










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.27 n.1 Bogotá jan./mar. 2007
Predicción computacional de la estructura terciaria de la iduronato 2-sulfato sulfatasa humana
Homero Sáenz 1, 2, Leonardo Lareo 3, Raúl A. Poutou 1,4, Ángela C. Sosa 1, Luis A. Barrera 1
1 Instituto de Errores Innatos del Metabolismo, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá D.C., Colombia.
2
Unidad de Biología Celular y Microscopía, Decanato de Medicina, Universidad Centroccidental "Lisandro Alvarado", Barquisimeto, Venezuela.3
Grupo de Investigación en Bioquímica Computacional, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá D.C., Colombia.4
Grupo de Biotecnología Ambiental e Industrial, Facultad de Ciencias, Pontificia Universidad Javeriana. Bogotá D.C., Colombia.Recibido: 09/03/06; aceptado: 23/10/06
Introducción. El síndrome de Hunter o mucopolisacaridosis tipo II (MC KUSIK 309900) es causado por la deficiencia de la iduronato 2-sulfato sulfatasa humana (E.C. 3.1.6.13). La enzima no ha sido cristalizada y por tanto sus estructuras no se conocen por deducción experimental.
Objetivo. Proponer un modelo computacional para la estructura tridimensional de la iduronato 2-sulfato sulfatasa humana.
Materiales y métodos. Se realizó un análisis computacional de esta enzima empleando programas de libre acceso en internet como Comput pI/MW, JaMBW Chapter 3.1.7, SWISS-MODEL, 3D-PSSM, ProSup. Los procesos de minimización de energía se realizaron con el programa Discover 3 del paquete Insight II (2004).
Resultados. Se propone un modelo tridimensional de la iduronato 2-sulfato sulfatasa humana que presenta 33,3% en hélice, 7,2% en hoja plegada y 59,5% en enrollamiento al azar (coil). Se hallaron valores de RMS (del inglés Root Mean Square) de 0,78 y 0,86Å al compararla con otras enzimas de la misma familia. El modelo revela cinco sitios potenciales de N-glicosilación y una entrada al bolsillo que contiene los aminoácidos que componen el sitio activo. Usando este modelo se encontró una buena correlación entre el tipo de mutaciones y la gravedad de la enfermedad en 20 pacientes analizados.
Conclusión. Los valores de RMS y la correlación genotipo-fenotipo en los pacientes analizados sugieren el modelo puede usarse para predecir ciertos aspectos del comportamiento biológico de la enzima.
Palabras clave: mucopolisacaridosis II, iduronato sulfatasa, estructura terciaria de proteína, metodologías computacionales.
Computational prediction of the tertiary structure of the human iduronate 2-sulfate sulfatase
Introduction. Hunter syndrome (MC KUSIK 309900) or mucopolysacharidosis type II is due to the deficiency of the enzyme iduronate 2 sulfate sulfatase (E.C. 3.1.6.13). This enzyme has not been crystallized, and therefore the experimental structures are not available.
Objectives. A computational three-dimensional model was proposed for the iduronate 2 sulfate sulfatase enzyme.
Materials and methods. A computational analysis of this enzyme used the following free internet software programs: Comput pI/MW, JaMBW Chapter 3.1.7, SWISS-MODEL, Geno3d, ProSup. Energy minimization was done with Discover 3 and Insight II version 2004.
Results. A three-dimensional conformational model was proposed. The model showed 33.3% of helix structure, 7.2% beta sheet, and 59.5% random coil. RMS values (Root Mean Square) (0.78 and 0.86Å) were found when compared with other enzymes of the same family. The model presented 5 exposed N-glycosylation potential sites and an entry to the pocket that contains the amino acids of the active site. A high correlation was found between the type of mutations and the severity of the phenotype in twenty patients analyzed.
Conclusion. The RMS values, as well as the high correlation between the type of mutation and the phenotype, indicated that the model predicts some aspects of the enzymes biological behavior.
Key words: mucopolysaccharidosis II, iduronate sulfatase, tertiary protein structure,computing methods.
El síndrome de Hunter o mucopolisacaridosis II (MPS II) (MC KUSIK 309900) es causado por la deficiencia de la enzima humana iduronato 2-sulfato sulfatasa (IDSh) (E.C. 3.1.6.13), lo que ocasiona la acumulación lisosómica del heparán y el dermatán sulfato con el consecuente aumento de su excreción urinaria. Las consecuencias de esta deficiencia enzimática se traducen en dos variantes de la enfermedad, la grave y la moderada; los cuadros clínicos más frecuentes son facies toscas, deformidades esqueléticas, hepato-esplenomegalia, estatura pequeña, hipertensión pulmonar, alteraciones en el miocardio, retardo mental y muerte prematura (1). En esta enferme-dad, el defecto se localiza a nivel del gen que codifica para la enzima, generando una patología intratable por vías convencionales. En Israel, la incidencia de la enfermedad se ha reportado con una frecuencia de 1:34.000 en hombres (2); en el Reino Unido se estimó en 1:132.000 (3) y en la Columbia Británica se reportó una incidencia de 1:111.000 (4), mientras que en Colombia no existen datos de incidencia o prevalencia de la enfermedad.
