteristics of tryparedoxin. Eur J Biochem 1997;248:913-8. [ Links ]
91. Montemartini M, Kalisz HM, Kiess M, Nogoceke E, Singh M, Steinert P et al. Sequence, heterologous expression and functional characterization of a novel tryparedoxin from Crithidia fasciculata. Biol Chem 1998; 79:1137-42. [ Links ]
92. Tetaud E, Fairlamb AH. Cloning, expression and reconstitution of the trypanothione-dependent peroxidase system of Crithidia fasciculata. Mol Biochem Parasitol 1998;96:111-23. [ Links ]
93. Levick MP, Tetaud E, Fairlamb AH, Blackwell JM. Identification and characterisation of a functional peroxidoxin from Leishmania major. Mol Biochem Parasitol 1998;96:125-37. [ Links ]
94. Haimeur A, Brochu C, Genest P, Papadopoulou B, Ouellette M. Amplification of the ABC transporter gene PGPA and increased trypanothione levels in potassium antimonyl tartrate (SbIII) resistant Leishmania tarentolae. Mol Biochem Parasitol 2000;108:131-5. [ Links ]
95. Dey S, Ouellette M, Lightbody J, Papadopoulou B, Rosen B. An ATP-dependent As(III) glutathione transport system in membrane vesicles of Leishmania tarentolae. Proc Natl Acad Sci USA 1996;93:2192-7. [ Links ]
96. Arana F, Pérez J, Repetto Y, Morello A, Castanys S, Gamarro F. Involvement of thiol metabolism in resistance to glucantime in Leishmania tropica. Biochem Pharmacol 1998;56:1201-8. [ Links ]
97. Haimeur A, Guimond C, Pilote S, Mukhopadhyay R, Rosen BP, Poulin R et al. Elevated levels of polyamines and trypanothione resulting from overexpression of the ornithine decarboxylase gene in arsenite-resistant Leishmania. Mol Microbiol 1999;4: 726-35. [ Links ]
98. Ferreira KC, Miranda AL, Anacleto C, Fernandes AP, Abdo MC, Petrillo-Peixoto ML, et al. Leishmania (V.) guyanensis: isolation and characterization of glucantime-resistant cell lines. Can J Microbiol 1996;42:944-9. [ Links ]
99. Medallo W, Horwitz S. Phosphorylation of the multidrug resistance associated glycoprotein. Biochemistry 1987; 26:6900-4. [ Links ]
100. Gottesman M, Hrycyna C. Genetic analysis of the multidrug transporter. Annu Rev Genet 1995;29:607-49. [ Links ]
101. Chambers T, Jacobs J, Eilon G. Protein kinase C phosphorylates P-glycoprotein in multidrug resistance human KB carcinoma cell. J Biol Chem 1990;265:7679-86. [ Links ]
102. Bates S, Currier S, Alvarez M, Fojo A. Modulation of P-glycoprotein phosphorylation and drug transport by sodium butyrate. Biochemistry 1992;31:6366-72. [ Links ]
103. Bates S, Lee J, Dickstein B, Spolyar M, Fojo A. Differential modulation of P-glycoprotein transport by protein kinase inhibition. Biochemistry 1993;32:9156-64. [ Links ]
104. Conseil G, Perez JM, Jault JM, Gamarro F, Goffeau A, Hofmann J et al. Protein kinase C effectors bind to multidrug ABC transporters and inhibit their activity. Biochemistry 2001;40:2564-71. [ Links ]
105. Borner C, Filipuzzi I, Wartman M, Eppenberger U, Fabbro D. Continuous synthesis of two protein kinase C related proteins after down-regulation by phorbol esters. Proc Natl Acad Sci USA 1988;85:2110-4. [ Links ]
106. Huang F, Yhosida Y, Cuhna M, Beaven M, Huang K. Differential down-regulation of protein kinase C isozymes. J Biol Chem 1989;264:4238-43. [ Links ]
107. Smith C, Zilfou J. Circumvention of P-glycoprotein mediated multidrug resistance by phosphorylation modulators is independent of protein kinases. J Biol Chem 1995;270:28145-52. [ Links ]
108. Goodfellow H, Sardini A, Ruetz S, Callaghan R, Gros P, McNaughton PA et al. Protein kinase C mediated phosphorylation does not regulate drug transport by the human multidrug resistance P-glycoprotein. J Biol Chem 1996;271:13668-74. [ Links ]
109. Ford J. Experimental reversal of P-glycoprotein mediated multidrug resistance by pharmacological chemosensitisers. Eur J Cancer 1996;32A:991-1001. [ Links ]
110. Chiba P, Holzer W, Landau M. Substituted 4-acylpyrazoles and 4-acylpyrazolones: synthesis and multidrug resistance modulating activity. J Med Chem 1998;41:4001-11. [ Links ]
111. Hofsli E, Nissen MJ. Reversal of multidrug resistance by lipophilic drugs. Cancer Res 1990;50:3997-4002. [ Links ]
112. Pawagi A, Wang J, Silverman R, Reithmeier F, Deber C. Transmembrane aromatic amino acid distribution in P-glycoprotein: a functional role in broad substrate specificity. J Mol Biol 1994;235:554-64. [ Links ]
