










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.27 n.1 Bogotá jan./mar. 2007
Generación de modelos matemáticos correlacionales entre afinidad y descriptores topológicos moleculares de antagonistas de glicina en receptores de glutamato
Guillermo Narváez 1, Leonardo Lareo 2, Javier Rincón 3
1 Facultad de Medicina, Maestría en Bioquímica, Universidad Nacional de Colombia, Bogotá, D.C., Colombia.
2
Facultad de Ciencias, Grupo de Bioquímica Computacional y Estructural y Bioinformática, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia.3
Facultad de Ciencias, Departamento de Farmacia, Universidad Nacional de Colombia, Bogotá, D.C., Colombia.Recibido:31/08/06; aceptado: 05/02/07
Introducción. Los modelos matemáticos que correlacionan la estructura química con la actividad biológica son útiles en el diseño de nuevos fármacos, y pueden emplearse para predecir el comportamiento biológico de moléculas nuevas químicamente relacionadas.
Objetivos. Generar un modelo matemático que correlacione la afinidad y la topología molecular de antagonistas de glicina en el receptor de glutamato subclase NMDA y proponer nuevas moléculas con actividad antagonista.
Materiales y métodos. Mediante los índices de conectividad molecular se codificó la estructura electrónica y el patrón de enlace atómico de 45 antagonistas de glicina. La correlación entre los índices de conectividad y la afinidad de los antagonistas se determinó mediante análisis de regresión.
Resultados. El índice de conectividad que mejor describió el comportamiento de la afinidad fue 4Xvpc, indicando la importancia de los heteroátomos, de la adyacencia de los sustituyentes sobre el anillo aromático y del delta de valencia de los sustituyentes. Las ecuaciones generadas pueden utilizarse para la predicción de la afinidad de nuevos antagonistas, y el modelo sugirió los requerimientos estructurales para diseñar compuestos con elevada afinidad. Con base en ello se proponen 12 moléculas nuevas, de las cuales tres parecen ser bastante promisorias, habiéndose calculado teóricamente su afinidad mediante las ecuaciones desarrollas. Como control de la metodología matemática desarrollada se utilizó el análisis de interacción energética .
Conclusión. Es posible analizar matemáticamente la estructura química de antagonistas de glicina mediante índices de conectividad, generando ecuaciones que modelan el comportamiento de unión al receptor y aportan información útil en el diseño de nuevos antagonistas.
Palabras clave: glicina/antagonistas e inhibidores, glutamatos, N-metilaspartato/farmacología.
Mathematical models to correlate molecular topology with substrate affinity of the glycine antagonist in glutamate receptors
Introduction. Mathematical models that correlate chemical structure with biological activity have been useful in the design of new drugs and can be used to predict biological behavior of new, chemically related molecules.
Objectives. A mathematical model was generated to correlate the substrate affinities with variations in the molecular topology of glycine antagonists in NMDA sub-class glutamate receptor and, subsequently, to propose new molecules with antagonist activity.
Materials and methods. By use of molecular connectivity indexes, the electronic structure and atomic bonding patterns of 45 glycine antagonists were coded. Correlation between connectivity indexes and antagonist affinity was determined by regression analysis.
Results. The connectivity index that best described affinity behavior was 4Xvpc, which indicates the relative importance of heteroatoms, the vicinity of aromatic ring substitutes, and valency gradient. The equations generated predicted new antagonist affinities, and the model was able to suggest structural requirements for designating compounds with increased affinity. Twelve new molecules were proposed, from which three appeared promising-based of the affinities previously calculated by means of the new equations. Energetic interaction analysis was developed as a control for the mathematical methodology.
Conclusion. Glycine antagonists structure were analyzed mathematically by means of connectivity indexes. The equations modeled receptor behavior and contributed useful information for new antagonist design.
Key words: glycine/antagonists & inhibitors, glutamate, N-methylaspartate/pharmacology.
En los últimos 15 años se han generado dos grandes aproximaciones para el descubrimiento de nuevas moléculas farmacológicamente activas: una basada en las características estructu-rales de la macromolécula biológica cuya actividad se quiere modificar y la otra basada en la estructura química de sustancias que se sabe interaccionan y alteran la funcionalidad de la macromolécula en cuestión (1,2). En esta última estrategia entra a jugar un papel decisivo la generación de modelos matemáticos que permitan relacionar cuantitativamente la estructura química de una molécula con su actividad biológica, evidenciando y perfilando de una manera racional el comportamiento biológico del sistema en estudio (3). En muchas ocasiones estos modelos permiten predecir desde un punto de vista teórico la alteración en la actividad biológica ocasionada por una determinada modificación estructural hecha sobre la molécula patrón que contiene el grupo farmacofórico de interés.
La topología molecular ha sido ampliamente utilizada como descriptor para caracterizar una estructura química y poderla incluir en modelos matemáticos cuantitativos (4). Entre los descriptores topológicos más versátiles se tienen los índices de conectividad molecular (5). Estos índices, introducidos inicialmente por Randic en 1975 (6) y posteriormente extensamente modi-ficados por Kier y Hall (7-12), codifican la información acerca del tamaño, la ramificación, la ciclización, la insaturación y el contenido de heteroátomos en las moléculas. Los algoritmos empleados para la obtención de estos índices se fundamentan básicamente en el conteo de electrones que participan en enlaces sigma (s), pi (p) y electrones no compartidos (n) de cada átomo en la molécula; y para aquellos elementos más allá del segundo período de la tabla periódica se involucra también la información del contenido de electrones internos. Adicionalmente, estos índices codifican el patrón de enlace entre los átomos. Ello implica que los índices de conecti-vidad codifican no solamente qué átomo está unido con otro, sino también la información electrónica de cada uno de ellos, información crítica en el momento de codificar numéricamente el comportamiento químico y biológico de una molécula. El cálculo de los mencionados índices es relativamente complejo y se basa en la fragmentación de la molécula en segmentos continuos desde un enlace atómico hasta seis enlaces, denominándose en cada caso, índice de conectividad de orden 1 (representado como 1X), orden 2 (2X), etc. Adicionalmente, se fragmenta la molécula en agrupaciones de átomos en forma de racimo (3Xc), en forma de ruta/racimo (4Xpc) o ciclos con tres enlaces o más. En la
figura 1 se ejemplifican algunos fragmentos estructurales de la conectividad. Para el cálculo de los índices de conectividad denominados como "simples" se tienen en cuenta solamente los electrones que participan en enlaces sigma (s) de cada uno de los átomos que conforman el fragmento estructural molecular que está siendo analizado, mientras que para los índices de conectividad denominados como "de valencia" se tienen en cuenta el conteo de electrones pi (p) , el conteo de electrones no compartidos (n) y el conteo de electrones de capas internas. La ecuación 1 (1X = å Ö(di dj)-1 describe el algoritmo utilizado para el cálculo del índice de conectividad simple de orden 1 (1X), donde di, dj corresponde al conteo de electrones sigma de los respectivos átomos que conforman cada fragmento estructural de la molécula que está siendo analizado. El símbolo 1X indica que la molécula se ha analizado en fragmentos de un enlace.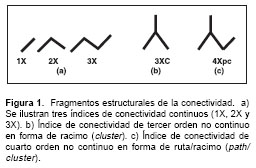
En varias investigaciones recientes se ha establecido que los índices de conectividad molecular son una herramienta bastante importante en el diseño y selección de moléculas farmaco-lógicamente activas tales como analgésicos, broncodilatadores, antihistamínicos, antivirales y antibióticos, entre otros (13-17).
