










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.25 no.3 Bogotá Sept. 2005
Detección de mutaciones de los genes hMLH1 y hMSH2 del sistema de reparación de malos apareamientos del ADN en familias colombianas sospechosas cáncer colorrectal no polipósico hereditario (síndrome de Lynch)
Andrea Gómez1, Gustavo Salguero1, Herbert García1, Fabio Aristizábal2, Óscar Gutiérrez1, Luis Alberto Ángel1, Jorge Padrón3, Carlos Martínez4, Humberto Martínez5, Omar Malaver6, Rosa Barvo7, Alejandro Giraldo1,8
1 Facultad de Medicina e Instituto de Genética, Universidad Nacional de Colombia, Bogotá, D. C., Colombia.
2
Departmento de Farmacia e Instituto de Biotecnología, Universidad Nacional de Colombia, Bogotá, D. C., Colombia.3
Facultad de Medicina, Universidad del Rosario, Bogotá, D. C., Colombia.4
Servicio de Coloproctología, Hospital Militar Central, Bogotá, D. C., Colombia.5
Departamento de Cirugía, Hospital de la Policía, Bogotá, D. C., Colombia.6
Departmento de Cirugía, Clínica San Pedro Claver, ISS, Bogotá, D. C., Colombia.7
Servicio de Gastroenterología, Clínica La Asunción, Barranquilla, Colombia.8
Fundación Arthur Stanley Gillow, Bogotá, D. C., Colombia.Introducción.
El cáncer colorrectal es la segunda causa de morbilidad y mortalidad por cáncer en los países desarrollados. En Colombia es la quinta causa de muerte entre los diferentes cánceres. Cerca del 75% de éstos corresponde a cánceres esporádicos, alrededor del 25% son familiares, y son claramente hereditarios el 5%. De éstos, el más importantes es el cáncer colorrectal no polipósico hereditario o síndrome de Lynch.Objetivo. Analizar los dos genes más importantes involucrados en el síndrome de Lynch, el hMLH1 y el hMSH2.
Materiales y métodos. En 17 familias colombianas que cumplían con los criterios de Ámsterdam II o las pautas de Bethesda, se analizaron por SSCP los 35 exones de estos dos genes y las variantes electroforéticas se secuenciaron.
Resultados. Se detectaron 8 mutaciones de línea germinal en las familias analizadas, 7 en el gen hMLH1 y 1 en hMSH2, y se encontró una tasa de detección de mutaciones del 47%. Seis de las 8 mutaciones encontradas en este estudio han sido previamente reportadas en la literatura. Un cambio de una base en el sitio donador de empalme en el exón 9 del gen hMLH1 (G>A) (dos familias), un cambio A>G en el codón 755 del exón 17, y un cambio G>A en el exón 18. Se detectaron dos nuevas mutaciones, una en el exón 17, un cambio C>T en el codón 640, y una deleción de TG en el codón 184 del exón 3 del gen hMSH2. También se detectó en dos familias un polimorfismo del intrón 13 del hMLH1.
Conclusión. Este es el primer estudio realizado en Colombia que detecta mutaciones en el síndrome de Lynch y pretende establecer un programa integral de manejo y prevención.
Palabras clave: síndrome de Lynch, neoplasmas colorrectales hereditarios sin poliposis, genes, reparación del ADN, disparidad de par base.
Detection mutations in the DNA mismatch repair genes of hMLH1 and hMSH2 genes in Colombian families with suspicion of hereditary non-polyposis colorectal carcinoma (Lynch syndrome)
Introduction. Colorectal cancer (CRC) is the second highest cause of cancer mortality in developed countries. In Colombia, CRC ranks fifth as a cause of cancer death. Approximately 75% of CRC appear to be spontaneous and 25% are familial, with 5% of the latter clearly hereditary. Of these, hereditary non-polyposis colorectal carcinoma (HNPCC)-or Lynch syndrome is the most important.
Objective. Herein, the two most important genes involved in Lynch syndrome, the hMLH1 and hMSH2 were analyzed for presence of mutations.
Materials and methods. Seventeen Colombian families that fulfilled the Amsterdam II criteria or Bethesda guidelines for Lynch syndrome were selected. The of 35 exons of hMLH1 and hMSH2 genes were screened by SSCP and those with electrophoretic variants were sequenced.