Para la IDSh se han reportado 333 mutaciones, de las cuales 176 son puntuales, 30 por corte y empalme, 61 deleciones pequeñas, 24 inserciones pequeñas, 5 inserciones/deleciones, 26 deleciones grandes, 10 rearreglos y una inserción grande (5-27) (
http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html ). Hasta el momento, la literatura no reporta estudios que establezcan el efecto de las mutaciones sobre la funcionalidad enzimática y el fenotipo de los pacientes con MPS II.En la actualidad existe un gran número de programas de computación desarrollados para el estudio de las biomoléculas. A través de ellos es posible hacer predicciones de estructura y función. Estos programas utilizan algoritmos estadísticos, neurales y genéticos, teniendo como base la información acumulada en bases de datos como Gen Bank, ExPASy y EMBL, entre otras. Los programas de computación se han convertido en herramientas útiles debido a que el conocimiento de aspectos relativos a la estructura y, por consiguiente, al comportamiento de una molécula facilita el diseño de experimentos y permite el ahorro de energía, tiempo y recursos. Dichas herramientas son de fácil acceso y, aunque su manejo es árduo, no son complicadas. Este trabajo se orientó a proponer un modelo computacional para la estructura tridimensional de la IDSh y su empleo en la predicción de la relación existente entre el genotipo y la gravedad de la enfermedad. El estudio se enmarca dentro de la línea de investigación que tiene como meta la producción de la enzima recombinante en la levadura metilotrófica Pichia pastoris con el fin de emplearla en la terapia de reemplazo enzimático en pacientes con el síndrome de Hunter.
Materiales y métodos
Análisis computacional con base en la estructura primaria de la IDSh
El número de acceso para la IDSh en Gen Bank es AAA63197, en Medline, 91046030 y en PubMed se ingresa con el código 2122463; allí se encuentra la secuencia aminoacídica de la IDSh en el formato FASTA necesario para los estudios computacionales; esta secuencia se obtuvo a partir de la traducción conceptual del ADNc y fue reportada por Wilson y colaboradores (28,29). El análisis computacional permitió la predicción de los perfiles de accesibilidad, hidrofobicidad y flexibilidad con el empleo de ProtScale de ExPASy (30).
El tamaño de ventana para los análisis con Protscale fue el básico de nueve aminoácidos recomendado por los programas para garantizar la óptima cobertura de la secuencia cuando se hace el recorrido sobre ella. En el caso del perfil de hidrofobicidad se empleó el algoritmo de Kite y Doolittle, cuya escala considera valores entre -3,0 y 4,0 (30). El punto isoeléctrico (pI) y el peso molecular se establecieron por medio del programa Comput pI/MW (31); los sitios potenciales de fosforilación se determinaron con Netphos-2.0 (32) y los de O- y N-glicosilaciones con los programas Net-Oglyc 3.1 (33) y Net-Nglyc 1.0 (34), respectiva-mente, mientras que los bloques e impresiones (prints) de similitud con otras moléculas se obtuvieron a través del programa Findermotif (35); para la determinación del perfil de antigenicidad se utilizó el programa JaMBW Chapter 3.1.7 (36) y los alineamientos de secuencias se realizaron con el programa BLAST de NCBI (37).
Modelos de estructura secundaria y terciaria y sitio activo
Para predecir la estructura secundaria se emplearon las Proteomics and Sequence Analysis herramientas de ExPASy y se obtuvo un consenso de las predicciones de los programas GOR, GOR 1, GOR 3, nnPred, Sspro, Casp, Meta-Predict, Fasta, SUB, rdb y Psi-Pred (38-44).
En la predicción de la estructura terciaria se utilizaron las Proteomics herramientas de ExPASy, para lo cual se remitió la secuencia a los siguientes programas de predicción de estructura terciaria: SWISS-MODEL, Geno3d, CPH models, 3D-PSSM, ProSup, SWEET (40,45-47), y el CPH models 2.0: X3m, un programa computacional para extraer modelos tridimensionales. Adicionalmente, múltiples alineamientos logrados con el programa BLAST permitieron obtener fragmentos de otras moléculas con secuencias similares a la IDSh. Para construir los segmentos peptídicos que no tuvieron predicción computacional se empleó el programa Swiss-PDBviewer (Spdbv) 3.7 (40), que permitió que los residuos que no se encontraban en ninguno de esos fragmentos fueran adicionados manualmente uno a uno, atendiendo a su orientación de acuerdo a la predicción secundaria. Finalmente, la estructura fue sometida a un proceso de minimización de energía empleando el programa Discover 3 del paquete de Insight II de 2004 (48) en un computador Silicon Graphics Octane. Para visualizar la estructura tridimensional se emplearon los programas RasMol 2.6 (49) y Spdbv 3.7 (40).
Un análisis teórico y computacional de la comparación de estructura y función de otras sulfatasas (50-54) permitió proponer un modelo para el sitio activo de la IDSh. Para la construcción del heparán sulfato (uno de los sustratos naturales de la IDSh), se empleó el programa WebLab Viewer Pro (55).
Cálculo de la raíz media cuadrática (RMS)
La RMS es una medida que permite establecer la diferencia en la distribución. Su expresión matemática
es: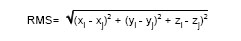
Los valores de RMS para todos los átomos menores o iguales a 1,0 representan moléculas con alta similitud estructural.
Se realizó un análisis teórico y computacional para 20 mutaciones puntuales de la IDSh (5-27).