Los receptores ionotrópicos de glutamato son canales iónicos activados por ligandos expresados principalmente a nivel del sistema nervioso central, los cuales median la mayoría de los procesos de neurotransmisión excitatoria y plasticidad sináptica (18,19). De los tres subtipos de receptores ionotrópicos de glutamato conocidos, el que más interés biomédico ha tenido es el receptor de glutamato activado por N-metil-D-aspartato (NMDA), ya que la estimulación excesiva de estos receptores bajo condiciones patológicas conduce a la denominada "excitotoxicidad neuronal", y tal estimulación se ha relacionado con la pérdida de neuronas que acompaña la isquemia cerebral y ciertas enfermedades neuro-degenerativas (20). Debido a que la isquemia cerebral, ya sea por trombosis o apoplejía, es una de las principales causas de muerte a nivel mundial y la primera causa de incapacidad a largo término, este evento patológico ha sido uno de los principales blancos terapéuticos hacia los cuales se ha enfocado la investigación con el fin de poder frenar, empleando para ello antagonistas del receptor NMDA, la cascada de eventos excitotóxicos generada por la excesiva estimulación glutamatérgica.
El receptor de glutamato subclase NMDA emplea como coagonista a la glicina, ligando tan importante como el ácido glutámico para la apertura del canal (21). La investigación preclínica y clínica ha evidenciado que los agonistas parciales o los antagonistas del sitio de unión de glicina poseen mejores perfiles de efectos colaterales que los bloqueadores directos del canal, aun cuando desde el punto de vista químico ha sido un verdadero reto desarrollar antagonistas potentes que compitan con este pequeño aminoácido dentro de los límites normales de dosificación (22).
En el presente trabajo se generaron ecuaciones matemáticas que relacionan la estructura química de antagonistas de glicina con sus propiedades de afinidad por el correspondiente dominio de unión en el receptor ionotrópico de glutamato subclase NMDA. Adicionalmente, la información aportada por el modelo se utilizó para proponer nuevos antagonistas de glicina, cuya afinidad fue predicha teóricamente con las ecuaciones desarrolladas.
Materiales y métodos
Selección de los antagonistas incluidos en la generación del modelo matemático
Para la construcción del modelo matemático se contó con una base de datos generada a partir de la revisión bibliográfica entre los años 1993 y 2004 (23), en la cual se tabularon aproximadamente 300 antagonistas del sitio de unión de glicina agrupados en 22 familias químicas, con sus correspondientes datos de afinidad expresados como CI50. El término CI50 (concentración inhibitoria 50) corresponde a la concentración molar del antagonista que inhibe en un 50% la unión de un ligando de referencia. Para el presente caso, cuanto menor sea el valor numérico de la CI50 para un antagonista, mayor será su afinidad por el dominio de unión a glicina. Como criterios de selección para los antagonistas que serían incluidos en el trabajo se tuvo en cuenta la homogeneidad experimental en la obtención de la CI50; el reporte de la precisión en la determinación experimental de la CI50, y variaciones estructurales sistemáticas sobre la región de interés del grupo farmacofórico.
Debido a que el modelo matemático tendría características semicuantitativas se definió el siguiente criterio de clasificación de los compuestos a ser empleados para construir el modelo:
Antagonistas positivos. Son aquellos con CI50 menor a 250 nM. Se considera que estos compuestos tienen una alta capacidad de unión al dominio de glicina del receptor NMDA y, por consiguiente, pueden competir y desplazar al ligando endógeno, antagonizando su efecto.
Antagonistas negativos. Son aquellos que tienen una CI50 mayor a 250 nM. Estos compuestos no presentan la suficiente capacidad de unión para ser utilizados farmacológicamente como anta-gonistas de glicina.
El umbral de 250 nM se eligió porque experimental-mente se ha encontrado que la afinidad de la glicina en receptores NMDA heteroméricos se encuentra entre 200 y 300 nM (24,25). Cualquier compuesto que deba competir con este coagonista debe tener la suficiente afinidad para contrarrestar las con-centraciones de glicina presentes en la hendidura sináptica, de tal forma que el objetivo es buscar antagonistas que posean una afinidad expresada en términos de CI50, inferior a 250 nM (26).
Generación de la conformación tridimensional energéticamente más favorable
Para generar la conformación molecular tridimensional energéticamente más favorable de cada antagonista seleccionado se utilizó el programa Molecular Modeling Pro CHEMSW® 2003, el cual emplea herramientas de mecánica molecular para llevar a cabo esta tarea.
Codificación numérica mediante índices de conectividad molecular de la estructura química de los antagonistas seleccionados para estudio
Se utilizaron los algoritmos ampliamente descritos en la literatura (7-12). Los índices de conectividad utilizados para el presente estudio fueron los siguientes.
Indices de conectividad simples: 0X, 1X, 2X, 3X, 4X, 5X, 6X, 3Xc, 4Xpc.
Indices de conectividad de valencia:0Xv, 1Xv, 2Xv, 3Xv, 4Xv, 5Xv, 6Xv, 3Xvc, 4Xvpc.
Diseño del modelo matemático y estadístico
Las CI50 y los diferentes índices de conectividad que codifican la estructura química de cada antagonista se utilizaron para generar el modelo matemático-estadístico mediante análisis de regresión simple y múltiple, expresando la afinidad como CI50, Ln CI50, 1/CI50, y evaluando la relación lineal, logarítmica, recíproca y multiplicativa a un nivel de probabilidad del 95%. Se utilizó el programa Excel de Microsoft Office 2003.
