










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.27 n.2 Bogotá abr./jun. 2007
DNA microarray analysis reveals metastasis-associated genes in rat prostate cancer cell lines
Ismael Reyes 1, Raj Tiwari 1, Jan Geliebter 1, Niradiz Reyes 2
1 Department of Microbiology and Immunology, New York Medical College, Valhalla, NY, USA
2
Department of Basic Sciences, School of Medicine, Universidad de Cartagena, Cartagena, ColombiaRecibido: 29/08/06; aceptado: 22/02/07
Introduction.
The molecular and cellular mechanisms involved in prostate cancer progression towards a hormone-independent and highly invasive, metastatic phenotype, are not well understood. Cell lines with different metastatic potential, when analyzed by microarray techniques, offer valuable tools for identifying genes associated with the metastatic phenotype.Objectives. Gene expression profiles were compared for two rat prostate cancer cell lines with differing metastatic abilities in order to better characterized molecular underpinnings of the prostate cancer metastatic process.
Materials and methods. Affymetrix arrays were used to analyze gene expression of two rat prostate cancer cell lines, MAT-LyLu and G. Microarray data were analyzed using pathway and functional group analysis. A selected set of genes was subjected to real-time polymerase chain reaction for validating the microarray data.
Results. Microarray data analysis revealed differential expression of genes from a number of signaling and metabolic pathways. Overexpression was detected in 48 genes and underexpression in 59 genes of the MAT-LyLu line compared to the standard G line. Genes were grouped into functional categories, including epithelial-extracellular matrix interaction, cell motility, cell proliferation, and transporters, among others. Many of these genes were not previously associated to prostate cancer metastasis.
Conclusions. Many genes with altered expression associated with a metastatic prostate cancer phenotype were identified. Further validation of these genes in human prostate samples will determine their usefulness as biomarkers for early diagnosis of recurrence or metastasis of prostate cancer, as well as potential therapeutic targets for this disease.
Keywords: carcinoma, neoplasm metastasis, gene expression, biological markers, extracellular matrix, microarray analysis.
Análisis de micromatrices de ADN revela genes asociados a metástasis en líneas celulares de cáncer de próstata de rata
Introducción. Los mecanismos moleculares y celulares involucrados en la progresión del cáncer de próstata hacia un fenotipo metastásico no son bien entendidos. El análisis molecular de líneas celulares con diferentes potenciales metastásicos ofrece un instrumento valioso para identificar genes asociados al fenotipo metastásico.
Objetivos. Comparar los perfiles de expresión genética de dos líneas celulares de cáncer de próstata de rata con diferentes capacidades metastásicas para un mejor entendimiento del proceso metastásico.
Materiales y métodos. Se utilizaron micromatrices de Affymetrix para analizar la expresión génica de dos líneas celulares de próstata de rata con diferentes propiedades metastásicas, MAT-LyLu y G. Los datos fueron analizados en el contexto de vías y grupos funcionales. Se utilizó reacción en cadena de la polimerasa en tiempo real para validación de genes seleccionados.
Resultados. Además de la expresión diferencial de genes en un número de vías de señalización y metabólicas, se detectó sobre-expresión de 48 genes y expresión disminuida de 59 genes en la línea MAT-LyLu comparado con la línea G. Los genes fueron agrupados en categorías funcionales, tales como interacción epitelial-matriz extracelular, motilidad celular, proliferación celular, y transportadores, entre otros. Muchos de estos genes no han sido asociados previamente a la metástasis del cáncer de próstata.
Conclusiones. Se identificaron genes con expresión alterada asociados al fenotipo metastásico del cáncer de próstata. La subsiguiente validación de estos genes en tejido prostático humano pudiera revelar su utilidad como marcadores biológicos para esta enfermedad.
Palabras clave: carcinoma, metástasis de la neoplasia, expresión génica, marcadores biológicos, matriz extracelular, análisis de micromatrices.
The American Cancer Society estimates that 234,460 new cases of prostate cancer were expected in the United States in 2006, representing the most common cancer in men and the third leading cause of male cancer-related deaths (1). Treatments for prostate cancer including surgical and hormonal therapies, have shown beneficial effects only for early-stage, hormone-dependent disease. However, for more advanced stages, the cancer cells become hormone-independent and highly invasive, until they reach an incurable clinical stage associated with an increased incidence of metastases, especially to the bone. The molecular and cellular mechanisms involved in this process are not well understood (2).