Results. Eight germinal mutations were detected, corresponding to a 47% detection mutation rate. Six of the eight mutations have previously been reported. These consisted of the following mutations: a single base substitution at the donor splicing site of exon 9, a single base substitution (A>G) at codon 755 of the exon 17, and another single base substitution (G>A) at codon 681 of exon 18. The two novel mutations consisted of a single base substitution (C>T) at codon 640 of exon 17 of the hMLH1 gene and a two-nucleotide deletion (TG) at codon 184 of exon 3 of hMSH2 gene. In addition, two families were observed with a polymorphism in the intron 13 (G>A) nt 1558+14, of hMLH1 gene.
Conclusions. This study represented the first survey for detecting mutations associated with Lynch syndrome in Colombia, and is intended to lead to the establishment of a management and prevention program.
Key words: Lynch syndrome, colorectal neoplasms, hereditary nonpolyposis, DNA repair, base pair mismatch, genes.
El cáncer colorrectal es una de las principales causas de morbilidad y mortalidad en los países desarrollados. Cerca del 75% corresponden a cánceres colorrectales esporádicos, mientras que alrededor del 25% son familiares, y son claramente hereditarios el 5%. De éstos, los más importantes son el cáncer colorrectal no polipósico hereditario o síndrome de Lynch y la poliposis adenomatosa familiar, el primero con una frecuencia de 80%, aproximadamente, de todos los cáncer colorrectal hereditarios y el segundo, con 20% (1,2).
En Colombia, el cáncer colorrectal es la quinta causa de muerte por cáncer (3,4). El síndrome de Lynch es una enfermedad autosómica dominante con un rango variable de inicio de la enfermedad de 25 a 60 años y una penetrancia de 85% (1,2,5). El síndrome de Lynch presenta carcinomas de colon localizados en el colon proximal y hacia el extremo derecho y es característica la presentación de sincronía y metacronía de los carcinomas. Además, se presentan cánceres extracolónicos, principalmente, carcinoma endometrial seguido por carcinoma de ovario, estómago, intestino delgado, tracto hepatobiliar, carcinoma de células de transición de uréter y pelvis renal y también adenoma sebáceo (1, 5). Las familias con cáncer colorrectal no polipósico hereditario presentan mutaciones de línea germinal en los genes de reparación del mal apareamiento de las bases en el ADN (MMR), principalmente, en hMLH1, hMSH2 y, también, en hMSH6, hPMS1 y hPMS2 (6-9). La identificación de las familias con cáncer colorrectal no polipósico hereditario es algunas veces difícil, ya que no existe un fenotipo clínico premórbido, como si lo existe en la poliposis adenomatosa familiar.
En 1991, el Grupo Colaborativo Internacional sobre cáncer colorrectal no polipósico hereditario estableció los criterios de Amsterdam para la identificación de estas familias, los cuales fueron modificados en 1999 (10-11). Se denomina ahora criterios de Amsterdam II, y son los siguientes: 1) por lo menos, tres pacientes histológicamente verificados con cáncer colorrectal, uno de ellos en pariente en primer grado de los otros dos, o con cáncer endometrial, cáncer gástrico y carcinoma de uréter y pelvis renal; 2) por lo menos, dos generaciones sucesivas afectadas; 3) por lo menos, un miembro afectado y diagnosticado antes de los 50 años; 4) se debe haber excluido la poliposis adenomatosa familiar.
Dado que estos criterios son muy estrictos y no incluían a muchas familias que sugerían el perfil de cáncer colorrectal no polipósico hereditario y con el fin de realizar estudios moleculares, en 1997 se establecieron las pautas de Bethesda que permiten la inclusión de un mayor número de familias (12) e incluyen: 1) individuos con cáncer que pertenecen a familias que cumplen los criterios de Ámsterdam; 2) individuos con dos carcinomas relacionados con cáncer colorrectal no polipósico hereditario incluido cáncer colorrectal metacrónicos y sincrónicos, o cánceres extracolónicos asociados; 3) individuos con cáncer colorrectal y un familiar con cáncer colorrectal en primer grado o cáncer colorrectal no polipósico hereditario con cáncer extracolónico o adenoma colorrectal (uno de los cánceres diagnosticados antes de los 45 años y el adenoma antes de los 40 años; 4) individuos con cáncer colorrectal o con cáncer endometrial antes de los 45 años; 5) individuos con cáncer colorrectal con histopatología no diferenciada diagnosticados antes de lo 45 años; 6) individuos con adenomas diagnosticados antes de los 40 años.