Resultados
Análisis computacional con base en la estructura primaria de la IDSh
La
figura 1 muestra la secuencia de aminoácidos de la IDSh (28), cuya composición es 29 A, 24 R, 39 D, 21 N, 6 C, 23 Q, 24 E, 29 G, 15 H, 22 I, 55 L, 19 K, 8 M, 28 F, 49 P, 41 S, 21 T, 7 W, 26 Y, 31 V. Presenta 63 residuos negativos y 58 positivos.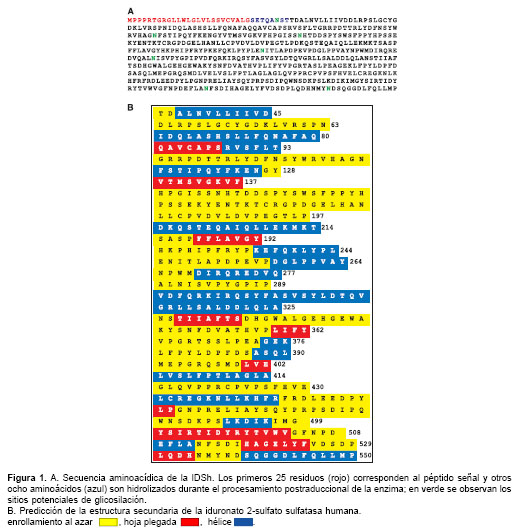
Los perfiles de hidrofobicidad (
figura 2A), accesibilidad (figura 2B) y flexibilidad (figura 2C) sugieren que la molécula es una estructura con un alto porcentaje de accesibilidad, poco hidrofóbica y bastante flexible.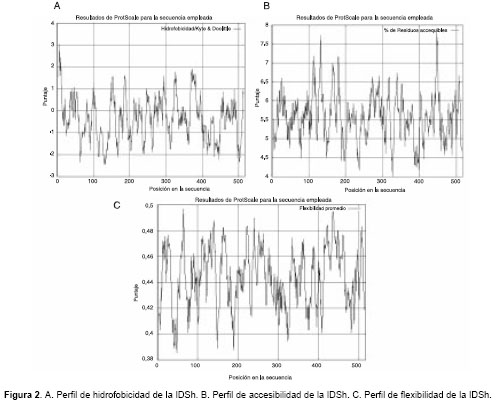
Los valores de predicción computacional del punto isoeléctrico y del peso molecular fueron 5,15 y 58,479 kDa; se encontraron 26 sitios potenciales de fosforilación (
figura 3A), 61 sitios potenciales de O-glicosilación (figura 3B) y 7 sitios potenciales de N-glicosilación (figura 3C).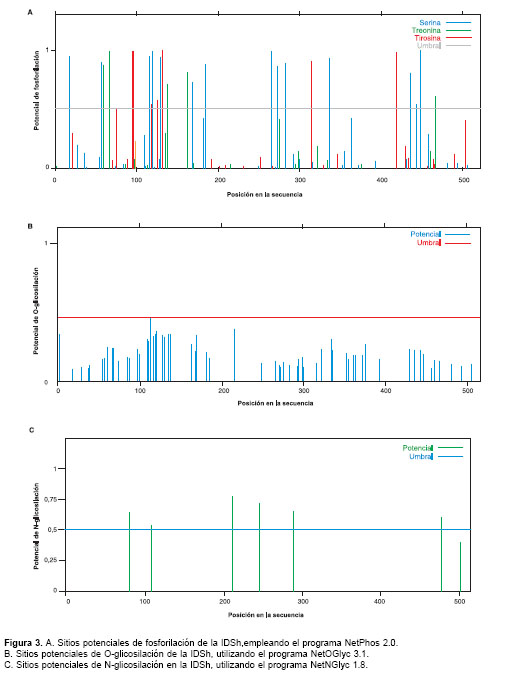
Tanto los bloques como las impresiones (prints) son secuencias con similitud; sin embargo, el tamaño de las impresiones es bastante más grande que el de los bloques. De las cinco moléculas con similitud en impresiones y de las 114 con bloques similares a secuencias de la IDSh, solamente dos sulfatasas tienen estructura tridimensional determinada experimentalmente. Sus números de acceso a SWISSPROT son PS00523 y PS00149, que en NCBI corresponden a P15289 y P15848. Estas moléculas son la ARSA (E.C. 3.1.6.8) y la ARSB (E.C. 3.1.6.12). Las secuencias de estas enzimas se alinearon con la secuencia de la IDSh y se encontraron porcentajes de similitud de 26 y 24%, respectivamente.
El perfil de antigenicidad mostró zonas altamente antigénicas entre los aminoácidos 50 a 60, 120 a 140, 230 a 240 y 400 a 420 (
figura 4).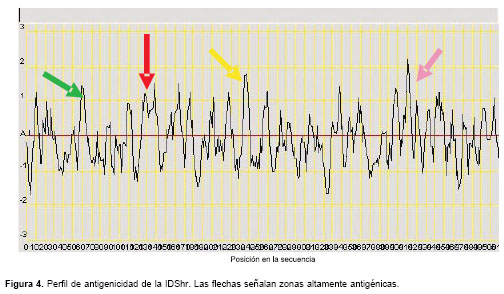
Modelos de las estructuras secundaria y terciaria y sitio activo
El consenso de las predicciones de los programas empleados, residuo por residuo, permitió la construcción de un modelo para la estructura secundaria de la IDSh. Los programas GOR, GOR-1, GOR-3 predicen para hélice, enrollamiento al azar, hoja plegada y vueltas beta (beta turn); los programas Sspro y Casp lo hacen para hélice, hoja plegada y enrollamiento al azar; los programas MetaPred, Fasta, SUB y rdb predicen para regiones en hélice, hoja plegada y lazos al azar (loops), mientras nnPredict solamente para hélice y hoja plegada. Los lazos y vueltas se homologaron con enrollamiento al azar, por lo cual la presentación del esquema se hace solamente con base en hélice, hoja plegada y enrollamiento al azar, correspondiendo a cada una de estas estructuras el 33, 13 y 54%, respectivamente. Las regiones sin predicción computacional se establecieron con base en los aminoácidos y las estructuras contiguas a estas regiones (
figura 1B).Para la predicción de la estructura terciaria, SWISS-MODEL proporcionó dos secuencias por similitud con las estructuras experimentalmente determinadas 1AUK e IFSU, que corresponden a la ARSA y la ARSB. La primera comprende los aminoácidos desde la posición 34 hasta la 108 y su secuencia es:
DALNVLLIIVDDLRPSLGCYGDKLVRSPNIDQLASHSLLFQNAFAQQAVCAPSRVSFLTGRRDTTRLY DFNSY, mientras que el segundo segmento con predicción comprende los aminoácidos de las posiciones 287 a 341 con la secuencia:
PIPVDFQRKIRQSYFASVSYLDTQVG RLLSALD DLAQLANSTIIAFTSDHGWAL.