Análisis de energías de interacción o "docking"entre los antagonistas de glicina seleccionados para estudio y su correspondiente sitio de unión
Con el fin de tener una metodología alterna que permitiera relacionar la afinidad con otro parámetro diferente a los índices de conectividad, se investigó si existía alguna correlación entre la CI50 y la energía de interacción de los antagonistas con su correspondiente dominio de unión. Para ello se utilizaron los segmentos S1S2 del dominio de unión de glicina del receptor de glutamato NMDA, cuya estructura terciaria, determinada mediante cristalografía de rayos X, fue recientemente publicada (27,28). La estructura tridimensional empleada tenía como ligando acoplado al ácido dicloroquinurénico un antagonista de glicina prototipo. En la figura 2 se observan los residuos aminoacídicos involucrados en la interacción con el ácido dicloroquinurénico. Para determinar la energía de interacción de los antagonistas seleccionados con el dominio de unión de glicina se utilizó el programa especializado ArgusLab 4.0.1 (29). Las dimensiones de la celda de unión para estos análisis fueron de 21,37Å x 18,67Å x 14,44Å; sin embargo, se hicieron ensayos de acople con celdas de hasta 15 Å x 14 Å x 14 Å. Se emplearon celdillas con una resolución de 0,4 Å, y se activó la opción de alta resolución de acople con un número máximo de interacciones de 450 poses.
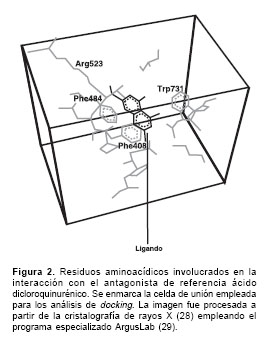
Los análisis de energía de interacción o docking se utilizaron para validar los resultados obtenidos con el modelo matemático-estadístico desarrollado con base en índices de conectividad cuando éste se aplicara a moléculas nuevas cuyas propiedades de unión al dominio de glicina se deseara conocer. De esta forma el análisis de docking se convirtió en un control del modelo propuesto mediante los índices de conectividad.
Aplicación del modelo matemático-estadístico basado en índices de conectividad para el diseño de nuevos antagonistas y predicción de su afinidad
Con base en el comportamiento de los índices de conectividad que más influencia mostraron sobre la afinidad, se diseñaron nuevos antagonistas del dominio de unión de glicina, prediciendo sus propiedades de unión a través de las ecuaciones matemáticas generadas. Los datos obtenidos con el modelo se confirmaron mediante el análisis de interacción bioquímica o docking.
Todos los programas computacionales mencionados fueron ejecutados en un equipo DELL™ XPS M1710 con procesador Intel® Core™ 2 Duo T7200 (2 GHz/ 667 MHz FSB) y memoria de 1GB SDRAM DDR2 a 667MHZ, 2 DIMM (1G2D).
Resultados
Selección de los antagonistas incluidos en la generación de los modelos matemáticos
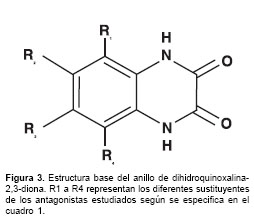
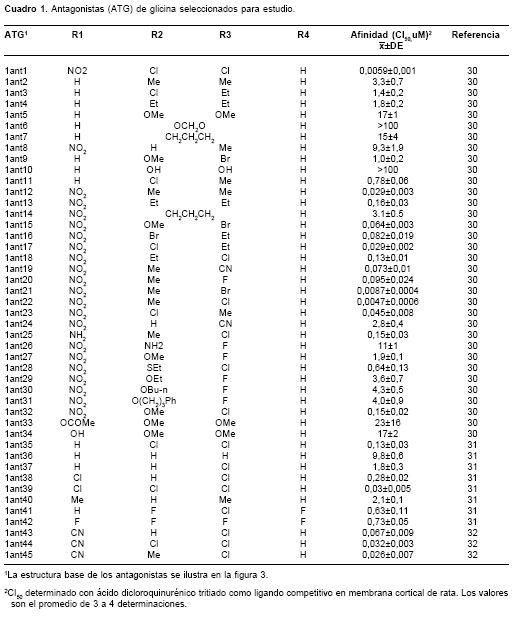
Con base en los criterios mencionados se seleccionaron para el estudio 45 compuestos (30-32) que presentan modificaciones estructurales sistemáticas sobre el anillo bencénico de la dihidroquinoxalina-2,3-diona ( figura 3 , cuadro 1 ), región que de acuerdo con el grupo farmacofórico propuesto hasta la fecha (24,33), influye significativamente sobre la afinidad de este tipo de antagonistas.
Generación de la conformación tridimensional energéticamente más favorable
La generación de la conformación estructural tridimensional energéticamente más favorable se llevó a cabo sin mayores complicaciones. Como se puede observar en la
figura 3 y en el cuadro 1 , los antagonistas seleccionados para el estudio tienen mucha restricción de rotación en torno a los enlaces de los átomos que conforman los anillos, lo cual disminuye significativamente la posibilidad de que tengan una amplia gama de conformaciones energéticamente favorables. Los resultados de esta etapa se emplearon posterior-mente durante los análisis de interacción energética o docking.Codificación numérica mediante índices de conectividad molecular de la estructura de los antagonistas seleccionados para estudio
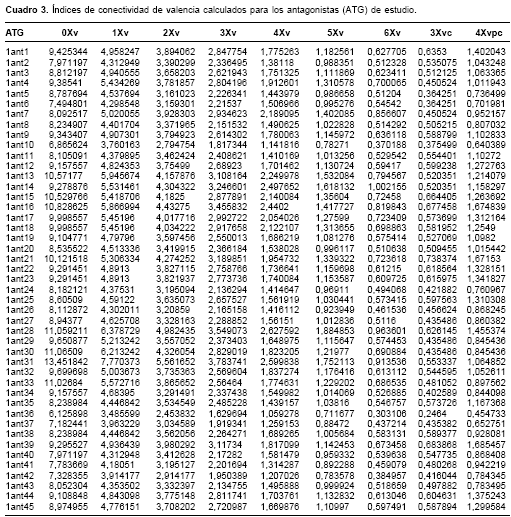
Los índices de conectividad simple y de valencia calculados para cada uno de los 45 antagonistas de estudio se presentan en los
cuadro 2 y cuadro 3 . Con estos resultados se procedió a efectuar el modelaje matemático y estadístico.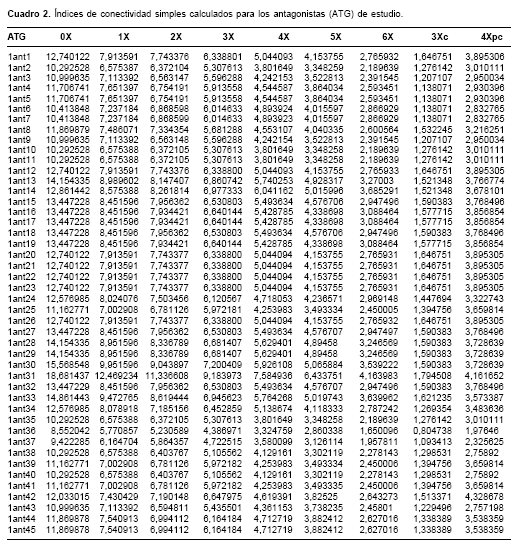
Modelaje matemático y estadístico
De los 18 índices de conectividad analizados, los que mejor se correlacionaron con la variable dependiente CI50 (afinidad) fueron los índices 3Xvc y 4Xvpc. Los coeficientes de determinación (r2) y de correlación (r) obtenidos con el análisis de regresión simple entre las variables CI50 y 3Xvc fueron de 0,606 y 0,778, respectivamente. Es decir que el índice 3Xvc explica en un 60,6% la variación en la afinidad. En forma similar, el análisis de regresión entre las variables CI50 y 4Xvpc condujo a un coeficiente de determinación (r2) de 0,557 y a un coeficiente de correlación (r) de 0,759. Para este caso, el índice 4Xvpc explica en un 55,7% la variación en la afinidad. Previa-mente se hicieron los mismos estudios utilizando los valores de CI50 como tal, o transformaciones como 1/CI50 o Ln CI50. Los mejores resultados se obtuvieron con el logaritmo natural. Se utilizó una significación del 95% (p<0,05).