Hasta la fecha se han descrito en la base de dados de cáncer colorrectal no polipósico hereditario http://www.nfdht.nl más de 400 mutaciones patogénicas, principalmente, en hMLH1 y hMSH2. Este el primer estudio molecular realizado en Colombia para detectar familias con cáncer colorrectal no polipósico hereditario e identificar mutaciones presentes en los genes hMLH1 y hMSH2. En este artículo se describen los resultados del estudio de 17 familias en las cuales se buscaron mutaciones de los genes hMLH1 y hMSH2.
Materiales y métodos
Familias
Se estudiaron 17 familias colombianas que cumplían con los criterios de Ámsterdam o las pautas de Bethesda, provenientes de la consulta de gastroenterología de hospitales de Bogotá, Medellín y Barranquilla, y de la práctica privada de varios especialistas. Estas familias fueron seleccionadas de un grupo de 36 familias de igual número de pacientes con carcinoma colorectal que informaron a sus médicos antecedentes familiares.
De las 17 familias seleccionadas, se estudió un caso índice, aunque en algunas familias se estudiaron dos o tres individuos afectados. De la misma manera, se estudiaron de dos a cuatro individuos sanos. En dos familias (UN-16 y UN- 17) no se estudiaron individuos afectados porque todos habían fallecido para la época del estudio, por lo que los casos índices aún no habían desarrollado tumores. Cada paciente del estudio firmó un consentimiento informado y, a través de ellos, se obtuvo la historia clínica y familiar.
Extracción de ADN
Para realizar la extracción se utilizaron 5 ml de sangre con EDTA. Se empleó la técnica de salting out (13); el ADN extraído se cuantificó con espectrofotómetro y se visualizó en un gel de agarosa al 1% teñido con bromuro de etidio.
Amplificación de los genes hMLH1 y hMSH2
Se amplificaron los 16 exones del gen hMSH2 y los 19 del gen hMLH1 utilizando los iniciadores descritos previamente por Weber et al. (14). Para los exones 1 y 11 del gen hMSH2 y 10 del gen hMLH1 se modificaron los iniciadores con el programa Primer 3 (http://www.genome.wi.mit.edu/cgibin/primer/primer3), ya que no se obtuvo la amplificación esperada.
Análisis de polimorfismo de conformación de cadena sencilla (SSCP)
Con el fin de detectar cambios de conformación del ADN sugestivos de mutaciones, se evaluaron los 35 exones de los dos genes con la técnica de polimorfismo de conformación de cadena sencilla (SSCP), para lo cual se estandarizaron las condiciones necesarias teniendo en cuenta la composición y el tamaño del fragmento. Se desnaturalizaron las muestras utilizando el tampón de SSCP (formamida al 95%, EDTA al 0,5M, NaOH 0,4N y 0,0025% de xilencianol y azul de bromofenol) en una proporción 1:9 (1 volumen de muestra por 9 volúmenes de tampón desnaturalizante). Los geles de SSCP se corrieron en una cámara The Dcode de BIO-RAD, a 4°C, en geles de poliacrilamida no desnaturalizantes del 12% al 15%, con un voltaje de 300-500 voltios y con un tiempo de corrido de 4 a 16 horas. Los geles se visualizaron utilizando la técnica de nitrato de plata convencional.
Secuenciación del ADN
Se secuenciaron las muestras que presentaron un patrón de migración anormal en el análisis de SSCP, comparado con el de los controles normales. Se utilizó el estuche Big Dye Terminator y la electroforesis capilar de las muestra se realizó en el analizador genético ABI Prism 310 (Applied Biosystems). Todas las mutaciones se confirmaron por duplicado y se secuenciaron en ambas direcciones. La secuencia obtenida se comparó con la base de datos del Grupo Colaborativo Internacional de Cáncer Colorrectal no polipósico hereditario (http://www.nfdht.nl/database/mdbchoice.htm) y la Human gene mutation database (http:// archive.uwcm.ac.uk/uwcm/mg/ search/203983. html).