Los alineamientos múltiples con el programa BLAST, empleando la Protein Database (PDB) como base de comparación para los fragmentos de otras moléculas con secuencias similares a la IDSh, permitió la utilización de 17 fragmentos con identidades entre 21 y 52%, los cuales fueron empleados para la elaboración del modelo. Utilizando el programa Spdbv 3.7, que permitió que los residuos que no se encontraban en ninguno de esos fragmentos se adicionaran manualmente uno a uno atendiendo a su orientación de acuerdo a la predicción secundaria, se completó el modelo de estructura terciaria (
figura 5A). Finalmente, el modelo presentó 33,3% en hélice, 7,2% en hoja plegada y 59,5% en enrollamiento al azar, porcen-tajes similares a los obtenidos en la predicción secundaria.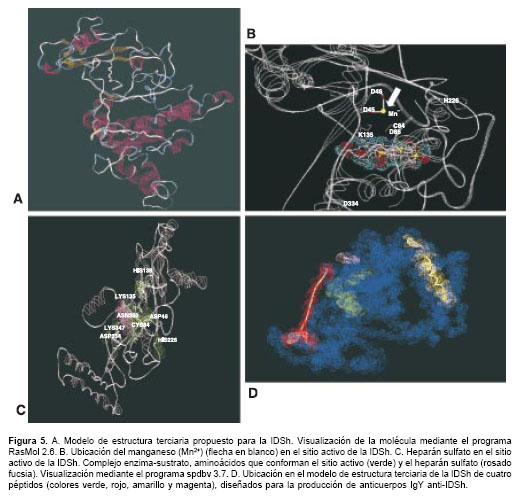
El análisis teórico y computacional de la compara-ción de estructura y función con otras sulfatasas (50-54) permitió proponer un modelo para el sitio activo de la IDSh. Se conoce que para su activación, las sulfatasas requieren una modifica-ción postraduccional que se realiza a nivel del retículo endoplásmico, consistente en la transformación de un residuo de cisteína en formilglicina (FGly) (50,53). En la IDSh, el residuo de cisteína que sufre la oxidación hasta FGly corresponde a la cisteína en posición 84. Los residuos ubicados cerca de la cisteína, que componen el centro activo de la ARSA (51) y la ARSB (54) según se ha demostrado experi-mentalmente, se comparan con los homólogos de la IDSh en el
cuadro 1. Con base en esta información se localizaron los residuos conserva-dos y su ubicación espacial se muestra en la figura 5B. Considerando la similitud de la organización del centro activo de la IDSh, la ARSA y la ARSB, se postula que así como estas dos enzimas requieren de iones magnesio (Mg2+) y calcio (Ca2+), respectivamente (50,54), para ser activa, la IDSh requiere la presencia de un catión divalente; se propone que en este caso sea el manganeso (Mn2+), dado que se demostró que cuando se presentan niveles bajos disminuyen también los niveles de actividad de la enzima ( http://www.brenda.uni-koeln). Su posible localización dentro del centro activo se muestra en la figura 5C.
Cálculo de la raíz media cuadrática (RMS)
El modelo de estructura terciaria de la IDSh se comparó con la ARSB y la ARSA y se encontraron valores de 0,78 y 0,86 Å, respectivamente.
Análisis de las mutaciones puntuales de la IDSh
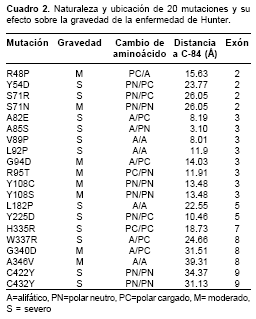
Con relación al análisis de las mutaciones puntuales, en este trabajo sólo se analizaron 20 de las 167 reportadas en la literatura. Para dicho análisis se estableció como referencia para el sitio activo la ubicación de la cisteína 84 (C-84) y una región circunscrita en un radio de 10 Å. Esta región se consideró cercana al sitio activo y los residuos fuera de la misma se catalogaron como lejanos. La clasificación de los aminoácidos se consideró según el pH fisiológico y se establecieron tres grupos de aminoácidos: hidrofóbicos (G, A, V, L, I, M, P, F, W); polares neutros (S, T, N, Q, Y, C), y polares cargados (K, R, D, H, E). El cuadro 2 resume los parámetros considerados en el análisis y los resultados obtenidos. Las mutaciones en la región cercana (distancia menor a 10 Å) a la C-84 (A82E, A85S) resultan en presentaciones graves de la enfermedad. Independientemente de la dis-tancia a la C-84, se presentan fenotipos graves cuando hay mutaciones por tirosina (C422Y, C432Y), pero si es mutada en Y por un residuo polar neutro (Y108C, Y108S), el fenotipo es moderado, mientras que si el cambio es por un aminoácido polar cargado (Y54D, Y225D), el fenotipo es grave. Cualquier aminoácido mutado por arginina genera un fenotipo grave (S71R, H335R, W337R); sin embargo, si muta R, el fenotipo es moderado (R48P, R95T). Si mutan aminoácidos alifáticos por aspartato (G94D, G340D), se producen fenotipos moderados. Las mutaciones por prolina son responsables de fenotipos graves (V89P, L92P, L182P). Los cambios de la misma naturaleza, es decir, aminoácido hidrofóbico por hidrofóbico (A346V) o polar neutro por polar neutro (S71N), resultan en fenotipos moderados.
