










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.26 n.3 Bogotá set. 2006
Factores solubles con actividad inhibitoria contra el virus de la inmunodeficiencia humana tipo 1
Wildeman Zapata, Carlos Julio Montoya, María Teresa Rugeles
Grupo de Inmunovirología-Biogénesis, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
La patogénesis de la infección por el VIH-1 es un proceso complejo que depende de múltiples factores, incluyendo los de origen viral, la respuesta inmune y los factores genéticos del hospedero. Debido a esto, se observa que en los individuos infectados por el VIH-1 existen diferentes patrones de progresión de la enfermedad, mientras que algunos individuos que se exponen repetidamente al virus permanecen sanos y seronegativos. Esta variabilidad luego de la exposición al VIH-1 sugiere que existen mecanismos de resistencia natural contra esta infección. Si bien en algunos individuos expuestos seronegativos y en infectados no progresores a largo término, se ha podido determinar que la resistencia se da por mecanismos genéticos o inmunológicos, existe un porcentaje importante de casos de resistencia para los cuales no ha sido posible establecer el mecanismo protector. Varios factores solubles, como las defensinas, las quimiocinas, los interferones y las ribonucleasas, entre otros, producidos por células del sistema inmune y por células de varios tejidos epiteliales, tienen una reconocida actividad antiviral que podría estar involucrada en la protección del hospedero durante la exposición al VIH-1. Conocer adecuadamente los mecanismos de acción de estos factores y su papel en la resistencia natural a la infección por el VIH-1 puede ser útil al momento de diseñar nuevas estrategias terapéuticas para combatir la morbilidad y mortalidad asociadas con la pandemia por el VIH-1.
Palabras clave: VIH-1, resistencia natural, ribonucleasas, interferones, defensinas, quimiocinas.
Soluble factors with inhibitory activity against type 1 Human Immunodeficiency Virus
The pathogenesis of HIV-1 infection is a complex process that depends on multiple factors, including viral and host immune and genetic characteristics. This leads to a variable pattern of disease progression among those HIV-1-exposed individuals who become infected, while there are a number of individuals who remain healthy and HIV-1 seronegative despite being serially exposed to HIV-1. These variable outcomes of HIV-1 exposure suggest that there are mechanisms of natural resistance to HIV-1 infection. Although several genetic and adaptive immune mechanisms of resistance have been reported in some exposed seronegative and long-term non-progressor individuals, the mechanisms involved in controlling the establishment and progression of HIV-1 infection are not fully understood. Several soluble factors, such as defensins, chemokines, interferons and ribonucleases, among others, produced by cells of the immune system and epithelial tissues, have a broad anti-viral activity that might play a role as protective mechanisms during HIV-1 exposure. A better understanding of the mechanisms and role of these soluble factors during the natural resistance to HIV-1 infection may have important implications for the design of novel therapeutic strategies to combat the morbidity and mortality associated with the HIV-1 pandemic.
Key words: HIV-1, immunity, natural, ribonucleases, interferons, defensins, chemokines.
El virus de la inmunodeficiencia humana (VIH) fue descubierto en 1983 y un año más tarde se identificó como la causa del síndrome de inmunodeficiencia adquirida (sida) (1). En la actualidad, 40,3 millones de personas están infectadas con el VIH alrededor del mundo y anualmente ocurren 4,9 millones de nuevas infecciones, de las cuales casi el 50% corresponde a infecciones en mujeres (2). En Colombia se estima que el número de individuos infectados con el VIH es de 190.000, para una prevalencia de la infección del 0,4 al 1,2% (3).
La infección por el VIH-1 se caracteriza por la disminución progresiva en el número y función de linfocitos T CD4+, células fundamentales en la inducción de una respuesta inmune humoral y celular; estas células son el principal blanco del virus y su disminución induce una grave inmunodeficiencia que se asocia con la aparición de infecciones oportunistas que, en última instancia, conducen a la muerte (4).
El VIH-1 se transmite por contacto sexual no protegido con un individuo seropositivo (SP), por la exposición a sangre infectada o sus derivados, durante el embarazo, en el nacimiento, o por medio de la alimentación materna (2,5). Sin embargo, a pesar de que la transmisión sexual es de las menos eficientes, se estima que más del 80% de las infecciones por VIH-1 se adquieren por esta vía (6).
La patogénesis de la infección por el VIH-1 es un proceso complejo y variable. Los individuos infectados exhiben diferentes velocidades de progresión hacia el sida; el 10% desarrolla sida en menos de cinco años, mientras que el 80% desarrolla la inmunodeficiencia en un promedio de 8 a 10 años. De otro lado, entre 8 y 10% de los individuos infectados se consideran progresores lentos o no progresores (LTNP, del inglés Long Term Non-Progressors) y se caracterizan porque en ausencia de antirretrovirales permanecen asintomáticos por más de 10 años, sin deterioro inmunológico y con cargas virales bajas o indetectables (7,8). Adicionalmente, existen individuos que a pesar de haber estado expuestos en múltiples ocasiones sin protección al VIH-1 no muestran evidencia clínica ni serológica de la infección; estas personas se conocen como expuestos seronegativos (ESN) (9). La existencia del grupo de ESN, así como de LTNP, hace evidente la existencia de mecanismos de resistencia natural a la infección y a su progreso.
Los mecanismos de resistencia dependen tanto del virus como del hospedero; las cepas virales defectuosas o atenuadas (10) y la exposición a inóculos virales muy bajos reducen la posibilidad de infección (11). En el hospedero, el principal mecanismo que confiere un alto grado de resistencia a la infección por el VIH es la mutación D32 en el gen que codifica para el correceptor viral CCR5 (12,13). Sin embargo, el genotipo D32/D32 está presente únicamente en 2 a 4% de las personas caucasoides y ESN (14-16). Otras mutaciones en genes del sistema de las quimiocinas se relacionan con una progresión lenta de la infección más que con resistencia a la misma (17,18). El grado de concordancia del complejo mayor de histocompatibilidad (CMH) clase I entre la madre y el feto es un factor determinante de la incidencia de la transmisión vertical (19), al igual que la presencia de ciertos alelos del CMH I y II (10). La respuesta inmune celular adaptativa es el principal mecanismo antiviral mediante la actividad citotóxica de células T CD8+ (CTL), las cuales son más efectivas en el control de la replicación viral que los anticuerpos específicos para el VIH-1 (4). Recientemente han cobrado importancia factores solubles que son secretados por diferentes células durante el proceso de respuesta inmune y que muestran gran capacidad antiviral, bien sea como función primaria o por efecto "accidental". Esto ha llevado a proponer un nuevo modelo de inmunidad antiviral que implica la supresión de la replicación viral sin eliminar la célula infectada y que podría potenciar los meca-nismos celulares específicos anti-VIH-1. Entre los factores solubles con actividad antiviral reportada se destacan las b-quimiocinas, como CCL5 (RANTES), CCL3 (MIP1-a) y CCL4 (MIP1-b) (20), la CXCL12 o factor 1 derivado de células estromales (SDF-1) (18), el factor antiviral de linfocitos T CD8+ (CAF) (21), los interferones (IFN) (22), el factor inhibidor de leucemia (LIF) (23), lisozimas y RNasas asociadas a la gonadotropina coriónica humana (24,25), un factor estimulado por aloantígenos (ASF) (26), las defensinas (27,28), el inhibidor de proteasas secretado por leucocitos (SLPI) (29) y otros más recientemente definidos como las proteínas con motivos TRIM (30).