Resultados
Se estudiaron 17 familias colombianas en un periodo comprendido entre febrero de 2000 y abril de 2003; 8 familias cumplían los criterios de Ámsterdam II y 9 familias las pautas de Bethesda. Las características generales de cada familia se observan en el cuadro 1. De las 17 familias analizadas, 6 presentaron mutaciones patogénicas y dos familias, una variante polimórfica (cuadro 2). Un alto porcentaje de éstas corresponde a mutaciones en el gen hMLH1 88% (7/8) frente al 12% (1/8) en el gen hMSH2. Tres mutaciones ya están reportadas en la literatura como patogénicas; dos familias (UN-1 y UN-6) presentaron la misma mutación, un cambio de una base en el sitio donador de empalme en el exón 9 del gen hMLH1 (G>A) (15); una familia (UN-16) presentó un cambio de A>G en el codon 755 del exón 17 del gen hMLH1 que produce un cambio de isoleucina/ valina (16), y una familia (UN-3) presentó cambio de G>A en el exón 18 del gen MLH1 que produce un cambio de alanina/treonina (17) (figura 1). Se detectaron 2 nuevas mutaciones patogénicas, una en el exón 17, un cambio de C>T en el codón 640 (UN-4) del gen hMLH1 que produce un cambio de prolina/serina y una deleción de TG en el codón 184 del exón 3 del gen (UN-2) hMSH2 que produce un cambio de cisteína/stop. También se detectó en dos familias, un polimorfismo del intrón 13 del gen hMLH1 (UN-8 y UN-17) un cambio de G>A en la posición 1558 +14 (18) y se determinó una frecuencia en población colombiana del 6,5% (figura 2).
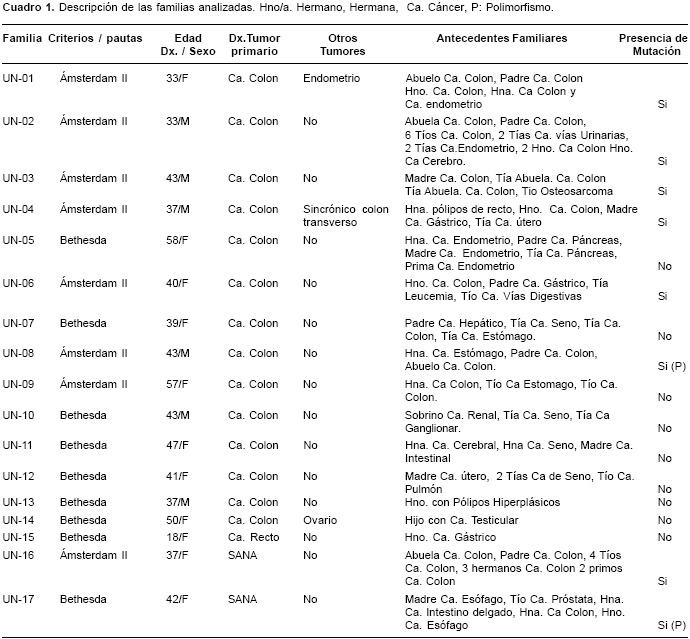
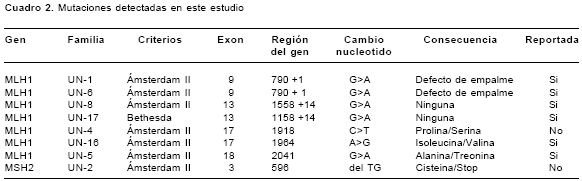
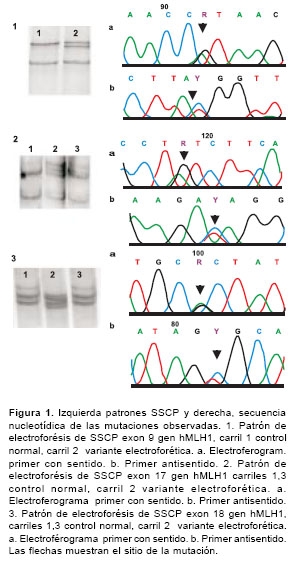
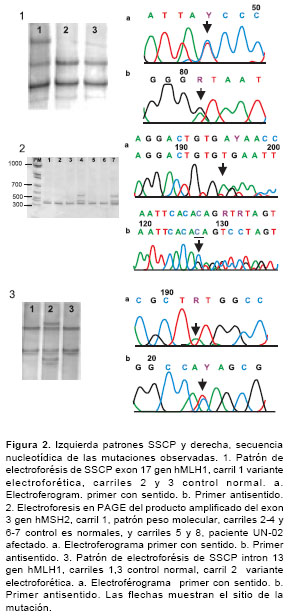
Al considerar las mutaciones con respecto a los criterios de Ámsterdam II y a las pautas de Bethesda, se observó que, de las mutaciones encontradas, 7 se presentaron en familias que cumplieron los criterios de Ámsterdam II (88%, 7/8), mientras que sólo 1 mutación se encontró en una familia que cumplió las pautas de Bethesda (11%, 1/9); esta diferencia fue estadísticamente significativa (prueba exacta de Fisher, p=0,005).