113. Klopman G, Shi L, Ramu A. Quantitative structure activity relationship of multidrug resistance reversal agents. Mol Pharmacol 1997;52:323-34. [ Links ]
114. Tsuruo T, Lida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981;41:1967-72. [ Links ]
115. Tsuruo T, Lida H, Tsukagoshi S, Sakurai Y. Increased accumulation of vincristine and adriamicine in drug resistance P388 tumor cells following incubation with calcium antagonist and calmodulin inhibitors. Cancer Res 1982;42:4730-3. [ Links ]
116. Yusa K, Tsuruo T. Reversal mechanism of multidrug resistance by verapamil: Direct binding of verapamil to P-glycoprotein on specific sites and transport of verapamil outward across the plasma membrane of K562/ADM cells. Cancer Res 1989;49:5002-6. [ Links ]
117. Merlin J, Guerci A, Marchal S. Comparative evaluation of S9788, verapamil and cyclosporine A in K562 human leukemia cell lines and in P-glycoprotein expressing samples from patients with hematologic malignancies. Blood 1994;84:262-9. [ Links ]
118. Krogstad DJ, Gluzman IY, Kyle DE, Oduola AM, Martin SK, Milhous WK et al. Efflux of chloroquine from Plasmodium falciparum: mechanism of chloroquine resistance. Science 1987;238:1283-5. [ Links ]
119. Martin S, Oduola A, Milhous W. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 1987;235:899-901. [ Links ]
120. Menezes C, Kirchgatter K, Di SS. In vitro evaluation of erythromycin in chloroquine resistant Brazilian P. falciparum freshly isolates: modulating effect and antimalarial activity evidence. Rev Inst Med Trop S Paulo 1999;41:249-53. [ Links ]
121. Guan J, Kyle D, Gerena L, Zhang Q, Milhous W, Lin A. Design, synthesis, and evaluation of new chemosensitizers in multidrug resistant Plasmodium falciparum. J Med Chem 2002;45:2741-8. [ Links ]
122. Ford J, Prozialeck W, Hait W. Structural features determining activity of phenothiazine and related drugs for inhibition of cell gowth and reversal of MDR. Mol Pharmacol 1989;35:105-15. [ Links ]
123. Ford JM, Bruggesman EP, Pastan I, Gotesmann MM, Hait WN. Cellular and biochemical characterization of thioxanthenes for reversal multidrug resistance in human and murine cell lines. Cancer Res 1990;50:1748-56. [ Links ]
124. Pajeva I, Wiese M. Molecular modelling of pheno-thiazines and related drugs as multidrug resistance modifiers: a comparative molecular field analysis study. J Med Chem 1998;41:1815-26. [ Links ]
125. Twentyman P. Cyclosporins as drug resistance modifiers. Biochem Pharmacol 1992;43:109-17. [ Links ]
126. Gruol D, Bourgeois S. Chemosensitizing steroids: Glucocorticoid receptor agonists capable of inhibiting P-glycoprotein function. Cancer Res 1997;57:720-7. [ Links ]
127. Ecker G, Chiba P, Hitzler M, Schmid D, Visser K, Cordes HP et al. Structure activity relationship studies on benzofuran analogs of propafenone type modulators of tumor cell multidrug resistance. J Med Chem 1996;39:4767-74. [ Links ]
128. Kaur J, Dey C. Putative P-glycoprotein expression in arsenite-resistant Leishmania donovani down-regulated by verapamil. Biochem Biophys Res Commun 2000;271:615-9. [ Links ]
129. Ford J, Haith W. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev 1990; 42:155-99. [ Links ]
130. Kosugi K, Sakai J, Zhang S, Watanabe Y, Sasaki H, Suzuki T et al. Neutral taxoids from Taxus cuspida as modulators of multidrug-resistant tumor cells. Phytochemistry 2000;54:839-45. [ Links ]
131. Ma X, Wang T, Yin L, Pan Y. Two pimarane diterpenoids from Ephemerantha lonchophylla and their evaluation as modulators of the multidrug resistance phenotype. J Nat Prod 1998;61:112-5. [ Links ]
132. Kim S, Kim Y, Lee J. A new sesquiterpene ester from Celastrus orbiculatus reversing multidrug resistance in cancer cells. J Nat Prod 1998;61:108-11. [ Links ]
133. Hasegawa H, Sung JH, Matsumiya S, Uchiyama M, Inouye Y, Kasai R et al. Reversal of daunomycin and vinblastine resistance in multidrug-resistant P388 leukemia in vitro through enhanced cytotoxicity by triterpenoids. Planta Med 1995;61:409-13. [ Links ]
134. Kim S, Kim Y, Kim C, Lee J. Torilin, a sesquiterpene from Torilis japonica, reverses multidrug resistance in cancer cells. Planta Med 1998;64:332-4. [ Links ]
135. Kim S, Kim Y, Kim C, Lee J. Mode of action of torilin in multidrug-resistant in cancer cell lines. Planta Med 1998;64:335-8. [ Links ]