Esta revisión se centrará en los factores solubles con actividad antiviral mejor caracterizados y hará especial énfasis en aquellos que demuestran una clara actividad anti-VIH, describiendo los mecanismos antivirales que se han propuesto y las evidencias reportadas hasta el momento. En la
figura 1 se señalan los sitios potenciales de acción de los diferentes factores solubles.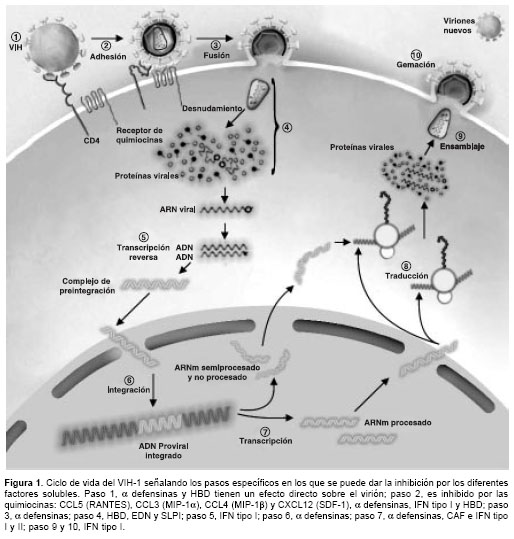
Defensinas
Las defensinas son péptidos pequeños de 18 a 42 aminoácidos y de 3 a 5 kDa, catiónicos, anfipáticos y con actividad microbicida contra un amplio grupo de microorganismos, incluidas bacterias gram-positivas, gram-negativas, protozoos, hongos y virus (31,32). Las defensinas son péptidos ricos en cisteina que se unen mediante tres enlaces disulfuro intramoleculares; son producidas principalmente por células epiteliales en diferentes mucosas, así como por células del sistema inmune, ya sea en forma constitutiva o en respuesta a productos microbia-nos o citocinas proinflamatorias (31,33,34). Con base en su tamaño y su secuencia de aminoácidos y en la distribución de los puentes disulfuro entre cisteínas, las defensinas en mamíferos se clasifican en tres grupos: a, b y q (31); en humanos sólo se ha descrito la presencia de las defensinas a y b (35).
Mecanismos inhibitorios
La actividad microbicida de las defensinas está dada por la capacidad de unirse a los componentes cargados negativamente de la membrana de los microorganismos, como el lipopolisacarido (LPS), el ácido teicoico y el fosfatidil-glicerol, entre otros (36). Esta interacción altera la integridad de la membrana o crea poros que llevan a la muerte del microorganismo (36).
Adicionalmente, las defensinas cumplen otra función importante, diferente a su acción microbicida directa, y es su capacidad quimiotáctica reportada ya hace 17 años (37); esto indica que las defensinas potencian la respuesta inmune innata y adaptativa (38).
Las evidencias mostradas hasta el momento de los posibles mecanismos de acción antiviral de las defensinas, y específicamente anti-VIH-1, son variadas y dependen del tipo de defensina; a continuación se describirán las principales características de las a y b defensinas humanas, incluyendo su papel antiviral.
a defensinas
Hasta el momento hay 6 a defensinas humanas identificadas: las HNP-1-4 son abundantes en neutrófilos y macrófagos, pero también son producidas por células NK, linfocitos B y células T gd activadas por citocinas (39). Las a defensinas humanas 5 y 6 (HD-5 y HD-6) son producidas principalmente por las células Paneth del intestino delgado (36).
Mecanismos inhibitorios
En 1993 se reportó por primera vez la inhibición in vitro de la replicación del VIH-1 por a defensinas, lo que sugiere su papel protector durante la infección in vivo (39-42). En esos estudios se observó que estos péptidos inhiben la infección por el VIH-1 después de la entrada viral, y que esta actividad no se ve afectada por la presencia de suero (40). Posteriormente se demostró que las HNP-1-3 purificadas de neutrófilos humanos, así como las HNP recombinantes, bloquean la infección por el VIH-1 después de la entrada viral (36). Adicionalmente, el tratamiento previo de células con HNP-1 y lavado posterior antes de la infección con VIH-1 bloqueó la infección por el VIH-1, indicando que no es necesario un efecto directo sobre el virus para inhibir la replicación viral (39); en este estudio los autores proponen que el efecto de las a defensinas sobre el ciclo viral ocurre después de la entrada del virus y probablemente después de la integración del ADN viral (39). Otros estudios demostraron un mecanismo dual para la acción de las a defensinas, con el cual éstas pueden inhibir la infección por un mecanismo directo sobre el virus o por un mecanismo sobre la célula que va a ser infectada (43-45). Chang y colaboradores reportaron un efecto directo de HNP-1 sobre el VIH-1 a un bajo título viral y en ausencia de suero (28). Además, encontraron que HNP-1 inhibe varios pasos después de la transcripción reversa y de la integración del virus, así como la actividad de la proteína cinasa C (PKC) en células T CD4+ (28). Teniendo en cuenta que PKC participa en el proceso de la transcripción viral (36, 46), la inhibición de su activación puede ser uno de los principales mecanismos de regulación de la infección ejercida por las a defensinas sobre la célula infectada (28). De otro lado, el efecto directo sobre el virión podría ser mediado por la unión de alta afinidad entre a defensinas y gp120 viral o el receptor viral CD4, ya que se ha reportado que las a defensinas pueden actuar como lectinas (44). Otro mecanismo antiviral puede ser el aumento en la expresión de CC quimiocinas en macrófagos infectados con VIH-1 previamente tratados con a defensinas 1 y 2. Los altos niveles de CCL3 (MIP-1a) y CCL4 (MIP-1b) pueden prevenir la infección de nuevas células al ocupar los receptores de quimiocinas necesarios para la entrada del virus (47).
Evidencias epidemiológicas
Las primeras evidencias epidemiológicas del papel antiviral de las a defensinas fueron obtenidas por Trabattoni y colaboradores, quienes evaluaron la expresión de las a defensinas en mononucleares de sangre periférica (MNSP) y células mono-nucleares cervicovaginales de individuos expuestos sexualmente al VIH-1 pero no infectados, en SP y en controles sanos (48). Este estudio señaló que la producción de HNP-1-3 por células T CD8+ fue 10 veces mayor en ESN que en controles sanos, y que esas mismas células contenían dos veces más RNAm de a defensinas (48). Toda esta evidencia sugiere que las a defensinas juegan un papel muy importante en la inmunidad innata contra el VIH-1 y que podría mediar el efecto protector en algunos de los individuos ESN.
b defensinas
Hasta la fecha sólo se han caracterizado bien seis. Las b defensinas humanas HBD (49), pero con base en estudios genómicos se conocen por lo menos 28 HBD (50). Las HBD se expresan en varios tejidos, tales como músculo esquelético, tracto respiratorio, esófago, lengua, intestino y piel (35). HBD-1 se expresa constitutivamente, mientras HBD-2 y 3 son inducidas por productos virales y bacterianos, así como por citocinas como la IL-1b o el TNF-a (34, 51-55). HBD-4 se expresa constitutivamente en el testículo, aunque puede ser inducida por una infección bacteriana y su producción parece no verse influenciada por citocinas proinflamatorias (34,56). Por último, las HBD más recientemente caracterizadas son la 5 y 6, las cuales se expresan en el epidídimo y en las vías aéreas (53,57).
Mecanismos inhibitorios
La primera evidencia del mecanismo anti-VIH-1 ejercido por las HBD fue reportada por Quiñones y colaboradores (27); ellos encontraron que la infección por el VIH-1 induce la expresión, en células de epitelio oral humano, del RNAm y de proteínas de HBD-2 y 3, pero no de HBD-1, y que las HBD-2 y 3 recombinantes, pero no la HBD-1, inhiben la replicación del virus dependiendo de la concentración incluso después de cinco días de infección y sin inducir efecto citotóxico visible (27). La inhibición de la replicación viral fue irreversible y dependió en parte de la unión directa de la HBD a la partícula viral. Adicionalmente, las HBD inducen la regulación negativa del correceptor viral CXCR4, pero no de CCR5, en células mononucleares y en linfocitos T (27). El mecanismo de interacción entre las HBD y la partícula viral no se ha establecido; una posibilidad es que este proceso sea similar al de las a defensinas, en el cual el péptido tiene actividad tipo lectina e interactuaría con la gp120 o el CD4 en forma similar a lo reportado para compuestos polianiónicos que son capaces de unirse a los sitios cargados positivamente del asa V3 de gp120 bloqueando la unión del virus con su receptor (58,59).
La evidencia más reciente de actividad antiviral presentada por Sun y colaboradores sugiere que las HBD-2 y 3 inhiben la infección de MNSP por cepas R5 y X4 del VIH-1 dependiendo de la dosis; es interesante que la dosis utilizada en los experimentos (100ug/ml) es similar a la encontrada en la mucosa oral de individuos sanos (60). En el trabajo citado se mostró que la HBD-2 tiene un efecto directo sobre el virión, con acumulación de productos virales tempranos de la transcripción reversa, y no se observó modulación de la expresión de correceptores virales (60).