El tipo y el porcentaje de tumores presentes en las familias estudiadas se observa en el
cuadro 3. El cáncer más frecuente es el cáncer colorrectal, seguido por el cáncer de endometrio/ ovario y el cáncer de estómago. También se observaron cánceres poco frecuentes en el espectro de cáncer colorrectal no polipósico hereditario como el de riñón/vías urinarias, cerebro e intestino delgado. Cánceres como los trastornos linfoproliferativos, seno, próstata, pulmón y cara que, usualmente, no se incluyen dentro del espectro de tumores de cáncer colorrectal no polipósico hereditario, se presentaron en algunas de las familias estudiadas.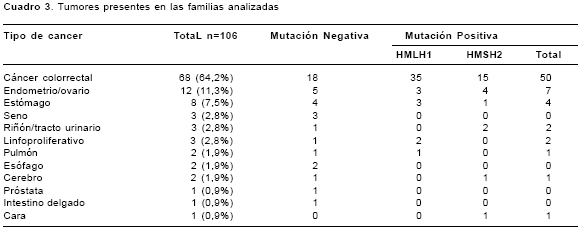
En cuanto al cáncer de colon, se presentaron 52 casos entre los familiares en los que se conoció la edad de diagnóstico del cáncer en las 17 familias estudiadas. De éstos, 36 estuvieron presentes en familias que cumplían con los criterios de Amsterdam y cuya edad de diagnóstico fue, en promedio, de 43,9 años. En las familias que cumplieron los criterios de Bethesda se presentaron 16 casos con una edad promedio de diagnóstico a los 46,7 años. No hubo diferencias entre la presencia de cáncer de colon y el sexo de los pacientes.
Discusión
El presente estudio es el resultado del análisis del ADN genómico para los genes hMLH1 y hMSH2 en 17 familias colombianas no relacionadas, sugestivas de presentar cáncer colorrectal no polipósico hereditario. Se detectaron 8 mutaciones en 17 familias lo que correspondió a una tasa de detección del 47%. Esta tasa de detección es semejante a lo publicado por diferentes autores en varios países de Europa, Norteamérica y Latinoamérica (14-21). La aplicación de los criterios de inclusión juegan un papel importante en la identificación de mutaciones en las familias con cáncer colorrectal no polipósico hereditario. La selección de familias utilizando los criterios de Amsterdam II pueden generar una probabilidad de detección de la mutación de cerca del 60%; cuando los criterios son menos estrictos como las pautas de Bethesda, la tasa de detección de mutación disminuye a valores cercanos al 34% (22). En el presente estudio, 8 de las 17 familias satisfacían los criterios de Ámsterdam II, por lo que al analizar por separado estas familias, la tasa de detección de mutaciones se elevó al 88% (7/8 familias), lo cual es mucho mayor al valor reportado por otros estudios (14-25). Al analizar las familias que cumplían las pautas de Bethesda, sólo se detectó el 11% de las mutaciones (1/9 familias). Esta diferencia resultó altamente significativa (P=0,005) Por lo anterior, se puede concluir que los criterios de Ámsterdam II son claves en la identificación de familias sugestivas de sufrir cáncer colorrectal no polipósico hereditario, pero las pautas de Bethesda también permiten identificar mutaciones.