Aunque estos análisis iniciales mostraron que el índice de conectividad 3Xvc explicaba en un porcentaje ligeramente mayor la variación en la afinidad, se decidió trabajar con el índice 4Xvpc debido a que según la literatura (9) este índice ha demostrado ser más rico en información, sobre todo en cuanto anillos aromáticos sustituidos se refiere, pues codifica información acerca del número de sustituyentes sobre el anillo, los patrones de sustitución y su longitud (hasta tres enlaces), así como el tipo de sustituyente heteroatómico.
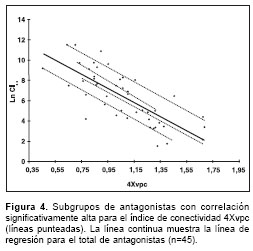
Analizando el diagrama de dispersión para el índice de conectividad 4Xvpc se observó que había un comportamiento diferencial entre los antagonistas evaluados, generándose subgrupos de antagonistas que correlacionaban muy bien entre sí. Es así como en la figura 4 se observaron cuatro subgrupos de antagonistas (líneas punteadas) cuya correlación entre afinidad, expresada como Ln CI50, y su correspondiente índice 4Xvpc era significativamente alta. Advirtién-dose este comportamiento, se decidió dividir el total de los antagonistas analizados en dos grupos: aquellos que se ubicaban por debajo de la línea de regresión inicial (línea continua en la figura 4 ) (grupo A); y aquellos que se ubicaban por encima de esta línea (grupo B). Se encontró que en el grupo A se ubicaron preferencialmente aquellos compuestos que tenían valores de afinidad menores de 250 nM (15 de 23 compuestos, correspondiente al 65,2%), es decir, aquellos que sí presentaban una unión significativa al receptor; mientras que en el grupo B, el 81,8% (18 de 22) de los compuestos tenía una afinidad por encima de 250 nM, es decir, los considerados sin afinidad por el sitio de unión de glicina. Posteriormente, mediante regresión lineal simple se analizaron los dos grupos de antagonistas generados (A y B) para la variable 4Xvpc, obteniéndose los resul-tados ilustrados en la figura 5 . Para el grupo A de antagonistas ( figura 5 , panel izquierdo), el coeficiente de determinación (r2) y de correlación (r) para la regresión efectuada fue de 0,8589 y de 0,9267, respectivamente, cumpliéndose todos los supuestos de normalidad estadística. La ecuación de la curva de regresión para este modelo fue la siguiente (ecuación 1):
Ln (CI50) = 12,7839 (+/-0,71732) – 7,1602 (+/-0,6490) (4Xvpc).
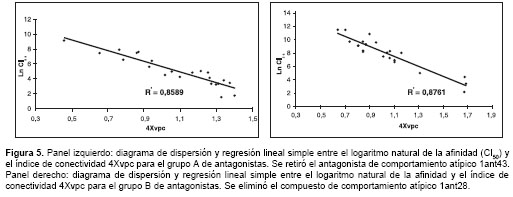
Es importante mencionar que la anterior ecuación se obtuvo a partir de la regresión lineal para el grupo A de antagonistas, en la cual se excluyó el compuesto 1ant43. Este compuesto correspondió a un punto atípico de regresión, el cual se alejaba significativamente de la línea de regresión en el análisis estadístico. Al efectuar la regresión con este compuesto se obtuvo un coeficiente de determinación (r2) de 0,7687, un coeficiente de correlación (r) de 0,8768, y un error típico de 0,981, mientras que al retirarlo se obtuvo un r2 de 0,8589, un coeficiente de correlación (r) de 0,9267 y un error típico de 0,7822. Desde el punto de vista químico no se evidenció una diferencia estructural significativa con relación a los otros compuestos del grupo que explicara este comportamiento atípico. Al aplicar la ecuación 1 a cada uno de los 22 compuestos del grupo A, se encontró que ésta predice correctamente la afinidad semicuantitativa para la totalidad de los integrantes del grupo, teniendo como punto de corte una CI50 de 250 nM.
Para el grupo B de compuestos (
figura 5 , panel derecho), el coeficiente de determinación (r2) y de correlación (r) para la regresión fue de 0,8761 y de 0,9359, respectivamente, cumpliéndose los supuestos de normalidad estadística. Para este análisis estadístico se excluyó el compuesto 1ant28, el cual correspondía a un punto atípico de residuales. Este compuesto también podría considerarse como un "atípico químico", pues es el único que dentro de su estructura involucra un tioeter como sustituyente del anillo bencénico. La ecuación de la curva de regresión para este modelo fue la siguiente (ecuación 2):Ln (CI50) = 15,860 (+/-0,715) – 7,597 (+/-0,655) (4Xvpc)
Al aplicar la ecuación 2 al grupo B de compuestos, se encontró que esta ecuación predice correctamente la afinidad semicuantitativa para 21 de los 22 compuestos con los cuales se construyó el modelo. Es decir, el modelo acertó en la predicción en un 95,24%.
El comportamiento de los grupos A y B sugiere que deben manejarse como grupos con comportamiento diferencial y no como un solo grupo de compuestos. Para confirmar esto se decidió realizar un análisis de regresión múltiple incluyendo como variables regresoras el índice de conectividad 4Xvpc y una variable dummy denominada "Grupo", la cual tomó el valor de 1 si el compuesto pertenecía al grupo A o el valor de 0 si el compuesto pertenecía al grupo B. Esta nueva regresión produjo como resultado para los 45 antagonistas un coeficiente de determinación (r2) de 0,839 y un coeficiente de correlación múltiple (r) de 0,916. Lo más importante de este análisis fue que la variable dummy "Grupo" introducida fue estadísticamente significativa (p <0,05). Estos resultados confirmaron el hecho de que los grupos A y B presentaban comportamientos diferentes.