Evidencia epidemiológica
Estudios epidemiológicos que estamos realizando en colaboración con Miguel E. Quiñones (Center for AIDS Research, Cleveland, Estados Unidos), indican que la expresión del RNAm de HBD-2 y 3 en mucosa endocervical de individuos ESN presenta aumento en comparación con individuos SP y controles sanos (resultados sin publicar). Adicionalmente, estamos trabajando en la búsqueda de polimorfismos en el gen DEFB-1, que codifica para HBD-1, asociados con resistencia/susceptibilidad; los reportes previos de Braida y colaboradores sugieren que el SNP (del inglés Single-Nucleotide Polymorphism) -44 C/G en una región altamente conservada entre especies en este mismo gen se asoció significativamente con susceptibilidad a la infección por el VIH-1 en niños italianos nacidos de madres VIH-1 positivas (61). Nuestros resultados muestran una frecuencia significativamente mayor de otros polimorfismos en esta misma región entre los individuos ESN, lo que sugiere la asociación de este gen con el fenómeno de resistencia/susceptibilidad a la infección por el VIH-1 (resultados sin publicar). Todas estas evidencias resaltan el posible papel de las HBD en la protección de la mucosa oral y de otras superficies mucosas durante la exposición al VIH-1.
Quimiocinas
Éstas son citocinas de bajo peso molecular (8-17 Kd) con actividad quimiotáctica, que participan activamente en los procesos inflamatorios y de reparación tisular (62). Las quimiocinas se clasifican en cuatro subgrupos, CL (g), CCL (b), CXCL (a) y CX3CL (d), con base en la secuencia de aminoácidos y especialmente en la posición de los dos residuos de cisteina en el extremo amino terminal (63).
Algunos miembros de las quimiocinas CCL (RANTES, MIP-1a y MIP-1b) y las CXCL (SDF-1) juegan un papel crucial durante la patogénesis de la infección por el VIH-1 (64), dado que para entrar en la célula blanco el virus necesita no sólo de su receptor, la molécula CD4, si no de un correceptor que corresponde a las moléculas CCR5 o CXCR4, receptores fisiológicos de estas quimiocinas b y a, respectivamente (65). El mecanismo por el cual estas quimiocinas inhiben la infección viral es el bloqueo del correceptor y por la regulación negativa inducida por la unión de los respectivos ligandos (66).
Factor 1 derivado del estroma (SDF-1)
Esta a quimiocina, conocida actualmente como CXCL12, es el ligando natural de CXCR4, uno de los correceptores virales; es secretada por células estromales de médula ósea, células epiteliales, monocitos y por células T vírgenes (64); tiene actividad quimiotáctica principalmente sobre neutrófilos y linfocitos (67). Esta quimiocina es esencial para mantener la viabilidad del embrión, para realizar una adecuada linfopoyesis de células B, la hematopoyesis de médula ósea y durante la cardiogénesis (68).
In vitro esta citocina une y regula negativamente la expresión de CXCR4 en la membrana celular (69), inhibiendo la fusión y la entrada del virus, lo cual ha llevado a sugerir su papel protector in vivo durante la infección por el VIH-1 (1).
Evidencia epidemiológica
Las células epiteliales de la mucosa rectal y cervicovaginal expresan constitutivamente altas concentraciones de CXL12 (SDF-1) y niveles fácilmente detectables de RNAm que pueden bloquear la unión del virus al correceptor CXCR4, pero no a CCR5, lo que explicaría parcialmente la mayor incidencia de transmisión de cepas R5 a través del contacto sexual (70, 71) y el bajo riesgo de adquirir la infección en un solo acto sexual (72). Un estudio reciente evaluó la concentración de quimiocinas en la leche materna de madres VIH-1 positivas y madres control sanas, encontrando un incremento en la producción de CXCL12 (SDF-1) que se asoció con un menor riesgo de transmisión vertical del VIH-1 (73).
CCL5 (RANTES), CCL3 (MIP-1a) y CCL4 (MIP-1b)
Son b quimiocinas producidas por células tanto de la inmunidad innata como adquirida: macrófagos, células dendríticas, células NK, células T gd, y linfocitos T CD8+ (66). Son los ligandos naturales de CCR5, correceptor de cepas R5 del VIH-1, que corresponden a la forma de transmisión primaria del virus (66). Estas quimiocinas cumplen varias funciones biológicas; la principal es su actividad quimiotáctica sobre linfocitos, mastocitos, eosinófilos y monocitos (67). Adicionalmente, estimulan la migración transendotelial, la liberación de elastasas por neutrófilos, de la ribonucleasa EDN por eosinófilos y de histamina por basófilos; así mismo, induce la polimerización de actina, incrementa el calcio intracelular y la actividad citotóxica de las células NK (67, 74).
La actividad anti-VIH de las b quimiocinas radica en bloquear la unión viral con el CCR5 y en regular negativamente la expresión de esta molécula en la superficie celular (20). In vitro se demostró que a través de estos mecanismos las CC quimiocinas inhiben la infección, lo que se traduce en una menor tasa de replicación viral (75).
Evidencia epidemiológica
La mayoría de estudios en individuos adultos VIH-1 positivos sugieren que el aumento en los niveles de b quimiocinas retrasa el desarrollo del sida (76). En forma similar, en pacientes pediátricos también se ha observado una correlación positiva entre los niveles de CC quimiocinas y una progresión lenta de la infección por VIH-1 (77). Adicional-mente, estudios recientes demuestran que, comparadas con trabajadoras sexuales infectadas, las trabajadoras sexuales resistentes al VIH-1 expresan recuentos elevados de linfocitos T CD4+ y CD8+, así como cantidades elevadas de RANTES en mucosa cervical (78).
Interferones
Los IFN se dividen en dos clases según las células que los secretan: los IFN tipo I, que pueden ser a,b u w, secretados por cualquier célula nucleada infectada por virus y por las CD plasmacitoides en respuesta al reconocimiento de PAMP por varios receptores tipo Toll (TLR) (79), y el tipo II o IFN g, que es secretado por las células del sistema inmune (80). Existe solo un gen miembro de la familia de IFN-b o IFN-w, pero se han reportado un gran número de genes para IFN-a (79). Los IFN tipo I poseen un receptor común, IFNAR (del inglés IFN-a/b Receptor) (22). A pesar de que la infección viral es la causa biológica más común de inducción de IFN tipo I, otros agentes como bacterias, micoplasmas y protozoos, o algunos de sus componentes, pueden inducir su síntesis (81).
Mecanismos inhibitorios
Los IFN tipo I tienen diversas funciones inmunomoduladoras como la activación de las células NK y NKT invariantes, la inducción de iNOS (del inglés inducible nitric oxide synthase) y de moléculas del CMH-I y II que favorecen la presentación antigénica (82, 83). Los IFN tipo I suprimen la replicación viral por varios mecanismos (82): inducen la expresión de proteínas como la 2'-5' oligoadenilato sintasa y la RNasa L, las cuales degradan el ARN viral y celular; activan la proteína cinasa dependiente de ARN de doble cadena (PKR) que inhibe la traducción del RNAm; estimulan la producción de la proteína MxA, proteína unidora de guanilato que inhibe la síntesis de ARN, e inducen la deaminasa adenosina específica de ARN (ADAR) que edita el ARN. A través de todas estas proteínas impiden la diseminación de la infección (84).
Adicionalmente, los IFN tipo I regulan la producción de anticuerpos in vivo e in vitro, inhiben las células T supresoras/reguladoras, tienen actividad antitumoral (85, 86) y promueven la respuesta de citocinas del patrón Th1 (84).
El IFN-a ha mostrado actividad anti-VIH in vitro, inhibiendo varios pasos del ciclo de vida del virus (66). Inicialmente se sugirió que podía inhibir el ensamblaje viral (87) y reportes posteriores señalan su actividad sobre pasos previos a la transcripción reversa (88) y sobre la transcripción reversa, la expresión del provirus y la liberación de viriones a partir de células infectadas crónicamente (22).