En el presente estudio se encontró una proporción mayor de mutaciones en hMLH1 con respecto a hMSH2, 88% vs. 12%, respectivamente. Estos resultados concuerdan con lo reportado recientemente para la población brasilera en los que se observó una frecuencia de mutaciones en hMLH1 de 80% contra 20% en hMSH2 (22). Las mutaciones patogénicas detectadas en el gen hMLH1 fueron del tipo de cambio de sentido; dos familias presentaron la misma mutación, una substitución en la posición 790 +1 de G/A en el sitio aceptor de empalme del exón 9 del gen hMLH1 (15); estas familias no están relacionadas aparentemente entre sí, pero provienen de la misma región geográfica (departamento de Boyacá), lo que podría sugerir un posible efecto fundador).
Se detectaron dos nuevas mutaciones, la primera en la familia (UN-04) en el exón 17 del gen hMLH1 consistió de un reemplazo de C>T en el codón 640 que produce un cambio del aminoácido de Pro/Ser. Esta mutación podría ser sólo un polimorfismo, pero se considera patogénica por las siguientes razones: 1) se produce un cambio en aminoácidos de polaridades diferentes que puede alterar considerablemente la estructura de la proteína, lo cual podría interferir la interacción de la proteína con sus homólogos como hMLH3 o hPMS2, ya que la mutación se encuentra en la región de interacción con estos genes; 2) el cambio se encuentra en una posición muy conservada entre diferentes organismos, como S. cervisiae y M. musculus, y está en la primera posición del codón CCC/TCC, lo que es inusual en las mutaciones polimórficas; 3) al analizar la familia, la mutación se segrega con la enfermedad ya que dos hermanos afectados del caso índice tienen la mutación, y una hermana sana no la presenta; 4) en un análisis adicional realizado en el presente en 150 individuos, no se encontró esta variante en la población.
La segunda mutación patogénica es una deleción de 2 nt (TG) en el exón 3 del gen hMSH2 en la posición 596-597 que provoca un cambio de Cis/ Stop en el codón 184, y genera una proteína truncada.
En este estudio se detectó una variante polimórfica en el gen hMLH1 en dos familias (UN-8 y UN-17) que consiste en un cambio de G>A en el intrón 13, en la posición 1558 + 14. Aunque este polimorfismo ya se había reportado en población sueca (17), se consideró conveniente realizar un estudio de población para determinar si realmente se trataba de un polimorfismo o podría ser una variante patogénica ya que, en un trabajo reciente, se describe una mutación patogénica en la posición 1558 + 13 que genera un sitio de empalme críptico (23).
Se realizó un estudio en una muestra de 150 individuos provenientes de la región cundiboyacense, la misma región de origen de las familias estudiadas; se determinó que la frecuencia de esta variante en esta población colombiana es de 6,5% (alelo G, 0,94; alelo A, 0,06) y que, al parecer, no está asociada con el cáncer colorrectal no polipósico hereditario, al menos, directamente.
La frecuencia de cáncer colorrectal en las familias analizadas en este estudio fue alta como era de esperarse (68 tumores, 64,2%), seguida de tumores de endometrio y de estómago. Este último es de gran interés debido a que, a pesar de que este cáncer es de muy baja frecuencia en familias con cáncer colorrectal no polipósico hereditario, en este estudio se encontró en 7,5%, muy cercana a la de cáncer de endometrio que fue del 11,3%. Este hallazgo es muy importante si se tiene en cuenta que, en Colombia, la principal causa de muerte por cáncer es la de cáncer gástrico (3,4). También la presencia de cánceres extracolónicos de baja frecuencia, como cáncer de riñón o vías urinarias (2/3, 2,8%) y de cerebro (1/2, 1,9%), se asoció con la presencia de mutaciones en el gen hMSH2, como ya se ha descrito (24).