Dunning rat tumor cell sublines of variable metastatic potential: G (non metastatic) and MAT-LyLu (highly metastatic) were used to investigate the differences in gene expression associated to metastatic prostate cancer. These two cell lines were derived from the same parental tumor, the Dunning R-3327 from Copenhagen rats (3). The R3327-G, or simply G, cell line is a poorly differentiated, androgen-dependent tumor line obtained in Dunnings laboratory after in vitro passaging of the original tumor. The MAT-LyLu (Metastatic At-Lymph Node and Lung) cell line was developed at Johns Hopkins School of Medicine and characterized by Isaacs et al (4,5). The G and MAT-LyLu cell lines exhibit many characteristics seen during the progression of human prostate cancer, including androgen responsiveness and differential metastatic ability, representing a well characterized model to study the metastatic process (6,7).
In the current study, the gene expression profiles of the two rat cell lines were determined with Affymetrix DNA microarrays (GeneChip Rat Genome U34A array). There was altered expression (overexpression and underexpression) of 107 genes in the MAT-LyLu cell line was compared to the G cell line. Several of the genes were not previously associated with prostate cancer metastasis. Validation of these metastasis-associated genes in human prostate samples have a potential application as biomarkers for early diagnosis of recurrence or metastasis or as therapeutic targets for this disease.
Materials and methods
Cell lines
Dunning rat tumor cell sublines, G (non metastatic) and MAT-LyLu (highly metastatic) were grown using RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin-streptomycin at 37oC in a humidified atmosphere containing 5% CO2. Cells were grown in T-75 culture flasks with 25 cc of growth media. All tissue culture work was done in a Class IIA cell culture hood with 20 min of UV light and ethanol wash between handling of the different cell lines.
Total RNA isolation
Total RNA was prepared from MAT-LyLu and G cells grown to approximately 75% confluence using Trizol solution (Invitrogen) and stored as an ethanol precipitate. The resulting RNA concentration was measured spectrophotometrically, and quality was assessed by the 260/280 ratio and agarose gel electrophoresis. Two biological replicates (duplicates)consisted of RNA samples obtained from cells grown at different dates and processed independently for microarray experiments (no sample pooling was done before microarray hibridizations). In this manner, microarray hybridization for each cell line was performed in duplicate.
Microarrays
To study the gene expression profile of these rat cell lines, the Rat Genome U34A Array (Affymetrix, Santa Clara, CA) was used. This array represented approximately 7,000 full-length gene sequences and approximately 1,000 EST clusters consisting of expressed sequence tags-transcribed sequences of unknown function. Microarray procedures
Total RNA was processed at the University DNA Microarray Facility of the Center for Molecular Medicine at the SUNY Stony Brook School of Medicine, according to protocols described in Affymetrix GeneChip Expression Analysis technical manual (Affymetrix, Santa Clara, CA). The manual can be downloaded from the Affymetrix website (http://www.affymetrix.com/support/technical/manual/expression_manual.affx ). RNA quality was first assessed by electrophoresis using the Agilent Bioanalyzer 2100 and spectrophotometric analysis. Then, total RNA from each sample was used to generate high fidelity cDNA. Briefly, first-strand cDNA was reverse transcribed from 15 µg of total RNA using 600 U of SuperScript II RT (Invitrogen) in a 20µl total volume containing T7-(dT)24 primer, 10 mM DTT, 500 µM of each dNTP, and 1x first-strand cDNA buffer for 1 h at 42°C. Second-strand cDNA was synthesized in 1x reaction buffer containing 200 µM of each dNTP, 10 U of DNA ligase, 40 U of DNA polymerase I, and 2 U of RNase H (final volume = 150 µl) for 2 h at 16°C. Samples were then treated with 20 U of T4 DNA polymerase for 5 min at 16°C and incubated in 10 µl of 0.5 M EDTA. Double-stranded cDNA was purified with phase lock gel electrophoresis (Eppendorf Scientific, Westbury, NY) and phenol-chloroform extraction, followed by ethanol precipitation. Upon completion of cDNA synthesis, 1µg of product was converted to biotinylated cRNA using the Enzo BioArray HighYield RNA Transcript Labeling Kit. Labeled cRNA was fragmented and hybridized to the Affymetrix oligonucleotide GeneChip rat U34A arrays. Twelve micrograms of biotinylated target cRNA were hybridized per microarray. Then, the gene chips were automatically washed, to remove non-specifically bound cRNA, and stained with streptavidin-phycoerythrin conjugate in the GeneChip Fluidics Station. Washed arrays were automatically scanned using Hewlett-Packard GeneArray Scanner equipped with an argon ion laser. The methodology used for the microarray procedure follows the MIAME (Minimum Information About a Microarray Experiment) guidelines (www.mged.org/ Workgroups /MIAME/miame.html).