Teniendo en cuenta los anteriores resultados, sumado a la observación de que en el grupo A se ubicaron preferencialmente aquellos antagonistas con una afinidad inferior a 250 nM, en tanto que en el grupo B se agruparon principalmente aquellos con afinidades superiores a 250 nM, se sugirió que el motivo de este comportamiento diferencial se originaba en el hecho de que el compuesto presentara o no afinidad por el receptor con un punto de corte de 250 nM, tal como se especificó desde el comienzo. Con esta información se decidió modificar el modelo de regresión anterior para incluir una variable dummy diferente denominada "PosNeg" que tomaría el valor de 1 para los compuestos con afinidad menor de 250 nM y el valor de 0 para los compuestos con afinidad mayor de 250 nM. Con este modelo (ecuación 3) nuevamente se observa no solamente un coeficiente de correlación múltiple (r) elevado (0,874), sino que la variable dummy "PosNeg" es significativa (p<0,05). Además, resulta interesante que la predicción semicuantitativa es exacta para el total de los antagonistas evaluados (n=45).
Ln (CI50) = 10,763 (+/- 0,919) – 3,6572 (+/- 0,560) (PosNeg) – 2,545 (+/- 0,978) (4Xvpc) (ecuación 3)
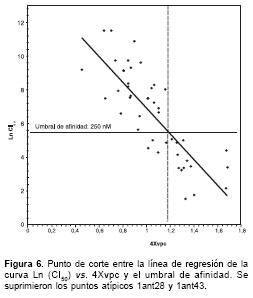
Sin embargo, aun cuando la predicción de la afinidad mediante la ecuación 3 es correcta para todos los antagonistas, se tenía el problema de que la variable dummy "PosNeg" dependía del valor de la afinidad, valor éste que en la práctica no se tiene para una molécula nueva. Por consiguiente, el interés se centró en construir una función que se aproximara de la mejor manera posible a la diferenciación hecha con las anteriores variables dummys, pero que ya no dependiera de la afinidad. Esta nueva variable se construyó teniendo como referencia la línea de regresión entre las variables Ln(CI50) y el índice de conectividad 4Xvpc, y el umbral de afinidad de 250 nM. Como se observa en la
figura 6 , a partir de un valor de 1,18 de la variable 4Xvpc, todos los compuestos son positivos (afinidad menor a 250 nM), y por debajo de 1,18, todos los compuestos son negativos (afinidad mayor a 250 nM), quedando solamente cuatro compuestos mal caracterizados. Con estos nuevos datos se decidió construir un nuevo modelo de regresión múltiple empleando para ello una nueva variable dummy denominada "Factor de corrección de grupo, FCG", la cual toma un valor de 1 si la variable 4Xvpc es mayor a 1,18 o de 0 si esta variable es menor o igual a 1,18. La regresión para las 43 observa-ciones arrojó un coeficiente de correlación múltiple (r) de 0,8109, un valor F de 38,43, un error típico de 1,66, siendo las dos variables regresoras (4Xvpc, FCG) significativas (p<0,05). Así mismo, la regresión cumplió con los supuestos de normalidad estadística. La ecuación de la curva de regresión para este modelo fue la siguiente:Ln (CI50) = 11,9853 (+/-1,4130) – 4,3776 (+/-1,5223)(4Xvpc) – 2,3691(+/-0,9089)(FCG) (ecuación 4).
Al aplicar la ecuación 4 a los 43 antagonistas con los cuales fue construido el modelo, se encontró que el pronóstico semicuantitativo de la afinidad fue correcto para 39 compuestos; es decir, el modelo predice correctamente la afinidad del 90,7% de los antagonistas evaluados.
Análisis de las energías de interacción o "docking" entre los antagonistas de glicina seleccionados para estudio y su correspondiente sitio de unión
El análisis de las energías de interacción entre antagonistas de glicina y su correspondiente sitio de unión o docking se llevó a cabo para los 45 compuestos seleccionados utilizando las estructuras moleculares con la conformación tridimensional energéticamente más favorable generadas según lo descrito. Para evaluar el desempeño del programa de docking en el equipo donde fue instalado se hicieron controles de acople con proteínas y ligandos no relacionados (transportador de ácido retinoico y receptor de estradiol). Los RMSD promedio obtenidos fueron de 0,42 Å y 0,55 Å, respectivamente, con una formación de puentes de hidrógeno idéntica a la del ligando de referencia contenido en la correspondiente cristalografía de rayos X (datos no mostrados). Una vez obtenidas las energías de interacción para los antagonistas de glicina en estudio, se construyó el diagrama de dispersión ilustrado en la
figura 7 . En esta figura se observa la generación de dos grupos de puntos separados por aproximadamente 400 calorías/mol, lo cual sugiere claramente que para obtener una afinidad inferior a 250 nM se requiere de una energía de interacción menor a –8,0 Kcal/mol. Desde el punto de vista estadístico, estos resultados se analizaron mediante la prueba de Fisher, con la cual se obtuvo un valor de p<0,05, indicando así que la energía de acople o de docking está estadísticamente asociada con la afinidad. Por otro lado, el coeficiente de correlación de Spearman obtenido fue de 0,999, con un valor de p<0,05 para los 32 compuestos analizados. Esto significa que el 99% de los compuestos cuya energía de interacción es menor a –8,0 Kcal/mol tendrán un valor de afinidad (Ln CI50) menor a 5,52, es decir menor a 250 nM.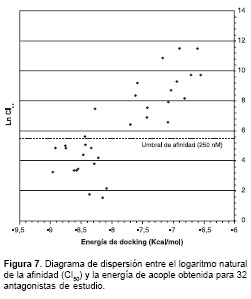
Es importante mencionar que para poder catalogar el análisis de docking como correcto se tuvo en cuenta el siguiente criterio: los anillos base de cada uno de los antagonistas analizados deben haber quedado superimpuestos a los anillos base del ácido dicloroquinurénico (ADCQ) de referencia, y en la posición correcta, es decir, el anillo bencénico de la quinolina del ADCQ debe superponerse al anillo bencénico de la dihidro-quinoxalina-2,3-diona, anillo base de todos los antagonistas analizados. De igual forma, los dos anillos heterocíclicos deben haberse super-impuesto. Sólo si se observaba esta condición, se tomaba en cuenta la energía de interacción que reportaba el programa. Con base en este criterio se obtuvo una posición correcta para 32 de los 45 antagonistas de estudio, de tal forma que la
figura 7 fue construida con estos 32 puntos.Aplicación del modelo matemático-estadístico propuesto basado en índices de conectividad para el diseño y predicción de la afinidad de nuevos antagonistas
Con base en el comportamiento mostrado por el índice de conectividad 4Xvpc en el modelo matemático y en las características estructurales que rigen su comportamiento, se propusieron nuevos antagonistas de glicina, de acuerdo con los siguientes criterios.