El uso de la técnica de SSCP para la detección de mutaciones tiene sus limitaciones, ya que tiene una sensibilidad de 80% y solamente con productos amplificados hasta de 250 a 350 pb. Con el uso de esta técnica en este estudio, la tasa de detección de mutación sería relativamente alta al compararla con un estudio reciente (25), en el cual también se emplea SSCP, aunque sólo analizan a familias que cumplen completamente con los criterios de Ámsterdam. En dicho estudio se logran detectar las mutaciones en el 50% de los casos. No obstante, en nuestro estudio se logró detectar la mutación en el 88% de las familias que cumplían con los criterios de Ámsterdam. Es importante aclarar que la tasa encontrada en este estudio podría estar subestimada, debido a que tres productos amplificados para hMLH1 tienen más de 350 pb, defectos como cambios en el promotor o en regiones enhancer, y cambios crípticos dentro de regiones no codificantes que puedan alterar el adecuado empalme de los exones, no se pueden detectar utilizando los iniciadores descritos en este trabajo, y existen otros genes del sistema de MMR que pueden tener mutaciones en familias con cáncer colorrectal no polipósico hereditario como el hMSH6, hPMS1, hPMS2 (8-9), en los que se ha encontrado un número importante de mutaciones de cambio de sentido, aunque no se han realizado estudios funcionales de las proteínas para determinar que tanto este tipo de mutaciones alteran la estructura o la funcionabilidad de las mismas.
En nuestro medio, hasta la fecha, no se aplican herramientas moleculares para orientar el diagnóstico y el manejo de pacientes con cáncer colorrectal ni determinar su pronóstico. Por otro lado, en los casos de cáncer hereditario, en los que los familiares tienen un alto riesgo de padecer el cáncer desde una edad temprana, los familiares deben someterse a procedimientos invasivos periódicos desde la juventud, con el fin de detectar el tumor en estadios iniciales. Por esta razón, la implementación de métodos diagnósticos como los realizados en el presente estudio (SSCPSecuencia) en el que se alcanzó una tasa de detección del 88% en aquellas familias que cumplen con los criterios de Ámsterdam II y 11% en familias con pautas de Bethesda, para una tasa de detección conjunta del 47%, es de vital importancia para la identificación de mutaciones en familias con un patrón sugestivo de cáncer colorrectal no polipósico hereditario. Además, la futura implementación de un método de tamizaje como el análisis de la inestabilidad de microsatélites permite realizar un exhaustivo trabajo de detección de mutaciones en aquellos casos que presenten una inestabilidad de microsatélites alta, ya que en 90% de los casos, se asocia a mutaciones en los genes del sistema MMR (principalmente, hMLH1 y hMSH2) en individuos que presentan una historia de cáncer familiar (26). El uso de estas técnicas diagnósticas como el SSCP y la inestabilidad de microsatélites ayudará a la detección de portadores de mutaciones en familias sugestivas de cáncer colorrectal no polipósico hereditario. En el presente estudio se analizaron diferentes miembros de las familias y se detectaron las mutaciones, excepto para el caso de la familia UN-3, en la que fue imposible contactar a los familiares del caso índice. Esto permitió realizar un asesoramiento genético en estas familias, y se sugiere que se incluya un manejo preventivo para el diagnóstico temprano del cáncer basado en la realización de colonoscopias, ya que varios estudios sugieren que el tamizaje por largos periodos con colonoscopias reduce el riesgo de muerte en pacientes con cáncer colorrectal no polipósico hereditario y portadores de mutación, en 56% a 62% (27). También debido a la alta frecuencia de cánceres extracolónicos, es necesario realizar otros exámenes como la biopsia de endometrio y las endoscopias, ya que los cánceres de endometrio y estómago tienen una frecuencia importante en las familias colombianas con cáncer colorrectal no polipósico hereditario. Esto representaría para el paciente y para el sistema de salud una relación costo-beneficio de gran provecho.
Conflicto de Intereses
Los autores declaran que no tienen intereses de ningún tipo con las empresas comerciales que puedan beneficiarse de la presente investigación.
Agradecimientos
A las familias que consintieron participar por la paciencia y cooperación. También agradecemos a Henry T. Lynch, Miguel Rodríguez-Bigas y Peggy Conrad, por su apoyo en las etapas iniciales del presente estudio.
Financiación
Esta investigación se realizó con el apoyo de Colciencias mediante el contrato con la Universidad Nacional, 114-2000.