Evidencias epidemiológicas
Diferentes estudios clínicos señalan que la pérdida de células productoras de IFN-a durante la infección por el VIH-1 se asocia con altas cargas virales y con una rápida progresión a sida (89). De otro lado, y en apoyo al papel antiviral de esta citocina, se ha descrito una mayor capacidad en la producción de IFN-a en individuos VIH-1 positivos que exhiben una progresión lenta de la infección (22). En individuos SP tratados con terapia HAART (del inglés Highly Active Antiretroviral Therapy), la disminución significativa de la carga viral se ha asociado con un aumento en el número de células dendríticas plasmaci-toides productoras de IFN-a (90, 91). Otros estudios señalan que el uso de esta citocina en pacientes con sida y procesos oncogénicos como el sarcoma de Kaposi disminuye la carga viral y potencia la respuesta antitumoral (92,93). Adicionalmente, se ha reportado que el uso de esta citocina mejora la actividad lítica e incrementa las concentraciones de granzimas y perforinas en las células NK (94, 95) y potencia la respuesta de las células dendríticas plasmacitoides contra antígenos virales (96). De los 21 subtipos de IFN-a descritos hasta el momento, el subtipo a2 se expresa en forma dominante (97), y probablemente sea el tipo de IFN responsable de la mayoría de los efectos antivirales antes mencionados.
Con respecto al IFN-g, estudios en macacos infectados con el virus de la inmunodeficiencia simiana (SIV) sugieren que esta citocina disminuye la replicación viral; adicionalmente, se ha encontrado una asociación entre niveles altos de RNAm de IFN-g en MNSP, nódulos linfoides y linfocitos de lámina propia intestinal, y el control de la replicación viral (22). A pesar de que la mayoría de las evidencias respalda el papel antiviral del IFN-g, también se ha sugerido que esta citocina, conjuntamente con la IL-10, induce la replicación viral durante la interacción entre CD y células T infectadas, favoreciendo la diseminación de la infección (98).
Factor antiviral de linfocitos T CD8 (CAF)
CAF es un factor soluble con actividad antiviral producido por los linfocitos T CD8+ en respuesta a la infección por el VIH-1; no es citotóxico para células T CD4+ (21) y tiene la capacidad de inhibir la replicación de cepas X4 y R5 del VIH-1 (66). Hasta el momento no está claramente identificada la naturaleza del CAF, pero se sabe que el componente activo es estable al calor, resistente a altas temperaturas (86°C, 10 min), a pH ácido (hasta 2,0) y es resistente a la tripsina pero sensible a la proteasa V8 de Staphylococcus (99).
Esta actividad antiviral no citotóxica de los linfocitos T CD8+ se comprobó mediante el cocultivo de estas células con linfocitos T CD4+ infectados, separados físicamente mediante membranas semipermeables; en estos cultivos, la replicación viral disminuyó significativamente y se mantuvo la viabilidad de las células T CD4+ (43, 66). Buscando determinar la o las moléculas responsables de la actividad del CAF, diferentes estudios han sugerido la homología entre el CAF y distintas moléculas antivirales. Las b quimiocinas CCL5 (RANTES), CCL3 (MIP-1a) y CCL4 (MIP-1b) se han propuesto como las moléculas responsables de la actividad anti-VIH-1 del CAF (20); sin embargo, la capacidad de inhibir cepas X4 y R5 exhibida por el CAF descarta la posibilidad de que estas proteínas sean sus componentes activos, ya que las las b quimiocinas sólo tienen capacidad de inhibir cepas R5 (20). Estudios recientes afirman que el componente activo del CAF corresponde a una forma modificada por proteasas celulares de la antitrombina III bovina (ATIII), presente en el suero fetal bovino utilizado en cultivos celulares en los cuales se ha reportado la actividad del CAF (100). Aunque estudios posteriores confirmaron la actividad antiviral de ATIII, se reportó que la actividad del CAF se mantiene en condiciones libres de proteínas del suero fetal bovino (101). Inicialmente, se postuló a las a defensinas como las moléculas responsables de la actividad del CAF (102); sin embargo, los mismo autores se retractaron posteriormente de esta afirmación (103). Adicionalmente, se determinó que las a defensinas ejercen su actividad antiviral en un paso del ciclo viral diferente a aquel en que actúa el CAF (39), y los anticuerpos contra las a defensinas fueron incapaces de bloquear la actividad anti-VIH-1 del CAF (43).
El CAF inhibe un paso de la transcripción viral conducido por el LTR (del inglés Long-Terminal Repeat) (104); se ha propuesto que esta inhibición involucra la unión de un ligando a un receptor en la superficie celular y la activación de una vía de transducción de señales en la cual participan la proteína STAT1 (del inglés Signal Transducer and Activator of Transcription 1) y los factores de transcripción nuclear NF-kB y NFAT (del inglés Nuclear Factor of Activated T cells), así como el IRF-1 (del inglés Interferon Regulatory Factor 1) (105-107). Estudios recientes señalan que las células T CD8+ producen un precursor del CAF que necesita ser cortado por una proteasa celular para ser activo, ya que en presencia de inhibidores de proteasas como la leucopeptina se bloquea la actividad no citotóxica de los linfocitos T CD8+ (108). Se ha sugerido que este precursor es activo en la superficie celular, aunque no se descarta su entrada a la célula para generar el estado antiviral en las células T CD4+ (101).
Evidencias epidemiológicas
En 1986 se demostró que individuos infectados con el VIH-1 eran capaces de controlar la infección mediante una fuerte respuesta inmune celular (21, 101); estos estudios revelaron que las células T CD8+ tenían la capacidad de inhibir la replicación viral, sin necesidad de eliminar la célula infectada, por medio de un factor soluble que más tarde se conoció como CAF (21). Estudios posteriores encontraron esta misma actividad en linfocitos T CD8+ de sangre periférica y de nódulos linfoides en individuos VIH-1 positivos (109). Investiga-ciones recientes realizadas en individuos VIH-2 positivos demuestran que esta actividad antiviral no citotóxica se mantiene e inhibe de manera similar a cepas R5 de ambos virus, VIH-1 y 2, pero parece ser más fuerte para las cepas X4 del VIH-2 que del VIH-1 (110). Una de las mayores evidencias del papel antiviral del CAF fue hallar que la replicación viral in vitro del VIH-1 en MNSP de individuos ESN era significativamente menor, y que dicho efecto antiviral estaba mediado por la actividad no citotóxica de los linfocitos T CD8+ (111). Además, la actividad antiviral no citotóxica de los linfocitos T CD8+ en individuos SP se ha correlacionado negativamente con la progresión hacia el sida (112).
RNasas
Las ribonucleasas son enzimas reguladoras que participan en varios procesos fisiológicos, que van desde el procesamiento alternativo del ARN hasta la muerte celular (113). Son proteínas expresadas por diferentes tejidos, que exhiben especificidades variables contra diferentes substratos de ARN; están compuestas de un péptido señal de aproximadamente 25 aminoácidos y un péptido maduro de 130 aminoácidos (114). Las ribonu-cleasas están divididas en tres grandes familias: la superfamilia A, la familia T1 y la familia T2. La superfamilia de ribonucleasas A incluye las 13 RNasas humanas hasta el momento descubiertas (114). El potencial terapéutico de las RNasas se ha sugerido en procesos oncogénicos y en infecciones virales (115).
Mecanismos inhibitorios
Se ha postulado que las RNasas tienen actividad antiviral directa y, además, se encontró que algunas de ellas son capaces de estimular las células dendríticas para la producción de varios factores solubles e inducir su maduración in vitro (116) potenciando la respuesta inmune contra diferentes patógenos. Algunos autores proponen que las RNasas tienen la capacidad de unirse a la membrana celular, penetrar al interior de la célula utilizando vesículas acídicas y degradar el RNA, causando la muerte celular (117, 118). La capacidad antiviral de las RNasas está ampliamente reportada en la literatura: la RNasa 2 (EDN, neurotoxina derivada de eosinófilos) y la RNasa 3 (ECP, proteína catiónica derivada de eosinófilos) inhiben la replicación del virus respiratorio sincitial y de otros paramixovirus (119). También se ha encontrado que la EDN y otras RNasas inhiben la replicación del VIH-1 en líneas celulares crónicamente infectadas (120). Nuestro laboratorio, en colaboración con el laboratorio de Gene Shearer en el NCI (National Cancer Institutes, Nacional Institutes of Health, Estados Unidos), encontró que la EDN es la molécula responsable de la actividad anti-VIH-1 de un factor soluble que se produce en respuesta a los aloantígenos (ASF), el cual se obtiene in vitro durante el cultivo mixto de linfocitos (26) y cuya actividad supresora parece darse sobre un paso previo a la transcripción reversa (121). El uso de un conjugado de RNasa pancreática y albúmina de suero humano inyectado en ratones infectados con virus influenza A e influenza B mostró alta actividad antiviral (122). Nosotros también observamos recientemente que cuatro RNasas recombinantes inhiben la replicación del VIH-1 en un cultivo primario de linfocitos T activados (Bedoya VI BA, Hardy A, Rybak SM, Shearer GM, Rugeles MT. Ribonucleases in HIV- 1 inhibition: effect of recombinant RNases on infection of primary T cells and immune activation-induced RNase gene and protein expression. AIDS Res Hum Retroviruses 2006;in press), lo que refuerza el potencial antiviral de estas enzimas.