Valor numérico del índice de conectividad 4Xvpc: como se mencionó antes, el índice de conectividad 4Xvpc es directamente proporcional a la afinidad, y su valor debe ser superior a 1,18.
Delta de valencia de los sustituyentes hetero-atómicos halogenados: en el contexto de los índices de conectividad, los valores del delta de valencia empleados para el fluor, cloro, bromo y yodo son, respectivamente, 7,000, 0,777, 0,259 y 0,155. Debido a que cuanto menor sea el delta de valencia del heteroátomo sustituyente mayor es el valor numérico del índice 4Xvpc (9), los probables candidatos a sustituyentes del anillo aromático de los nuevos antagonistas serían el bromo y el yodo. Sin embargo, se decidió excluir el yodo, ya que en el grupo de antagonistas con los cuales se construyó el modelo no había ninguna molécula que tuviera este heteroátomo como sustituyente.
Número de sustituyentes y adyacencia entre ellos: se evaluaron anillos mono, di y trisustituidos, con todos sus posibles arreglos de adyacencia, teniendo en mente que el incremento en el número de sustituyentes, así como el incremento en su adyacencia, aumenta el valor numérico de 4Xvpc.
De acuerdo con las anteriores condiciones, se sugirieron para la evaluación de afinidad a través del modelo matemático propuesto, las nuevas moléculas enumeradas en el
cuadro 4 . En este punto es donde entran a jugar un papel decisivo los resultados obtenidos con los análisis de energías de interacción ( docking), metodología esta que se utilizó para validar los resultados obtenidos mediante el modelo de índices de conectividad. Es así como los datos generados por el modelo en cuanto a las características de unión de estas nuevas moléculas, se confirmaron a través de la determinación de la energía de interacción entre ellas y el dominio de unión de glicina para ratificar o no de esta forma los resultados obtenidos con el modelo matemático generado (cuadro 4 ).La predicción semicuantitativa mediante la ecuación 4 del modelo matemático-estadístico desarrollado estuvo acorde con las correspon-dientes energías de interacción para 11 de las 12 nuevas moléculas evaluadas, lo cual sustenta sólidamente la validez del modelo propuesto. De acuerdo con este modelo, los compuestos P5, P6 y P9 son moléculas promisorias, candidatas para síntesis y evaluación biológica. Si bien las moléculas P3 y P4 tienen una energía de docking favorable y el valor de su índice de conectividad 4Xvpc cumple el requisito de ser superior a 1,18, éste se sale de la línea de regresión del grupo de compuestos analizados, lo cual no hace tan confiable la predicción de su afinidad como sí lo es para las moléculas P5, P6 y P9, cuyos valores del índice mencionado caen sobre la línea de regresión mostrada en la
figura 6 .Discusión
Uno de los principales problemas que se tienen para generar y construir modelos matemáticos y estadísticos en farmacia química es la hetero-geneidad experimental con la cual se evalúa el comportamiento biológico in vitro de una serie de sustancias, restringiéndose así el número de moléculas que pueden analizarse mediante la meto-dología propuesta. Tal como lo expresa la Unión Internacional de Farmacología (34), el término CI50 es dependiente de las condiciones experimen-tales, de tal forma que esta expresión solamente puede utilizarse para la comparación de fármacos bajo condiciones de igualdad experimental.
Existe además el inconveniente de que muchos reportes experimentales se publican sin la suficiente rigurosidad estadística que permita evaluar la calidad de los datos presentados y, por tanto, sobre las conclusiones basadas en ellos. Es así como gran cantidad de moléculas no fueron incluidas en el estudio, pues carecían de una desviación estándar que permitiera evaluar la precisión en la obtención de la CI50. Este aspecto es importante, ya que no se le puede exigir al modelo matemático generado que prediga datos de CI50 con mayor precisión y exactitud que los que experimentalmente pueden obtenerse. El promedio de las desviaciones estándar reportadas para la determinación de la CI50 in vitro de los antagonistas de estudio (
cuadro 1 ) fue de alrededor de 16%, siendo los valores mínimos y máximos de 5% y 26%, respectivamente. Esta deficiente precisión en la evaluación de la afinidad, propia de la metodología experimental actual, puede explicar el hecho de que la estructura de un compuesto, codificada en los índices de conectividad, no explique por sí sola un mayor porcentaje de la variación en la afinidad. Pese a lo anterior, se considera que el modelo mate-mático generado es de una buena calidad estadística. Adicionalmente, el hecho de que se haya logrado seleccionar 45 moléculas que cumplieron los criterios de selección dio una base confiable para el desarrollo del presente trabajo.Es de resaltar el poder de codificación químico-estructural que encierran los índices de conectividad. El hecho de que se haya obtenido una ecuación que relacione la afinidad con un sólo índice de conectividad, de un total de 18 índices, deja entrever la gran cantidad de información que codifica el índice 4Xvpc. Si bien en los análisis iniciales de correlación, el índice 3Xvc también explicaba en un buen porcentaje la variación en la afinidad observada para los antagonistas, este índice ha demostrado no ser tan rico en información, sobre todo en cuanto a anillos aromáticos sustituidos se refiere. Los intentos por realizar un análisis de regresión lineal múltiple entre estos dos descriptores mostraron que el índice 3Xvc no era estadísticamente significativo, lo cual se explica por el alto grado de colinealidad entre él y el índice 4Xvpc. El coeficiente de correlación (r) entre ellos fue de 0,911, lo cual sugiere que en el índice 4Xvpc se incluye también la información codificada por el índice 3Xvc.
Por otro lado, es interesante observar el comportamiento de los antagonistas clasificados como grupo A y grupo B; sin embargo, no se logró evidenciar alguna característica estructural que pudiese ser la responsable del comportamiento diferencial observado. Estudios posteriores deben tratar de dilucidar desde un enfoque cuántico, por ejemplo, cuáles pueden ser las características que diferencian a los dos grupos. De encontrar una variable que diferencie los mencionados grupos de antagonistas, ésta se podría incluir en una nueva ecuación que mejore aun más el poder de predicción semicuantitativo de la afinidad.