136. Kim S, Kim H, Hong Y, Kim Y, Lee J. Sesquiterpene esters from Celastrus orbiculatus and their structure activity relationship on the modulation of multidrug resistance. J Nat Prod 1999;62:697-700. [ Links ]
137. Muñoz-Martinez F, Lu P, Cortes-Selva F, Perez-Victoria JM, Jimenez IA, Ravelo AG et al. Celastraceae sesquiterpenes as a new class of modulators that bind specifically to human P-glycoprotein and reverse cellular multidrug resistance. Cancer Res 2004;64:7130-8. [ Links ]
138. Kennedy ML, Cortes F, Perez JM, Jimenez IA, Gonzalez AG, Munoz OM et al. Chemosensitization of a multidrug resistant Leishmania tropica line by new sesquiterpenes from Maytenus magellanica and Maytenus chubutensis. J Med Chem 2001;44:4668-76. [ Links ]
139. Perez-Victoria JM, Tincusi BM, Jimenez IA, Bazzocchi IL, Gupta MP, Castanys S et al. New natural sesquiterpenes as modulators of daunomycin resistance in a MDR Leishmania tropica line. J Med Chem 1999;42:4388-93. [ Links ]
140. Nissler L, Gebhardt R, Berger S. Flavonoid binding to a multi-drug-resistance transporter protein: an STD-NMR study. Annal Bioanal Chem 2004;379:1045-9. [ Links ]
141. Shapiro A, Ling V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem Pharmacol 1997;53:587-96. [ Links ]
142. Bois F, Beney C, Boumendjel A, Mariotte A, Conseil G, Di PA. Halogenated chalcones with high affinity binding to P-glycoprotein: potential modulators of multidrug resistance. J Med Chem 1998;41:4161-4. [ Links ]
143. Ferte J, Kuhnel J, Chapuis G, Rolland Y, Lewin G, Schwaller M. Flavonoid related modulators of multidrug resistance: synthesis pharmacological activity, and structure-activity relationships. J Med Chem 1999;42:478-89. [ Links ]
144. Dayan G, Jault JM, Baubichon H, Baggetto LG, Renoir JM, Baulieu EE et al. Binding of steroid modulators to recombinant cytosolic domain from mouse P-glycoprotein in close proximity to the ATP site. Biochemistry 1997;36:15208-15. [ Links ]
145. Conseil G, Cortay H, Dayan G, Jault J, Barron D, Di PA. Flavonoids: a class of modulators with bifunctional interaction at vicinal ATP and steroid-binding sites on mouse P-glycoprotein. Proc Natl Acad Sci USA 1998;95:9831-6. [ Links ]
146. Di Pietro A, Conseil G, Perez JM, Dayan G, Baubichon H, Trompier D et al. Modulation by flavonoids of cell multidrug resistance mediated by P-glycoprotein and related ABC transporters. Cell Mol Life Sci 2002;59:307-22. [ Links ]
147. Perez-Victoria JM, Chiquero MJ, Conseil G, Dayan G, Di Pietro A, Barron D et al. Correlation between the affinity of flavonoids binding to the cytosolic site of Leishmania tropica multidrug transporter and their efficiency to revert parasite resistance to daunomycin. Biochemistry 1999;38:1736-43. [ Links ]
148. Perez-Victoria JM, Perez-Victoria FJ, Conseil G, Maitrejean M, Comte G, Barron D, et al. High affinity binding of silybin derivatives to the nucleotide binding domain of a Leishmania tropica P-glycoprotein like transporter and chemosensitization of a multidrug-resistant parasite to daunomycin. Antimicrob Agents Chemother 2001;45:439-46. [ Links ]
149. Martin C, Berrdge G, Higgins C, Mistry P, Charlton P, Callaghan C. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol 2000; 58:624-32. [ Links ]
150. Rho M, Totoshima M, Hayashi M, Subramaniam G, Kam T, Komiyama K. Reversal of multidrug resistance by kopsiflorine isolated from Kopsia dasyrachis. Planta Med 1999;65:307-10. [ Links ]
151. Frappier F, Jossang A, Soudon J, Calvo F, Rasoanaivo P, Ratsimamanga S et al. Bisbenzyl-isoquinolines as modulators of chloroquine resistance in Plasmodium falciparum and multidrug resistance in tumor cells. Antimicrob Agents Chemother 1996;40: 1476-81. [ Links ]
152. Frédérich M, Hayette M, Tits M, Mol P, Angenot L. Reversal of chloroquine and mefloquine resistance in Plasmodium falciparum by the two monoindole alkaloids, Icajine and Isoretuline. Planta Med 2001;67: 523-7. [ Links ]
153. Pajeva I, Wiese M. Molecular modeling of phenothiazines and related drugs as multidrug resistance modifiers: a comparative molecular field analysis study. J Med Chem 1998;41:1815-26. [ Links ]
154. Ecker G, Hubber M, Schmid D, Chiba P. The importance of a nitrogen atom in modulators of multidrug resistance. Mol Pharmacol 1999;56:791-6. [ Links ]