Inhibidor de proteasas secretado por leucocitos (SLPI)
SLPI es una proteína de 11,7 kDa, no-glicosilada, altamente básica y estable en medio ácido (123), lo que le permite ser funcional en ambientes como el de las mucosas (29). Fue originalmente caracterizada en parótidas humanas (124) y es secretada por células epiteliales no ciliadas que recubren las superficies mucosas (123), por la piel (125), por neutrófilos (126) y por macrófagos estimulados con LPS (127). El blanco fisiológico de SLPI son las proteasas que actúan sobre residuos de serina, como la elastasa del neutrófilo y la catepsina G liberadas durante la respuesta inflamatoria (128). De esta manera, SLPI contribuye en la defensa de las mucosas mediante la regulación de la inflamación (129). Además, posee una amplia actividad antimicrobiana inhibiendo bacterias, hongos y virus (130). Otras funciones incluyen la promoción de la fecundación protegiendo a los espermatozoides para que no sufran la reacción acrosomal por la acción de las elastasas (131) y la reparación tisular en la piel (132).
Mecanismos inhibitorios
McNeely y colaboradores reportaron la primera actividad inhibitoria de la SLPI, recombinante o derivada de saliva, contra diferentes cepas del VIH-1 en monocitos humanos, en cultivo primario de linfocitos y en líneas de células T transformadas (133,134). En estos estudios se observó que el efecto inhibitorio se produce en las etapas tempranas de la infección, ya que la SLPI inhibió la replicación viral sólo cuando se adicionó simultáneamente con el virus al cultivo de células (133). Estudios posteriores señalan que la SLPI no afecta la unión del virión a la molécula CD4, ni los pasos posteriores a la transcripción reversa (29,134), lo que sugiere que su actividad se da en un paso intermedio (134). Investigaciones recientes demuestran que el efecto inhibidor de la la SLPI se da por medio de la interacción de alta afinidad con una molécula de superficie celular conocida como anexina II; esta unión no permite la interacción de residuos de fosfatidilserina presentes en la envoltura viral con la proteína unidora anexina II en la membrana celular de macrófagos susceptibles (135). Existen evidencias que indican que el VIH-1 induce la producción de RNAm y proteína de la SLPI en queratinocitos orales humanos inmortalizados por medio de la interacción con gp120 en ausencia de infección (136).
Evidencias epidemiológicas
Investigaciones de Pillay y colaboradores señalan que madres VIH-1 positivas que no transmitieron el virus a sus hijos expresaban mayores cantida-des de la proteína SLPI en fluidos vaginales que las madres VIH-1 positivas transmisoras (137). Farquhar y colaboradores evaluaron el papel de la SLPI en la transmisión del VIH-1 a través de la leche materna, encontrando mayores niveles de SLPI en la saliva de bebés que recibieron alimentación materna y no fueron infectados que en los bebés que adquirieron la infección (138). Otros estudios evaluaron la expresión de la SLPI en saliva de parótida, glándula sublingual y submandibular de individuos SP y controles sanos, encontrando una mayor expresión de la SLPI en la saliva de individuos SP; adicionalmente, los individuos SP que recibían terapia HAART presentaban un aumento en los niveles de SLPI comparados con individuos sin tratamiento, lo que podría reducir el riesgo de transmisión oral del VIH-1 (130).
Conclusiones
Los elementos claves para resistir a la infección por el VIH y al progreso posterior hacia sida residen principalmente en el sistema inmune del hospedero. Tanto el sistema inmune innato como el adaptativo participan en forma coordinada en la respuesta a los procesos infecciosos por medio de componen-tes celulares y de factores solubles con actividad microbicida. Diversos factores solubles se han postulado como elementos importantes de la respuesta antiviral. Estos factores ejercen su actividad por medio de mecanismos no citolíticos, lo que los hace un blanco importante de investigación y destaca su potencial terapéutico, particularmente como complemento de la terapia antirretroviral actualmente en uso. Aunque se ha comprobado la actividad antiviral in vitro de estos factores solubles y existe evidencia epidemioló-gica que respalda su papel protector in vivo durante la infección por VIH-1, hasta el momento no se tiene claramente identificado el mecanismo exacto por medio del cual muchos de estos factores ejercen su acción antiviral. Tampoco existen estudios controlados y en modelos animales que evalúen su actividad in vivo, y que permitan establecer las condiciones óptimas para su uso terapéutico. La intensa investigación en esta área muy posiblemente logrará la identificación de nuevos factores solubles con actividad anti-VIH-1 y sus mecanismos de acción, lo que permitirá establecer nuevas estrategias terapéuticas para pacientes VIH-1 positivos.
Agradecimientos
Al Comité para el Desarrollo de la Investigación (CODI) de la Universidad de Antioquia por su apoyo económico; al personal de salud de la Institución Prestadora de Salud (IPS), HERES Salud, Empresa Unipersonal de la ciudad de Santa Marta, por su participación y colaboración, y al estudiante de medicina de la Universidad de Antioquia Mario Archila Duarte por el diseño de la figura.
Conflicto de intereses
Las autores de esta revisión manifestamos que no existe ningún conflicto de intereses en la realización de este trabajo.
Financiación
Este trabajo fue financiado por el Comité para el Desarrollo de la Investigación (CODI) de la Universidad de Antioquia.
Correspondencia:
María Teresa Rugeles, Grupo de Inmunovirologia-Biogénesis, Facultad de Medicina, Universidad de Antioquia, Calle 62 # 52-59, Laboratorio 532, Medellín, Colombia.
Teléfono: (4) 2106551, fax: (4) 2106481.