La comparación del índice de conectividad 4Xvpc con su homólogo 4Xpc, el cual, a diferencia del primero, no codifica información acerca de la presencia de heteroátomos e insaturaciones, arrojó resultados estadísticos bastante pobres. Lo anterior sugiere que las insaturaciones, y en especial la presencia de heteroátomos, son factores decisivos para la adecuada interacción entre esta región y los aminoácidos presentes en el dominio de unión del antagonista en el receptor NMDA. El índice 4Xvpc es especialmente útil para codificar información acerca del número de sustituyentes sobre anillos bencénicos, los patrones de sustitución y su longitud (hasta tres enlaces), así como el tipo de sustituyente heteroatómico. Adicionalmente, este índice codifica información acerca de la orientación de los sustituyentes en el anillo: en cuanto mayor sea la adyacencia de los sustituyentes, mayor es el valor numérico del índice (9). Lo anterior puede observarse por ejemplo para los compuestos 1ant38 y 1ant35, ambos con dos halógenos como sustituyentes, donde el valor del índice 4Xvpc es mayor para el compuesto que tiene los halógenos vecinales. El incremento en el número de sustituyentes sobre el anillo aromático también incrementa el valor de este índice, lo cual se evidencia en los compuestos 1Ant36, 1Ant37, 1Ant38, 1Ant39 (
cuadros 1 y cuadro3 ).Adicionalmente, el índice 4Xvpc tiene la capacidad de discriminar entre compuestos cuya única diferencia es el tipo de heteroátomo sustituyente sobre el anillo aromático. El índice de conectividad simple homólogo 4Xpc carece de esta capacidad (ver los compuestos 1Ant15, 1Ant32 y 1Ant27 en los
cuadros 1 y cuadro3 ). Es importante anotar que cuanto mayor sea el delta de valencia para los heteroátomos en la molécula, menor es el valor numérico del índice 4Xvpc. De la misma forma, el índice 4Xvpc puede diferenciar entre sustituyentes que contienen los mismos átomos, pero en diferente posición (ver compuestos 1Ant17, 1Ant18). De allí la importancia de este descriptor para codificar la estructura química en esta clase de compuestos.Con base en lo anterior, y debido a que el índice de conectividad 4Xvpc es directamente proporcional a la afinidad (inversamente proporcional a la CI50), se sugiere tener en cuenta las siguientes características estructurales al desarrollar nuevos antagonistas de glicina derivados de la 1,4-dihidroquinoxalin-2,3-diona con elevada afinidad por el sitio de unión de glicina: valor numérico del índice de conectividad 4Xvpc, pues el índice 4Xvpc debe tener un valor numérico superior a 1,18; adyacencia de sustituyentes sobre el anillo aromático, ya que entre mayor sea la adyacencia, mayor es el valor del índice 4Xvpc; delta de valencia de sustituyentes halogenados, pues cuanto menor es el delta de valencia del halógeno sustituyente mayor es el valor numérico del índice 4Xvpc; número de sustituyentes sobre el anillo, ya que el incremento en el número de sustituyentes incrementa el valor de 4Xvpc; tamaño de sustituyentes, pues esta región no acepta sustituyentes voluminosos ni ciclos, tal como se observó para los compuestos 1ant6, 1ant7 y 1ant14, antagonistas con valores bajos del índice 4Xvpc.
Un paso importante en el desarrollo de metodologías numéricas que pretenden ser una herramienta predictiva es la etapa de validación del modelo. Por tal motivo, con el objeto de contar con una metodología alterna de control que permitiera confirmar y validar los resultados que se estaban observando con el modelo matemático propuesto basado en índices de conectividad, se desarrolló la metodología de evaluación de energías de interacción o docking aplicada a los antagonistas de estudio. Uno de los principales inconvenientes para evaluar la energía de interacción entre los antagonistas y el dominio de unión de glicina fue que para 13 compuestos, el programa computacional empleado no hizo un posicionamiento correcto de la molécula con respecto al compuesto de referencia ácido dicloroquinurénico, molécula con la cual se determinó la cristalografía de rayos X. Aún así, para los restantes 32 compuestos sí fue posible determinar las energías de interacción requeridas para el acople correcto con el receptor. El criterio para evaluar si hubo un posicionamiento correcto de la molécula en evaluación con el dominio de unión de glicina se especificó con anterioridad en los resultados; se tomó de esta forma debido a que una molécula a la cual se le han hecho modificaciones estructurales puede interactuar con residuos cercanos al dominio de unión de glicina, pero ello no significa que la consecuencia de tal interacción sea un antagonismo de los efectos del coagonista glicina. Debido a que se sabe con certeza cuáles son los residuos amino-acídicos relevantes con los que una molécula antagonista debe interactuar para ejercer su efecto farmacológico, se buscó evaluar las consecuencias de las modificaciones estructurales en la interacción con estos residuos y no con otros situados en la cercanía del sitio de unión. Aunque el número de moléculas cuya energía de interacción con el receptor fue posible determinar fue menor que el total de moléculas empleadas para construir el modelo matemático, se consideró que dicho nú-mero era suficiente para evaluar de forma confiable los requerimientos energéticos de interacción entre ellas y el dominio de unión de glicina.
Los resultados obtenidos son bastante interesantes teniendo en cuenta que solamente para una molécula hubo discrepancia entre la afinidad semicuantitativa reportada por el modelo matemático y el análisis de docking . Es bien importante observar que los 12 compuestos nuevos se pueden agrupar en tres grupos: mono-bromados, dibromados y nitrobromados. Cada grupo está constituido por moléculas que son isómeros de posición entre ellas. Por consiguiente, es de resaltar que el modelo generado es capaz de predecir la afinidad de moléculas que están estructuralmente muy relacionadas entre sí, lo cual es una ventaja sobresaliente de la metodología propuesta.
Con base en la revisión bibliográfica, el presente trabajo es el primer modelo cuantitativo que propone nuevas moléculas con alta afinidad por el dominio de unión de glicina basado en índices de conectividad molecular y que confirma su com-portamiento biológico a través de metodologías de interacción energética bioquímica o docking.
Finalmente, se concluye que es posible codificar matemáticamente la estructura química de antagonistas del dominio de unión a glicina mediante los índices de conectividad molecular, generando ecuaciones definidas que modelan el comportamiento de unión al receptor y aportan información estructural útil en el diseño de nuevos antagonistas, así como en la predicción de sus propiedades de unión.
Conflicto de intereses
Los autores declaran no haber tenido ningún conflicto de intereses con empresas públicas o privadas durante la realización de la investigación.
Financiación
Los programas y computadores fueron aportados por el Departamento de Nutrición y Bioquímica, Grupo de Bioinformática, Facultad de Ciencias, Pontificia Universidad Javeriana.
Correspondencia: Leonardo Lareo, Facultad de Ciencias, Departamento de Nutrición y Bioquímica, Grupo de Bioquímica Computacional y Estructural y Bioinformática, Pontificia Universidad Javeriana, Carrera 7 No. 43-82, oficina 107, Edificio Carlos Ortiz, Bogotá, D.C., Colombia.