Discusión
Del total de proteínas codificadas por los 30.000 genes humanos, un buen porcentaje de estructuras terciarias ha sido obtenido por la industria farmacéutica y gran parte de ellas no se encuentra reportada en PDB. De las 25.000 secuencias presentes en esta base de datos, sólo 5.100 se consideran de buena calidad y de ellas 1.100 corresponden a proteínas humanas. Sin embargo, la gran mayoría son simplemente fragmentos o dominios, es decir, menos del 1% del total de proteínas tienen estructura terciaria determinada experimentalmente (56-58); sin embargo, cerca del 20% de los dominios del total de proteínas se reportan como producto de la predicción computacional por homología (58). Lo anterior permite concluir que se tiene conocimiento de menos del 10% del total de proteínas, lo cual en gran medida es producto de los altos costos y dificultades técnicas para los procesos de cristalización y resonancia magnética nuclear (56). En el caso de las proteínas humanas, a lo anterior hay que sumar la dificultad para purificarlas a partir de sus fuentes naturales y la posibilidad de contraer enfermedades infectocontagiosas.
La predicción por modelación homóloga ofrece la posibilidad de hacer aproximaciones reales a la estructura tridimensional de una proteína de interés y su aplicación al diseño de experimentos y explicación de resultados. En general, el primer paso de estos procesos de predicción se realiza con algoritmos de minimización de energía que reportan un posible modelo en uno de los posibles mínimos de energía de la superficie de múltiples mínimos que es la solución completa a las ecuaciones funcionales de la energía potencial. Esto fue lo que se realizó para este trabajo y se obtuvo una geometría optimizada en un mínimo de potencial de energía con una sola estructura posible. Siguiendo el principio de estabilidad de las soluciones de un conjunto de ecuaciones diferenciales, como lo es la de energía potencial de una molécula, se da por entendido que si existen dos soluciones estables que representan geometrías para las proteínas en cada caso, éstas deben ser muy similares, de lo contrario no serían soluciones estables y menos aun cumplirían el criterio de estabilidad asintótica según Liapunov.
Está bien establecido que las proteínas homólogas han evolucionado de un ancestro común, que conservan su función a través de la escala biológica y que adoptan una estructura tridimensional similar. Estas proteínas generalmente tienen secuencias similares en cerca del 20%, mientras que las proteínas análogas llegan a tener alrededor del 10% de similitud (57,58). Dado que las secuencias similares decrecen en proteínas homólogas distantes y análogas, el reconoci-miento de homologías y analogías es importante para una aproximación a la estructura tridimensional. Los alineamientos de la IDS con la ARSA y la ARSB humanas mostraron similitudes del 24 y 26%, valores que están dentro de los rangos establecidos para considerar que la predicción tiene una alta probabilidad de ocurrir.
Dado que se están trabajando alineamientos locales y no globales, un puntaje de similitud del 50% y uno de positividad superior (> 50%) es lo suficientemente significativo para realizar los procedimientos de mutaciones virtuales y minimización energética de ajuste de la geometría óptima. Dicho puntaje no se empleó para esta-blecer la evolución ni las genealogías, para lo cual sí se requiere que sea mayor.
Los resultados obtenidos para la predicción del perfil de accesibilidad sugieren que la IDSh es una molécula con un alto porcentaje de zonas de alta accesibilidad (figura 2B), lo cual está acorde con el alto porcentaje de estructura terciaria en forma de enrollamiento al azar (figura 5A). La alta flexibilidad (figura 2C) de la molécula se explica porque es una enzima y requiere de esta propiedad para cumplir su función. El bajo perfil de hidrofobicidad (figura 2A) en gran parte de la molécula está acorde con el alto porcentaje de zonas expuestas en la estructura terciaria y su naturaleza de enzima lisosomal en constante interacción con fases polares.
Computacionalmente se determinaron valores de 5,15 para el punto isoeléctrico y de 58,479 kDa para el peso molecular. Experimentalmente se han determinado valores similares en cuanto al pI; sin embargo, en la determinación del peso molecular por otros métodos se han reportado pesos entre 43 y 49 kDa para la proteína madura en diferentes tipos celulares (28,59-61). Esta diferencia se debe posiblemente a que las formas maduras de la IDSh han perdido en el procesamiento un fragmento de 18 kDa (28), lo que muestra que la secuencia madura se extiende hasta el residuo 456.
En cuanto a las glicosilaciones, se sabe que pueden ser de dos tipos fundamentalmente, N y O-glicosilación. Por su naturaleza de enzima lisosómica, la IDSh sufre una serie de modifica-ciones postraduccionales entre las cuales el proceso de glicosilación es uno de los más importantes. El componente computacional indica que existen en el péptido precursor ocho sitios potenciales para la glicosilación de residuos de asparagina; el primero es eliminado al escindirse el péptido señal y los dos últimos se pierden al eliminarse un fragmento de 18 kDa (59-61). Cinco de estos sitios tienen entre el 65 y 80% de proba-bilidad de glicosilarse, dos con probabilidades entre 50 y 60%, y uno presenta una probabilidad inferior al 50% (figura 3C). De los 61 sitios potenciales de O-glicosilación, ninguno tiene una probabilidad superior al 50%, por lo cual no se considera como potencial sitio de glicosilación (figura 3B).
El modelo de estructura terciaria permite observar la ubicación de los potenciales sitios de glicosilación. Los mismos se encuentran expuestos, lo que facilita su ocupación por glicósidos. Igualmente, se puede apreciar una entrada al bolsillo que contiene el sitio activo (figura 5B), lo cual apoya la validez del modelo tridimensional propuesto. Adicionalmente, el modelo computacional se utilizó como una herramienta clave para el diseño de péptidos antigénicos de IDS (figura 5D), empleados para la producción en gallinas de anticuerpos IgY anti-IDSh (62). Los anticuerpos obtenidos han sido de utilidad en la realización de pruebas de inmuno-detección de la IDSh como dot blot, Elisa y Western blot, las cuales se emplean actualmente en nuestro laboratorio para el diagnóstico de la enfermedad de Hunter y la detección de la enzima requerida en investigación básica.