Recibido: 28/03/06; aceptado: 16/08/06
Referencias
1. Dybul M, Connors M, Fauci AS. Immunology of HIV infection. In: Paul WE, editor. Fundamental Immunology. 5th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2003.p. 1285-318. [ Links ] 2. UNAIDS U, WHO. Informe sobre la epidemia mundial de SIDA 2005. Resumen analítico. Geneve: WHO; 2005. [ Links ] 3. UNAIDS U, WHO. Colombia: epidemiological fact sheets on HIV/AIDS and sexually transmitted infections. Geneve: WHO; 2004. [ Links ] 4. Sierra S, Kupfer B, Kaiser R. Basics of the virology of HIV-1 and its replication. J Clin Virol 2005; 34: 233-44. [ Links ] 5. Summers T. Public policy for health care workers infected with the human immunodeficiency virus. Jama 2001; 285: 882. [ Links ] 6. Mann JM, Tarantola DJ. HIV 1998: the global picture. Sci Am 1998; 279: 82-3. [ Links ] 7. Munoz A, Kirby AJ, He YD, Margolick JB, Visscher BR, Rinaldo CR, et al. Long-term survivors with HIV-1 infection: incubation period and longitudinal patterns of CD4+ lymphocytes. J Acquir Immune Defic Syndr Hum Retrovirol 1995; 8: 496-505. [ Links ] 8. Cao Y, Qin L, Zhang L, Safrit J, Ho DD. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med 1995; 332: 201-8. [ Links ] 9. Paxton WA, Martin SR, Tse D, OBrien TR, Skurnick J, VanDevanter NL, et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nat Med 1996; 2: 412-7. [ Links ] 10. Mikhail M, Wang B, Saksena NK. Mechanisms involved in non-progressive HIV disease. AIDS Rev 2003; 5: 230-44. [ Links ] 11. Endo Y, Igarashi T, Nishimura Y, Buckler C, Buckler-White A, Plishka R, et al. Short- and long-term clinical outcomes in rhesus monkeys inoculated with a highly pathogenic chimeric simian/human immunodeficiency virus. J Virol 2000; 74: 6935-45. [ Links ] 12. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996; 382:722-5. [ Links ] 13. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horu k R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996; 86: 367-77. [ Links ] 14. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996; 381: 661-6. [ Links ] 15. Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med 1996; 2: 1240-3. [ Links ] 16. Rugeles MT, Solano F, Diaz FJ, Bedoya VI, Patino PJ. Molecular characterization of the CCR 5 gene in seronegative individuals exposed to human immunodeficiency virus (HIV). J Clin Virol 2002; 23: 161-9. 17. Smith MW, Dean M, Carrington M, Winkler C, Huttley GA, Lomb DA, et al. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science 1997; 277: 959-65. 18. Winkler C, Modi W, Smith MW, Nelson GW, Wu X, Carrington M, et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC). Science 1998; 279: 389-93. [ Links ] 19. MacDonald KS, Fowke KR, Kimani J, Dunand VA, Nagelkerke NJ, Ball TB, et al. Influence of HLA supertypes on susceptibility and resistance to human immunodeficiency virus type 1 infection 20. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995; 270: 1811-5. [ Links ] 21. Walker CM, Moody DJ, Stites DP, Levy JA. CD8+ lymphocytes can control HIV infection in vitro by suppre-ssing virus replication. Science 1986; 234: 1563-6. [ Links ] 22. Alfano M, Poli G. Role of cytokines and chemokines in the regulation of innate immunity and HIV infection. Mol Immunol 2005; 42: 161-82. [ Links ] 23. Patterson BK, Behbahani H, Kabat WJ, Sullivan Y, OGorman MR, Landay A, et al. Leukemia inhibitory factor inhibits HIV-1 replication and is upregulated in placentae from nontransmitting women. J Clin Invest 2001; 107: 287-94. [ Links ] 24. Lee-Huang S, Huang PL, Sun Y, Kung HF, Blithe DL, Chen HC. Lysozyme and RNases as anti-HIV components in beta-core preparations of human chorionic gonadotropin. Proc Natl Acad Sci U S A 1999; 96: 2678-81. [ Links ] 25. Lunardi-Iskandar Y, Bryant JL, Blattner WA, Hung CL, Flamand L, Gill P, et al. Effects of a urinary factor from women in early pregnancy on HIV-1, SIV and associated disease. Nat Med 1998; 4: 428-34. [ Links ] 26. Rugeles MT, Trubey CM, Bedoya VI, Pinto LA, Oppenheim JJ, Rybak SM, et al. Ribonuclease is partly responsible for the HIV-1 inhibitory effect activated by HLA alloantigen recognition. AIDS 2003; 17: 481-6. [ Links ] 27. Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 2003; 17: F39-48. [ Links ] 28. Chang TL, Vargas J Jr, DelPortillo A, Klotman ME. Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J Clin Invest 2005; 115:765-73. [ Links ] 29. Shugars DC, Alexander AL, Fu K, Freel SA. Endogenous salivary inhibitors of human immunodeficiency virus. Arch Oral Biol 1999; 44: 445-53. [ Links ] 30. Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol 2005; 3: 799-808. [ Links ] 31. Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol 2002; 23: 291-6. [ Links ] 32. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol 2005; 6: 551-7. [ Links ] 33. Zaballos A, Villares R, Albar JP, Martinez AC, Marquez G. Identification on mouse chromosome 8 of new beta-defensin genes with regionally specific expression in the male reproductive organ. J Biol Chem 2004; 279: 12421-6. [ Links ] 34. Garcia JR, Krause A, Schulz S, Rodriguez-Jimenez FJ, Kluver E, Adermann K, et al. Human beta-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J 2001; 15: 1819-21. [ Links ] 35. Froy O. Regulation of mammalian defensin expression by Toll-like receptor-dependent and independent signalling pathways. Cell Microbiol 2005; 7: 1387-97. [ Links ] 36. Ganz T. Defensins: antimicrobial peptides of vertebrates. C R Biol 2004; 327: 539-49. [ Links ] 37. Territo MC, Ganz T, Selsted ME, Lehrer R. Monocyte-chemotactic activity of defensins from human neutrophils. J Clin Invest 1989; 84: 2017-20. [ Links ] 38. Lehrer RI. Primate defensins. Nat Rev Microbiol 2004; 2: 727-38. [ Links ] 39. Chang TL, Francois F, Mosoian A, Klotman ME. CAF-mediated human immunodeficiency virus (HIV) type 1 transcriptional inhibition is distinct from alpha-defensin-1 HIV inhibition. J Virol 2003; 77: 6777-84. [ Links ] 40. Nakashima H, Yamamoto N, Masuda M, Fujii N. Defensins inhibit HIV replication in vitro. AIDS 1993; 7: 1129. [ Links ] 41. Ganz T, Lehrer RI. Antimicrobial peptides of vertebrates. Curr Opin Immunol 1998; 10: 41-4. [ Links ] 42. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L et al. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood 2000; 96: 3086-93. [ Links ] 43. Mackewicz CE, Yuan J, Tran P, Diaz L, Mack E, Selsted ME, et al. alpha-Defensins can have anti-HIV activity but are not CD8 cell anti-HIV factors. AIDS 2003; 17: F23-32. [ Links ] 44. Wang W, Owen SM, Rudolph DL, Cole AM, Hong T, Waring AJ, et al. Activity of alpha- and theta-defensins against primary isolates of HIV-1. J Immunol 2004; 173: 515-20. [ Links ] 45. Chang TL, Klotman ME. Defensins: natural anti-HIV peptides. AIDS Rev 2004; 6: 161-8. [ Links ] 46. Kagnoff MF, Roebuck KA. Human immunodeficiency virus type 1 (HIV-1) infection and expression in intestinal epithelial cells: role of protein kinase A and C pathways in HIV-1 transcription. J Infect Dis 1999; 179 (Suppl. 3): S444-7. [ Links ] 47. Guo CJ, Tan N, Song L, Douglas SD, Ho WZ. Alpha-defensins inhibit HIV infection of macrophages through upregulation of CC-chemokines. AIDS 2004; 18: 1217-8. [ Links ] 48. Trabattoni D, Caputo SL, Maffeis G, Vichi F, Biasin M, Pierotti P, et al. Human alpha defensin in HIV-exposed but uninfected individuals. J Acquir Immune Defic Syndr 2004; 35: 455-63. [ Links ] 49. Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol 2004; 22: 181-215. [ Links ] 50. Schutte BC, Mitros JP, Bartlett JA, Walters JD, Jia HP, Welsh MJ, et al. Discovery of five conserved beta-defensin gene clusters using a computational search strategy. Proc Natl Acad Sci U S A 2002; 99: 2129-33. [ Links ] 51. Bensch KW, Raida M, Magert HJ, Schulz-Knappe P, Forssmann WG. hBD-1: a novel beta-defensin from human plasma. FEBS Lett 1995; 368: 331-5. [ Links ] 52. Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature 1997; 387: 861. [ Links ] 53. Yamaguchi Y, Nagase T, Makita R, Fukuhara S, Tomita T, Tominaga T, et al. Identification of multiple novel epididymis-specific beta-defensin isoforms in humans and mice. J Immunol 2002; 169: 2516-23. [ Links ] 54. Lehrer RI, Ganz T. Defensins of vertebrate animals. Curr Opin Immunol 2002; 14: 96-102. [ Links ] 55. Donnarumma G, Paoletti I, Buommino E, Orlando M, Tufano MA, Baroni A. Malassezia furfur induces the expression of beta-defensin-2 in human keratinocytes in a protein kinase C-dependent manner. Arch Dermatol Res 2004; 295: 474-81. [ Links ] 56. Fahlgren A, Hammarstrom S, Danielsson A, Hammarstrom ML. Beta-defensin-3 and -4 in intestinal epithelial cells display increased mRNA expression in ulcerative colitis. Clin Exp Immunol 2004; 137: 379-85. [ Links ] 57. Kao CY, Chen Y, Zhao YH, Wu R. ORFeome-based search of airway epithelial cell-specific novel human beta -defensin genes. Am J Respir Cell Mol Biol 2003; 29: 71-80. [ Links ] 58. Schols D, Pauwels R, Desmyter J, De Clercq E. Dextran sulfate and other polyanionic anti-HIV compounds specifically interact with the viral gp120 glycoprotein expressed by T-cells persistently infected with HIV-1. Virology 1990; 175: 556-61. [ Links ] 59. Damonte E, Neyts J, Pujol CA, Snoeck R, Andrei G, Ikeda S, et al. Antiviral activity of a sulphated polysaccharide from the red seaweed Nothogenia fastigiata. Biochem Pharmacol 1994; 47: 2187-92. [ Links ] 60. Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra Maggiore GM, et al. Human beta-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection. J Virol 2005; 79: 14318-29. [ Links ] 61. Braida L, Boniotto M, Pontillo A, Tovo PA, Amoroso A, Crovella S. A single-nucleotide polymorphism in the human beta-defensin 1 gene is associated with HIV-1 infection in Italian children. AIDS 2004; 18: 1598-600. [ Links ] 62. Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood 2000; 95: 3032-43. [ Links ] 63. Schall TJ, Bacon KB. Chemokines, leukocyte trafficking, and inflammation. Curr Opin Immunol 1994; 6: 865-73. [ Links ] 64. Sell S. Immunology, immunopathology and immunity. Sixth ed. Washington, D.C.: ASM press; 2001. [ Links ] 65. Clapham PR, McKnight A. Cell surface receptors, virus entry and tropism of primate lentiviruses. J Gen Virol 2002; 83: 1809-29. [ Links ] 66. DeVico A L, Gallo RC. Control of HIV-1 infection by soluble factors of the immune response. Nat Rev Microbiol 2004; 2: 401-13. [ Links ] 67. Kalinkovich A, Weisman Z, Bentwich Z. Chemokines and chemokine receptors: role in HIV infection. Immunol Lett 1999; 68: 281-7. [ Links ] 68. Nagasawa T, Tachibana K, Kishimoto T. A novel CXC chemokine PBSF/SDF-1 and its receptor CXCR4: their functions in development, hematopoiesis and HIV infection. Semin Immunol 1998; 10: 179-85. [ Links ] 69. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996; 382: 829-33. [ Links ] 70. Agace WW, Amara A, Roberts AI, Pablos JL, Thelen S, Uguccioni M, et al. Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV transmission and propagation. Curr Biol 2000; 10: 325-8. [ Links ] 71. Bomsel M, David V. Mucosal gatekeepers: selecting HIV viruses for early infection. Nat Med 2002; 8: 114-6. [ Links ] 72. Shattock RJ, Moore JP. Inhibiting sexual transmission of HIV-1 infection. Nat Rev Microbiol 2003; 1: 25-34. [ Links ] 73. Farquhar C, Mbori-Ngacha DA, Redman MW, Bosire RK, Lohman BL, Piantadosi AL, et al. CC and CXC chemokines in breastmilk are associated with mother-to-child HIV-1 transmission. Curr HIV Res 2005; 3: 361-9. [ Links ] 74. Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev 2002; 13: 455-81. [ Links ] 75. Gong W, Howard OM, Turpin JA, Grimm MC, Ueda H, Gray PW, et al. Monocyte chemotactic protein-2 activates CCR5 and blocks CD4/CCR5-mediated HIV-1 entry/replication. J Biol Chem 1998; 273: 4289-92. [ Links ] 76. Cocchi F, DeVico AL, Yarchoan R, Redfield R, Cleghorn F, Blattner WA, et al. Higher macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels from CD8+ T cells are associated with asymptomatic HIV-1 infection. Proc Natl Acad Sci US A 2000; 97: 13812-7. [ Links ] 77. Wasik TJ, Wierzbicki A, Whiteman VE, Trinchieri G, Lischner HW, Kozbor D. Association between HIV-specific T helper responses and CTL activities in pediatric AIDS. Eur J Immunol 2000; 30: 117-27. [ Links ] 78. Iqbal SM, Ball TB, Kimani J, Kiama P, Thottingal P, Embree JE, et al. Elevated T cell counts and RANTES expression in the genital mucosa of HIV-1-resistant Kenyan commercial sex workers. J Infect Dis 2005; 192: 728-38. [ Links ] 79. Hengel H, Koszinowski UH, Conzelmann KK. Viruses know it all: new insights into IFN networks. Trends Immunol 2005; 26: 396-401. [ Links ] 80. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol 1997; 15: 749-95. [ Links ] 81. Vilcek JS. Interferons and other citokines. In: Fields BN, Knipe DM, Howley PM, editors. Fields virology. 3rd ed. Vol. 1. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. 82. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001; 14: 778-809. [ Links ] 83. Gao JJ, Filla MB, Fultz MJ, Vogel SN, Russell SW, Murphy WJ. Autocrine/paracrine IFN-alphabeta mediates the lipopolysaccharide-induced activation of transcription factor Stat1alpha in mouse macrophages: pivotal role of Stat1alpha in induction of the inducible nitric oxide synthase gene. J Immunol 1998; 161: 4803-10. [ Links ] 84. Bogdan C. The function of type I interferons in antimicrobial immunity. Curr Opin Immunol 2000; 12: 419-24. [ Links ] 85. Belardelli F, Gresser I. The neglected role of type I interferon in the T-cell response: implications for its clinical use. Immunol Today 1996; 17: 369-72. [ Links ] 86. Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13: 119-34. [ Links ] 87. Fernie BF, Poli G, Fauci AS. Alpha interferon suppresses virion but not soluble human immunodeficiency virus antigen production in chronically infected T-lymphocytic cells. J Virol 1991; 65: 3968-71. [ Links ] 88. Pinto LA, Blazevic V, Patterson BK, Mac Trubey C, Dolan MJ, Shearer GM. Inhibition of human immunodeficiency virus type 1 replication prior to reverse transcription by influenza virus stimulation. J Virol 2000; 74: 4505-11. [ Links ] 89. Levy JA, Scott I, Mackewicz C. Protection from HIV/AIDS: the importance of innate immunity. Clin Immunol 2003; 108: 167-74. [ Links ] 90. Servet C, Zitvogel L, Hosmalin A. Dendritic cells in innate immune responses against HIV. Curr Mol Med 2002; 2: 739-56. [ Links ] 91. Siegal FP, Fitzgerald-Bocarsly P, Holland BK, Shodell M. Interferon-alpha generation and immune reconstitution during antiretroviral therapy for human immunodeficiency virus infection. AIDS 2001; 15: 1603-12. [ Links ] 92. Haas DW, Lavelle J, Nadler JP, Greenberg SB, Frame P, Mustafa N, et al. A randomized trial of interferon alpha therapy for HIV type 1 infection. AIDS Res Hum Retroviruses 2000; 16: 183-90. [ Links ] 93. Lane HC, Kovacs JA, Feinberg J, Herpin B, Davey V, Walker R, et al. Anti-retroviral effects of interferon-alpha in AIDS-associated Kaposis sarcoma. Lancet 1988; 2: 1218-22. [ Links ] 94. Delhem N, Hadida F, Gorochov G, Carpentier F, de Cavel JP, Andreani JF, et al. Primary Th1 cell immunization against HIVgp160 in SCID-hu mice coengrafted with peripheral blood lymphocytes and skin. J Immunol 1998; 161: 2060-9. [ Links ] 95. Portales P, Reynes J, Rouzier-Panis R, Baillat V, Clot J, Corbeau P. Perforin expression in T cells and virological response to PEG-interferon alpha2b in HIV-1 infection. AIDS 2003; 17: 505-11. [ Links ] 96. Santini SM, Di Pucchio T, Lapenta C, Parlato S, Logozzi M, Belardelli F. The natural alliance between type I interferon and dendritic cells and its role in linking innate and adaptive immunity. J Interferon Cytokine Res 2002; 22: 1071-80. [ Links ] 97. Loseke S, Grage-Griebenow E, Wagner A, Gehlhar K, Bufe A. Differential expression of IFN-alpha subtypes in human PBMC: evaluation of novel real-time PCR assays. J Immunol Methods 2003; 276: 207-22. [ Links ] 98. Ludewig B, Gelderblom HR, Becker Y, Schafer A, Pauli G. Transmission of HIV-1 from productively infected mature Langerhans cells to primary CD4+ T lymphocytes results in altered T cell responses with enhanced production of IFN-gamma and IL-10. Virology 1996; 215: 51-60. [ Links ] 99. Levy JA, Mackewicz CE, Barker E. Controlling HIV pathogenesis: the role of the noncytotoxic anti-HIV response of CD8+ T cells. Immunol Today 1996; 17: 217-24. [ Links ] 100. Geiben-Lynn R, Brown N, Walker BD, Luster AD. Purification of a modified form of bovine antithrombin III as an HIV-1 CD8+ T-cell antiviral factor. J Biol Chem 2002; 277: 42352-7. [ Links ] 101. Levy JA. The search for the CD8+ cell anti-HIV factor (CAF). Trends Immunol 2003; 24: 628-32. [ Links ] 102. Zhang L, Yu W, He T, Yu J, Caffrey RE, Dalmasso EA, et al. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 2002; 298: 995-1000. [ Links ] 103. Zhang L, Lopez P, He T, Yu W, Ho DD. Retraction of an interpretation. Science 2004; 303: 467. [ Links ] 104. Chen CH, Weinhold KJ, Bartlett JA, Bolognesi DP, Greenberg ML. CD8+ T lymphocyte-mediated inhibition of HIV-1 long terminal repeat transcription: a novel antiviral mechanism. AIDS Res Hum Retroviruses 1993; 9: 1079-86. [ Links ] 105. Chang TL, Mosoian A, Pine R, Klotman ME, Moore JP. A soluble factor(s) secreted from CD8(+) T lymphocytes inhibits human immunodeficiency virus type 1 replication through STAT1 activation. J Virol 2002; 76: 569-81. [ Links ] 106. Copeland KF, McKay PJ, Rosenthal KL. Suppression of activation of the human immunodeficiency virus long terminal repeat by CD8+ T cells is not lentivirus specific. AIDS Res Hum Retroviruses 1995; 11: 1321-6. [ Links ] 107. Copeland KF, McKay PJ, Rosenthal KL. Suppression of the human immunodeficiency virus long terminal repeat by CD8+ T cells is dependent on the NFAT-1 element. AIDS Res Hum Retroviruses 1996; 12: 143-8. [ Links ] 108. Mackewicz CE, Craik CS, Levy JA. The CD8+ cell noncytotoxic anti-HIV response can be blocked by protease inhibitors. Proc Natl Acad Sci USA 2003; 100: 3433-8. [ Links ] 109. Blackbourn DJ, Mackewicz CE, Barker E, Hunt TK, Herndier B, Haase AT, et al. Suppression of HIV replication by lymphoid tissue CD8+ cells correlates with the clinical state of HIV-infected individuals. Proc Natl Acad Sci U S A 1996; 93: 13125-30. [ Links ] 110. Ahmed RK, Norrgren H, da Silva Z, Blaxhult A, Fredriksson EL, Biberfeld G, et al. Antigen-specific beta-chemokine production and CD8 T-cell noncytotoxic antiviral activity in HIV-2-infected individuals. Scand J Immunol 2005; 61: 63-71. [ Links ] 111. Stranford SA, Skurnick J, Louria D, Osmond D, Chang SY, Sninsky J, et al. Lack of infection in HIV-exposed individuals is associated with a strong CD8(+) cell noncytotoxic anti-HIV response. Proc Natl Acad Sci U S A 1999; 96: 1030-5. [ Links ] 112. Mackewicz CE, Ortega HW, Levy JA. CD8+ cell anti-HIV activity correlates with the clinical state of the infected individual. J Clin Invest 1991; 87: 1462-6. [ Links ] 113. Deshpande RA, Shankar V. Ribonucleases from T2 family. Crit Rev Microbiol 2002; 28: 79-122. [ Links ] 114. Cho S, Beintema JJ, Zhang J. The ribonuclease A superfamily of mammals and birds: identifying new members and tracing evolutionary histories. Genomics 2005; 85: 208-20. [ Links ] 115. Aravind L, Koonin EV. A natural classification of ribonucleases. Methods Enzymol 2001; 341: 3-28. [ Links ] 116. Yang D, Rosenberg HF, Chen Q, Dyer KD, Kurosaka K, Oppenheim JJ. Eosinophil-derived neurotoxin (EDN), an antimicrobial protein with chemotactic activities for dendritic cells. Blood 2003; 102: 3396-403. [ Links ] 117. Saxena SK, Sirdeshmukh R, Ardelt W, Mikulski SM, Shogen K, Youle RJ. Entry into cells and selective degradation of tRNAs by a cytotoxic member of the RNase A family. J Biol Chem 2002; 277: 15142-6. [ Links ] 118. Haigis MC, Raines RT. Secretory ribonucleases are internalized by a dynamin-independent endocytic pathway. J Cell Sci 2003; 116: 313-24. [ Links ] 119. Rosenberg HF, Domachowske JB. Eosinophils, eosinophil ribonucleases, and their role in host defense against respiratory virus pathogens. J Leukoc Biol 2001; 70: 691-8. [ Links ] 120. Youle RJ, Wu YN, Mikulski SM, Shogen K, Hamilton RS, Newton D, et al. RNase inhibition of human immunodeficiency virus infection of H9 cells. Proc Natl Acad Sci USA 1994; 91: 6012-6. [ Links ] 121. Pinto LA, Blazevic V, Shearer GM, Patterson BK, Dolan MJ. Alloantigen-induced anti-HIV activity occurs prior to reverse transcription and can be generated by leukocytes from HIV-infected individuals. Blood 2000; 95: 1875-6. [ Links ] 122. Zelepuga EA, Ponomareva RB, Kolikov VM, Pautov VD, Sheveleva TV, Medvedev ML, et al. Conjugates of pancreatic ribonuclease and ligand-free human serum albumin. Biomed Khim 2003; 49: 588-96. [ Links ] 123. Franken C, Meijer CJ, Dijkman JH. Tissue distribution of antileukoprotease and lysozyme in humans. J Histochem Cytochem 1989; 37: 493-8. [ Links ] 124. Thompson RC, Ohlsson K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc Natl Acad Sci USA 1986; 83: 6692-6. [ Links ] 125. Sorensen OE, Cowland JB, Theilgaard-Monch K, Liu L, Ganz T, Borregaard N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol 2003; 170: 5583-9. [ Links ] 126. Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol 1997; 61: 695-702. [ Links ] 127. Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell 1997; 88: 417-26. [ Links ] 128. Shugars DC. Endogenous mucosal antiviral factors of the oral cavity. J Infect Dis 1999; 179 (Suppl. 3): S431-5. [ Links ] 129. Hiemstra PS. Novel roles of protease inhibitors in infection and inflammation. Biochem Soc Trans 2002; 30: 116-20. [ Links ] 130. Lin AL, Johnson DA, Stephan KT, Yeh CK. Salivary secretory leukocyte protease inhibitor increases in HIV infection. J Oral Pathol Med 2004; 33: 410-6. [ Links ] 131. Ota Y, Shimoya K, Zhang Q, Moriyama A, Chin R, Tenma K, et al. The expression of secretory leukocyte protease inhibitor (SLPI) in the fallopian tube: SLPI protects the acrosome reaction of sperm from inhibitory effects of elastase. Hum Reprod 2002; 17: 2517-22. [ Links ] 132. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 2002;111:867-78. [ Links ] 133. McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM. Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J Clin Invest 1995; 96: 456-64. [ Links ] 134. McNeely TB, Shugars DC, Rosendahl M, Tucker C, Eisenberg SP, Wahl SM. Inhibition of human immunodeficiency virus type 1 infectivity by secretory leukocyte protease inhibitor occurs prior to viral reverse transcription. Blood 1997; 90: 1141-9. [ Links ] 135. Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, et al. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J Exp Med 2004; 200: 1337-46. [ Links ] 136. Jana NK, Gray LR, Shugars DC. Human immunodeficiency virus type 1 stimulates the expression and production of secretory leukocyte protease inhibitor (SLPI) in oral epithelial cells: a role for SLPI in innate mucosal immunity. J Virol 2005; 79: 6432-40. [ Links ] 137. Pillay K, Coutsoudis A, Agadzi-Naqvi AK, Kuhn L, Coovadia HM, Janoff EN. Secretory leukocyte protease inhibitor in vaginal fluids and perinatal human immunodeficiency virus type 1 transmission. J Infect Dis 2001; 183: 653-6. [ Links ] 138. Farquhar C, VanCott TC, Mbori-Ngacha DA, Horani L, Bosire RK, Kreiss JK, et al. Salivary secretory leukocyte protease inhibitor is associated with reduced transmission of human immunodeficiency virus type 1 through breast milk. J Infect Dis 2002; 186: 1173-6. [ Links ]