Correspondencia:
Alejandro Giraldo, Instituto de Genética, oficina 204, entrada calle 53, Universidad Nacional de Colombia, Bogotá, D.C., Colombia.
Teléfonos: 316 5485 y 620 6505; fax: 316 5532 y 620 0926.
Recibido: 14/12/04; aceptado: 11/05/05
Referencias
1. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003;348:919-32. [ Links ]
2. de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer 2004;4:769-80. [ Links ]
3. Espinosa G., Alandate J. Boletín Epidemiológico Distrital 1998;8:1-3.
4. Ángel LA, Giraldo A, Pardo CE. Mortalidad por cánceres del aparato digestivo en Colombia entre 1980 y 1998. Análisis de tendencias y comparación regional. Rev Fac Medicina (Universidad Nacional de Colombia) 2004;52:19-37. [ Links ]
5. Wijnen JT, Vasen HF, Khan PM, Zwinderman AH, van der Klift H, Mulder A, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998;339:511-8.
6. Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993;75:1215-25. [ Links ]
7. Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994;368:258-61. [ Links ]
8. Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994;371:75-80. [ Links ]
9. Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 1997;17:271-2 [ Links ]
10. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer (ICG-HNPCC), Dis. Colon Rectum 1991;34:424-5. [ Links ]
11. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453-6. [ Links ]
12. Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997;89:1758-62. [ Links ]
13. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acid Res 1988;16:1215. [ Links ]
14. Weber TK, Conlon W, Petrelli NJ, Rodriguez-Bigas M, Keitz B, Pazik J, et al. Genomic DNA-based hMSH2 and hMLH1 mutation screening in 32 Eastern United States hereditary nonpolyposis colorectal cancer pedigrees. Cancer Res 1997;57:3798-803. [ Links ]
15. Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 1995;9:48-55. [ Links ]
16. Froggatt NJ, Brassett C, Koch DJ, Evans DG, Hodgson SV, Ponder BA, et al. Mutation screening of MSH2 and MLH1 mRNA in hereditary non-polyposis colon cancer syndrome. J Med Genet 1996;33:726-30. [ Links ]
17. Tannergard P, Lipford JR, Kolodner R, Frodin JE, Nordenskjold M, Lindblom A. Mutation screening in the hMLH1 gene in Swedish hereditary nonpolyposis colon cancer families. Cncer Res 1995;55:6092-6. [ Links ]
18. Nystrom-Lahti M, Wu Y, Moisio AL, Hofstra RM, Osinga J, Mecklin JP, et al. DNA Mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet 1996;5:763-9. [ Links ]
19. Bartosova Z, Fridrichova I, Bujalkova M, Wolf B, Ilencikova D, Krizan P, et al. Novel MLH1 and MSH2 germline mutations in the first HNPCC families identified in Slovakia. Hum Mutat 2003;21:449. [ Links ]
20. Katballe N, Christensen M, Wikman FP, Orntoft TF, Laurberg S. Frequency of hereditary non-polyposis colorectal cancer in Danish colorectal cancer patients. Gut 2002;50:43-51. [ Links ]
21. Sarroca C, Peltomaki P, Alfano N, Tedesco G, Della Valle A, Dominguez A, et al. Three new mutations in hereditary nonpolyposis colorectal cancer (Lynch syndrome II) in Uruguay. Cancer Genet Cytogenet 2003;142:13-20. [ Links ]
22. Rossi BM, Lopes A, Oliveira Ferreira F, Nakagawa WT, Napoli Ferreira CC, Casali Da Rocha JC et al. hMLH1 and hMSH2 gene mutation in Brazilian families with suspected hereditary nonpolyposis colorectal cancer. Ann Surg Oncol 2002;9:555-61. [ Links ]
23. Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, et al. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet 2001;68:118-127. [ Links ]
24. Wang Q, Lasset C, Desseigne F, Saurin JC, Maugard C, Navarro C, et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum Genet 1999;105:79-85. [ Links ]
25. Beck NE, Tomlinson IP, Homfray T, Frayling I, Hodgson SV, Harocopos C, et al. Use of SSCP analysis to identify germline mutations in HNPCC families fulfilling the Amsterdam criteria. Hum Genet 1997;99:219-24. [ Links ]
26. Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997;57:4749-56. [ Links ]
27. Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829-34
. [ Links ]