El modelo de sitio activo incluye la propuesta de un átomo de manganeso (Mn2+) como catión divalente, necesario para la actividad catalítica de la IDSh, puesto que cuando aparece en bajos niveles disminuyen los niveles de actividad de la enzima (http://www.brenda.unikoeln). La posible localización de este catión dentro del centro activo se muestra en la figura 5C.
El modelo de estructura terciaria se comparó mediante el RMS con las estructuras experimen-tales de la ARSA y la ARSB, encontrándose diferencias no significativas (menores de 1,0 Å), lo que sugiere que la estructura propuesta constituye un modelo con un alto porcentaje de posibilidad de presentarse en la naturaleza, dado que se considera que un modelo altamente comparativo presenta una RMS de 2,0 (56).
La información obtenida computacionalmente, además de lo reportado en este trabajo, ha sido de utilidad en el diseño experimental y la explicación de resultados obtenidos en experimentos de expresión y purificación de la proteína recombinante (63). Igualmente, servirá para orientar el diseño de experimentos futuros en procura de mejorar los niveles de expresión y purificación de la enzima, así como para enrutarla hacia los tejidos de animales de experimentación.
Con relación a la variabilidad fenotípica de la enfermedad de Hunter, es claro que ésta es reflejo de la heterogeneidad mutacional de la IDSh (11). Sin embargo, los niveles de actividad enzimática no se correlacionan con la variabilidad fenotípica, ya que las pruebas de detección no son lo suficientemente sensibles. En este tipo de ensayo actúan como limitantes la baja concentración de sustrato y las condiciones in vitro que pretenden simular las condiciones del microambiente lisosomal. Se sabe que este tipo de ensayos no son capaces de detectar actividades residuales en el caso de pacientes con enfermedad moderada. En este sentido se inició un análisis que busca encontrar una relación genotipo-fenotipo. Aunque para las 20 mutaciones estudiadas se han encontrado algunas tendencias, un estudio más completo se adelanta actualmente con las 176 mutaciones puntuales reportadas, por lo que lo presentado en este artículo es un adelanto.
Con base en el aminoácido, la carga, el tamaño y la distancia a la C-84 como punto de referencia del centro activo, se han encontrado varias relaciones; algunas de ellas se discuten a continuación. Las nuevas interacciones locales por cambios electrostáticos y de estructura generados por los cambios de residuos cargados o aromáticos generalmente resultan en variaciones drásticas en la estructura de la proteína mutada, lo que genera que pierda o disminuya su capacidad catalítica, lo cual, a su vez, explicaría los fenotipos graves. En el caso de las presentaciones clínicas moderadas, las proteínas mutadas no deben sufrir cambios muy drásticos sino, por ejemplo, cambio de un aminoácido hidrofóbico por otro de la misma naturaleza. Independientemente del tipo de residuo que mute, las mutaciones presentes en zonas cercanas al sitio activo siempre resultan en fenotipos graves. El distanciamiento de la C-84 puede aminorar los cambios locales, de tal manera que la funcionalidad de la enzima puede verse afectada de forma mínima y se presenten fenotipos moderados de la enfermedad.
En conclusión, se ha propuesto un modelo computacional para la estructura tridimensional de la IDSh que presenta 33,3 en hélice, 7,2 en hoja plegada y 59,5% en enrollamiento al azar. En este modelo se aparecen expuestos cinco sitios potenciales de N-glicosilación, zonas de alta antigenicidad, una entrada al bolsillo que contiene los aminoácidos que componen el sitio activo en el cual encaja el sustrato heparán sulfato. Cuando se comparó con las enzimas ARSA y ARSBA se obtuvieron valores de RMS de 0,78 y 0,86A°, respectivamente, lo que significa que el modelo tiene una alta probabilidad de representar la realidad biológica.
A pesar de las limitaciones de los modelos computacionales, éstos constituyen la única alternativa factible para hacer predicciones estructurales y funcionales cuando no se dispone de la proteína purificada en cantidades suficientes para determinar su estructura experimentalmente. Además, las predicciones son muy útiles en el diseño de los procesos experimentales para la purificación de las proteínas y, en algunos casos, al realizar las comparaciones entre predicciones teóricas y datos experimentales se han encontrado altos valores de correlación. Cabe anotar que las estructuras derivadas de los análisis de rayos X y resonancia magnética nuclear presentan también limitaciones que hasta ahora no se han podido resolver.
Agradecimientos
Agradecemos a la Pontificia Universidad Javeriana por el apoyo logístico (proyecto registrado en la Vicerrectoría, número 1825).
Conflicto de Intereses
Los autores declaramos que no existen conflictos de intereses que puedan influir en forma alguna en los resultados presentados y discutidos en este trabajo.
Financiación
Este trabajo es parte del proyecto financiado por el Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología "Francisco José de Caldas" (Colciencias), titulado "Desarrollo de un modelo de expresión, purificación y estudio de patrones de glicosilación de proteínas recombinan-tes humanas en levaduras metilotróficas con fines terapéuticos" (proyecto 12010ER0101103).
Correspondencia:
Luis A. Barrera, Instituto de Errores Innatos del Metabolismo, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá D.C., Colombia.