Teléfono: 3208320, ext. 1437. l.lareo@javeriana.edu.co
Referencias
1. Neamati N, Barchi JJ Jr. New paradigms in drug design and discovery. Curr Top Med Chem 2002;2:211-27. [ Links ]
2. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004;3:935-49. [ Links ]
3. Brown N, Lewis RA. Exploiting QSAR methods in lead optimization. Curr Opin Drug Discov Devel 2006;9: 419-24. [ Links ]
4. García D, Gálves J. Topología Molecular. Su papel en el diseño de nuevos fármacos. Investigación y Ciencia 1993;197:86-7. [ Links ]
5. Gonzalbes R, Doucet JP, Derouin F. Application of topological descriptors in QSAR and drug design: history and new trends. Curr Drug Targets Infect Disord 2002;2:93-102. [ Links ]
6. Randic M. On characterization of molecular branching. J Am Chem Soc 1975;97:6609-15. [ Links ]
7. Kier LB, Hall LH. The meaning of molecular connectivity: A bimolecular accessibility model. Croatia Chem Acta 2002;75:371-82. [ Links ]
8. Hall LH, Kier LB. Issues in representation of molecular structure the development of molecular connectivity. J Mol Graph Model 2001;20:4-18. [ Links ]
9. Kier LB, Hall LH. Molecular connectivity in structure-activity analysis England: Research Studies Press; 1986. [ Links ]
10. Hall LH, Kier LB. Molecular connectivity and substructure analysis. J Pharm Sci 1978;67:1743-7. [ Links ]
11. Kier LB, Hall LH. General definition of valence delta-values for molecular connectivity. J Pharm Sci 1983;72:1170-3. [ Links ]
12. Kier LB, Hall LH. Molecular connectivity VII: specific treatment of heteroatoms. J Pharm Sci 1976;65:1806-9. [ Links ]
13. Rios-Santamarina I, Garcia-Domenech R, Galvez J, Cortijo J, Santamaria P, Morcillo E. New bronchodilators selected by molecular topology. Bioorg Med Chem Lett 1998;8:477-82. [ Links ]
14. de Gregorio Alapont C, Garcia-Domenech R, Galvez J, Ros MJ, Wolski S, Garcia MD. Molecular topology: a useful tool for the search of new antibacterials. Bioorg Med Chem Lett 2000;10:2033-6. [ Links ]
15. Agrawal VK, Sohgaura R, Khadikar PV. QSAR study on inhibition of brain 3-hydroxy-anthranilic acid dioxygenase (3-HAO): a molecular connectivity approach. Bioorg Med Chem 2001;9:3295-9. [ Links ]
16. Bajaj S, Sambi SS, Madan AK. Topological models for prediction of anti-HIV activity of acylthiocarbamates. Bioorg Med Chem 2005;13:3263-8. [ Links ]
17. Garcia-Garcia A, Galvez J, de Julian-Ortiz JV, Garcia-Domenech R, Munoz C, Guna R et al. New agents active against Mycobacterium avium complex selected by molecular topology: a virtual screening method. J Antimicrob Chemother 2004;53:65-73. [ Links ]
18. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 1999;51:7-61. [ Links ]
19. Candy S, Brickley S, Farrant M. NMDA receptor subunits: Diversity, development and disease. Curr Opin Neurobiol 2001;11:327-35. [ Links ]
20. Popescu G, Auerbach A. The NMDA receptor gating machine: lessons from single channels. Neuroscientist 2004;10:192-8. [ Links ]
21. Yun L, Juntian Z. Recent development in NMDA receptors. Chinese Med J 2000;113:948-56. [ Links ]
22. Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci 2002;5(Suppl):1039-42. [ Links ]
23. Yosa J. Desarrollo y validación de un modelo para identificar factores que definen el tipo de acción de un ligando. Trabajo de grado (Magíster en Ciencias biológicas). Bogotá D.C.: Facultad de Ciencias, Pontificia Universidad Javeriana; 2005. [ Links ]
24. Leeson PD, Iversen LL. The glycine site on the NMDA receptor: structure-activity relationships and therapeutic potential. J Med Chem 1994;37:4053-67. [ Links ]
25. Danysz W, Parsons AC. Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev 1998; 50:597-664. [ Links ]
26. Brown DG, Urbanek RA, Bare TM, McLaren FM, Horchler CL, Murphy M et al. Synthesis of 7-chloro-2,3-dihydro-2-[1-(pyridinyl)alkyl]-pyridazino[4,5-b]quinoline-1,4,10(5H)-triones as NMDA glycine-site antagonists. Bioorg Med Chem Lett 2003;13:3553-6. [ Links ]
27. Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. Embo J 2003;22: 2873-85. [ Links ]
28. RCSB Protein Data Bank. Crystal structure of the NR1 ligand binding core in complex with 5,7-dichlorokynurenic acid (DCKA) at 1.90 angstroms resolution. [Consultado: Mayo 27 de 2005]. Disponible en http://www.rcsb.org/pdb/explore/explore.do? structureId=1PBQ [ Links ]
29. Thompson M. ArgusLab version 4.0.1.. Seattle, WA: Planaria Software; 2004. [Consultado: mayo 27 de 2005] Disponible en http:// http://www.planaria-software.com/arguslab40.htm [ Links ]
30. Cai SX, Kher SM, Zhou ZL, Ilyin V, Espitia SA, Tran M et al. Structure-activity relationships of alkyl- and alkoxy-substituted 1,4-dihydroquinoxaline-2,3-diones: potent and systemically active antagonists for the glycine site of the NMDA receptor. J Med Chem 1997;40:730-8. [ Links ]
31. Cai SX, Zhou ZL, Huang JC, Whittemore ER, Egbuwoku ZO, Lu Y et al. Synthesis and structure-activity relationships of 1,2,3,4-tetrahydroquinoline-2,3,4-trione 3-oximes: novel and highly potent antagonists for NMDA receptor glycine site. J Med Chem 1996;39:3248-55. [ Links ]
32. Zhou ZL, Kher SM, Cai SX, Whittemore ER, Espitia SA, Hawkinson JE et al. Synthesis and SAR of novel di- and trisubstituted 1,4-dihydroquinoxaline-2,3-diones related to licostinel (Acea 1021) as NMDA/glycine site antagonists. Bioorg Med Chem 2003;11:1769-80. [ Links ]
33. Tikhonova IG, Baskin, II, Palyulin VA, Zefirov NS. CoMFA and homology-based models of the glycine binding site of N-methyl-d-aspartate receptor. J Med Chem 2003;46:1609-16. [ Links ]
34. Neubig RR, Spedding M, Kenakin T, Christopoulos A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev 2003;55:597-606. [ Links ]