Referencias
1. Neufeld E, Muenzer J. The Mucopolysaccharidoses. En: Scriver CH, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001.p.3421-52. [ Links ]
2. Schaap T, Bach G. Incidence of mucopolysac-charidoses in Israel: is Hunter disease a "Jewish disease"? Hum Genet 1980;56:221-3. [ Links ]
3. Young ID, Harper PS. Incidence of Hunters syndrome. Hum Genet 1982;60:391-2. [ Links ]
4. Lowry R, Applegarth D, Toone J, Macdonald E, Thunem N. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Hum Genet 1990;85:389-90. [ Links ]
5. Bunge S, Steligch C, Zuther C, Beck M, Morris CP, Schwinger E et al. Iduronate 2-sulfatase gene mutations in 16 patients with mucopolysaccharidosis type II. Hum Mol Genet 1993;2:1871-5. [ Links ]
6. Hopwood JJ, Bunge S, Morris CP, Wilson PJ, Steglich C, Beck M et al. Molecular basis of muco-polysaccharidosis type II: mutations in the iduronate-2-sulphatase gene. Hum Mutat 1993;2:435-42. [ Links ]
7. Jonsson JJ, Aronovich EL, Braun SE, Whitley CB. Molecular diagnosis of mucopolysaccharidosis type II (Hunter syndrome) by automated sequencing and computer-assisted interpretation: toward mutation mapping of the iduronate-2-sulfatase gene. Am J Hum Genet 1995;56:597-607. [ Links ]
8. Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. Mucopolysaccharidosis type II (Hunter syndrome): mutation "hot spots" in the iduronate-2-sulfatase gene. Am J Hum Genet 1996;59:1202-9. [ Links ]
9. Villani GR, Balzano N, Grosso M, Salvatore F, Izzo P, Di Natalie P. Mucopolysaccharidosis type II: Identification of six novel mutations in Italian patients. Hum Mutat 1997;10:71-5. [ Links ]
10. Bunge S, Rathmann M, Steglich C, Bondeson ML, Tylki-Szymanska A, Popowska E et al. Homologous nonallelic between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur J Hum Genet 1998;6:492-500. [ Links ]
11. Froissart R, Maire I, Millat G, Cudry S, Birot AM, Bonnet V et al. Identification of iduronate sulfatase gene alterations in 70 unrelated Hunter patients. Clin Genet 1998;53:362-8. [ Links ]
12. Gort L, Chabas A, Coll MJ. Hunter disease in the Spanish population: molecular analysis in 31 families. J Inherit Metab Dis 1998;21:655-61. [ Links ]
13. Isogai K, Sukegawa K, Tomatsu S, Fukao T, Song XQ, Yamada Y et al. Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease). J Inherit Metab Dis 1998;21:60-70. [ Links ]
14. Karsten S, Voskoboeva E, Tishkanina S, Pettersson U, Krasnopolskaja X, Bondenson M. Mutational spectrum if the iduronate-2-sulfatase (IDS) gene in 36 unrelated Russian MPS II patients. Hum Genet 1998;103:732-5. [ Links ]
15. Vafiadaki E, Cooper A, Heptinstall LE, Hatton CE, Thornley M, Wraith JE. Mutation analysis in 57 unrelated patients with MPS II (Hunters disease). Arch Dis Child 1998;79:237-41. [ Links ]
16. Li P, Bellows AB, Thompson JN. Molecular basis of iduronate 2-sulfatase gene mutations in patients with mucopolysaccharidosis type II (Hunter syndrome). J Med Genet 1999;36:21-7. [ Links ]
17. Vallance HO, Bernard L, Rashed M, Chiu D, Le G, Toone J et al. Identification of 6 novel mutation in the iduronate sulfatase gene. Mutation in brief 233. Online. Hum Mutat 1999;13:338. [ Links ]
18. Cudry S, Tigaud I, Froissart R, Bonnet V, Maire I, Bozon D. MPS II in females: Molecular basis of two different cases. J Med Genet 2000;37:E29. [ Links ]
19. Bonuccelli G, Di Natale P, Corsolini F, Villani G, Regis S, Filocamo M. The effect of four mutations on the expression of iduronate-2-sulfatase in mucopolysaccharidosis type II. Biochem Biophys Acta 2001;1537:233-8. [ Links ]
20. Filocamo M, Bonuccelli G, Corsolini F, Mazzotti R, Cusano R, Gatti R. Molecular analysis of 40 Italian patients with mucopolysaccharidosis type II: New mutation in the iduronate-2-sulfatase (IDS) gene. Hum Mutat 2001;21:164-5. [ Links ]
21. Moreira da Silva I, Froissart R, Marques dos Santos H, Caseiro C, Maire I, Bozon D. Molecular basis of mucopolysaccharidosis type II in Portugal: Identification of four novel mutations. Clin Genet 2001;60:316-8. [ Links ]
22. Ricci V, Regis S, Di Duca M, Filocamo M. An Alu-mediated rearrangement as cause of exon skipping in Hunter disease. Hum Genet 2003;112:419-25. [ Links ]
23. Kim CH, Hwang HZ, Song SM, Paik KH, Kwon EK, Moon KB et al. Mutational spectrum of the iduronate 2 sulfatase gene in 25 unrelated Korean Hunter syndrome patients: Identification of 13 novel mutations. Hum Mutat 2003;21:449-50. [ Links ]
24. Tomatsu S, Orii KO, Bi Y, Gutierrez MA, Nishioka T, Yamaguchi S et al. General implications for CpG hot spot mutations: methylation patterns of the human iduronate-2-sulfatase gene locus. Hum Mutat 2004;23:590-8. [ Links ]
25. Parkinson EJ, Muller V, Hopwood JJ, Brooks DA. Iduronate 2-sulphatase protein detection in plasma from mucopolysaccharidosis type II patients. Mol Genet Metab 2004;81:58-64. [ Links ]
26. Chang JH, Lin SP, Lin SC, Tseng KL, Li CL, Chuang CK et al. Expression studies of mutations underlying Taiwanese Hunter syndrome (mucopolysaccharidosis type II). Hum Genet 2005;116:160-6. [ Links ]
27. Tomatsu S, Sukegawa K, Trandafirescu GG, Gutierrez MA, Nishioka T, Yamaguchi S et al. Differences in methylation patterns in the methylation boundary region of IDS gene in Hunter syndrome patients: implications for CpG hot spot mutations. Eur J Hum Genet 2006;14:838-45. [ Links ]
28. Wilson PJ, Morris CP, Anson DS, Occhiodoro T, Bielick J, Clements PR et al. Hunter syndrome: Isolation of an Iduronate 2-sulfatase cDNA clone and analysis of patients DNA. Proc Natl Acad Sci USA 1990;87:8531-5. [ Links ]
29. Wilson PJ, Meaney CA, Hopwood JJ, Morris CP. Sequence of the human iduronate 2-sulfatase (IDS) gene. Genomics 1993;17:773-5. [ Links ]
30. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins M, Appel R et al. Protein identification and analysis tools on the expasy server. En: Walker JM, editor. The proteomics protocols handbook. New York, USA: Human Press; 2005. p.571-660. [ Links ]
31. Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sánchez JC et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 1993;14:1023-31. [ Links ]
32. Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999;294:1351-62. [ Links ]
33. Julenius K, Molgaard G, Gupta R, Brunak S. Prediction, conservation analysis and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 2005;15:153-64. [ Links ]
34. Blon N, Ponten T, Gupta R, Brunak S. Prediction of post-translational glycosilation and phosphorylation of proteins from aminoacid sequence. Proteomics 2004;4:1633-49. [ Links ]
35. Frith MC, Spoge JL, Hansen U, Weng Z. Statistical significance of clusters of motifs represented by position specific scoring matrices in nucleotide sequences. Nucleic Acids Res 2002;30:3214-24. [ Links ]
36. Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA 1981;78:3824-8. [ Links ]
37. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. Gapped Blast and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997;25:3389-402. [ Links ]
38. Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J Mol Biol 1990;214:171-82. [ Links ]
39. Garnier J, Gibrat J, Robson B, Doolittle R. Gor secondary structure prediction method version iv. Methods Enzymol 1996;266:540-53. [ Links ]
40. Guex N, Peitisch M. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997;18:2714-23. [ Links ]
41. Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. JPred: A consensus secondary structure prediction server. Bioinformatics 1998;14:892-3. [ Links ]
42. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999;292:195-202. [ Links ]
43. Shindyalov IN, Bourne PE. A database and tools for 3-D protein structure comparison and alignment using the Combinatorial Extension (CE) algorithm. Nucleic Acids Res 2001:29:228-9. [ Links ]
44. Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 2002;47:228-35. [ Links ]
45. Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J Mol Biol 1994;245:275-96. [ Links ]
46. Combet C, Jambon M, Deléage G, Geourjon C. Geno3d: Automatic comparative molecular modeling of protein. Bioinformatics 2002;18:213-4. [ Links ]
47. Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3d: Prediction of proteins 3D structures. Bioinformatics 2002;18:1250-6. [ Links ]
48. Discover©. Accelrys software Inc. San Diego, USA: Accelrys software Inc; 2001-2006. [ Links ]
49. Sayle RA, Milner-White EJ. RASMOL: Biomolecular graphics for all. Trends Biochem Sci 1995;20:374. [ Links ]
50. Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency cell. Cell 1995;82:271-8. [ Links ]
51. Bond CS, Clements PR, Ashby SJ, Collyer CA, Harrop SJ, Hopwood JJ et al. Structure of human lysosomal sulfatase. Structure 1997;5:277-89. [ Links ]
52. Parenti G, Meroni G, Ballabio A. The sulfatase gene family. Curr Opin Genet Dev 1997;7:386-91. [ Links ]
53. Dierks T, Lecca M, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J 1999;18:2084-91. [ Links ]
54. Waldow A, Schmidt B, Dierks T, Von Bulow R, von Figura K. Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem 1999;274:12284-8. [ Links ]
55. Web Lab Viewer Pro. Web Lab, Molecular Simulation. Getting Starting. Ca, USA: Web Lab Viewer Pro; 2000. [ Links ]
56. Baker D, Sali A. Protein structure prediction and structural genomics. Science 2001;294:93-6. [ Links ]
57. Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J 1986;5:823-6. [ Links ]
58. Stemberg M. Protein structure prediction. Oxford: IRL Press; 1996. p.298. [ Links ]
59. Froissart R, Millat G, Mathiu M, Bozon D, Maire I. Processing of iduronate 2-sulfatase in human fibroblasts. Biochem J 1995;309:425-30. [ Links ]
60. Millat G, Froissart R, Maire I, Bozon D. Characterization of iduronate sulfate sulfatase mutants affecting N-glycosylations sites and cysteine-84 residue. Biochem J 1997;326:243-7. [ Links ]
61. Millat G, Froissart R, Maire I, Bozon D. IDS transfer from overexpressing cell to IDS-deficient. Exp Cell Res 1997;230:362-7. [ Links ]
62. Sosa A, Barrera L. Production of specific chicken egg yolk antibodies to Iduronate 2-sulphate sulphatase (IDS) and its applicability in an Enzime-Linked Immunosorbent Assay (Elisa). J Inherit Metab Dis 2005;28(Suppl.1);185. [ Links ]
63. Sáenz H. Expresión, purificación parcial y estudios computacionales de la IDShr producida en Pichia pastoris. (Tesis Doctoral). Bogotá D.C.: Facultad de Ciencias, Pontificia Universidad Javeriana; 2005. p.149. [ Links ]