Microarray data analysis
Scanned output files were analyzed with dChip v1.3 software (www.dchip.org.) and Affymetrix MicroArray Suite 5.0 (MAS 5.0) software. First, arrays were normalized by dChip v1.3 using the invariant set normalization method (8). After normalization, model-based gene expression estimates and outlier detection algorithm were obtained according to the perfect-match-only model performed by Li-Wong (9); transcripts regarded as outliers were excluded for further analysis. Affymetrix MAS 5.0 software was used to determine if the transcripts were detected as present, absent, marginal, or no call. For significant expression level, a cut off value of 500 units was used. DNA microarray data generated from MAT-LyLu samples were compared to microarray data from G samples using dChip v1.3 software and the resulting expression analysis files were subjected to biological pathway and functional group analysis to determine the significance of changes at the biological context.
Pathway analysis
To identify biological pathways playing a role in the metastatic process, the microarray data were analyzed with the GenMAPP (Gene Map Annotator and Pathway Profiler) and MAPPFinder programs developed at the Gladstone Institutes at the University of California at San Francisco (10) (www.genmapp.org). Criteria used for GenMAPP/ MAPPFinder analysis for increased expression were a minimum average intensity of 500 units, a % present (P) call of 100, and a fold change >1.8 in the MAT-LyLu cell line compared to the G cell line. Criteria for decreased expression were a minimum intensity of 500 units, a % P call of 100, and a fold change of < -1.8 in the MAT-LyLu cell line compared to the G cell line. The programs generated a Z score based on the hypergeometric distribution.
Functional group analysis
The catalog of characterized genes is incomplete in the context of biological pathways, and many genes are of unknown function. Therefore a functional group analysis was performed in order to have a more complete picture of genes associated to the metastatic phenotype and to increase the possibilities of finding genes not previously associated to prostate cancer metastasis. For functional group analysis, the dChip v1.3 software provided a means for comparing the MAT-LyLu microarray data vs. G microarray data., It generated a gene list that contained genes with P< 0.05 (from t-test), a fold change >+ 2.0, and a % P call of 100.These genes were thereby identified as having significantly altered expression (either overexpression or underexpression). This set of genes was subjected to hierarchical clustering (dChip v1.3) using the Pearson correlation metric. Before clustering, the expression value for a gene across all samples was standardized to have a mean of 0 and standard deviation of 1. dChip v1.3 also determined in all branches the presence of local clusters enriched by genes having a particular function (functional clusters), according to gene ontology (GO) terms (11). In this way, co-expressed genes were detected that shared similar functions relevant to prostate cancer metastasis. dChip v1.3 also generated a p value for functional clusters with significance set at p<0.01.
Real time PCR
Quantitative real time PCR (qRT-PCR) was performed for selected genes with the same individual RNAs used in microarray experiments in order to validate the technique (12). Gene-specific primers were designed with Primer3 software for kelch repeat 1 (krp1) (forward primer = aaggagatggc-gcta gacaa; reverse primer = cacggcctc-aaat accactt), endothelial cell-specific molecule 1(ESM-1) (forward primer = ctactttg gacggctgcttc; reverse primer = cgtcacc gaaatcatcttcc) and caveolin-1 (CAV-1) (forward primer = tgaagatgtgat tgcgg aac; reverse primer = atctctt cctgcgtgctgat).
One microgram total RNA was reverse transcribed using oligo-dT and the Reverse Transcription System kit (Promega). Two microliters of reverse transcribed product was subjected in triplicate to qRT-PCR using the LightCycler DNA Master SYBR Green system (Roche Molecular Biochemicals, Mannheim, Germany). PCR was performed in a LightCycler (Roche, Mannheim, Germany) with a 2 min pre-incubation at 95°C followed by 40 cycles of amplification steps. PCR products were subjected to melting curve analysis to verify that no amplification of non-specific products had occurred. In each qRT-PCR run, a standard curve was generated using a serial dilution of a cDNA standard identical in size and sequence to the target gene fragment to be amplified. Serial dilution ranged from 0.01 to 100 pg of the standard cDNA fragment. The standard curve was calculated by the Light Cycler 5.32 software (LC-Run Version 5.32, Roche) by regression of the crossing points of the PCR curves from the dilution series of the standard cDNA fragment. Changes in gene expression were quantified by calculating the average value of the triplicate reactions from the MAT-LyLu cell line and comparing that to the average derived from the triplicate reactions from the G cell line. The GAPDH gene was chosen as a control housekeeping gene for normalization, since no change in the GAPDH mRNA levels occurred across the entire sample set (MAT-LyLu and G samples). Statistical significance of changes was determined by t-test.
Statistical methods
For DNA microarray and qRT-PCR analyses, a p <0.05 from t-test (two-sided) was considered statistically significant. For hierarchical clustering, significant functional cluster p values were calculated using the hyper-geometric distribution. For pathways analysis with GenMAPP/MAPPFinder programs, the Z score based on the hypergeometric distribution was used. The Z score reflected the significance of changes and it falls in a range of positive to negative values. A cut-off value of 2.0 was selected for the Z score, representing the equivalent of p= 0.05 for the hypergeometric distribution. Z scores were generated automatically by the programs.
Results
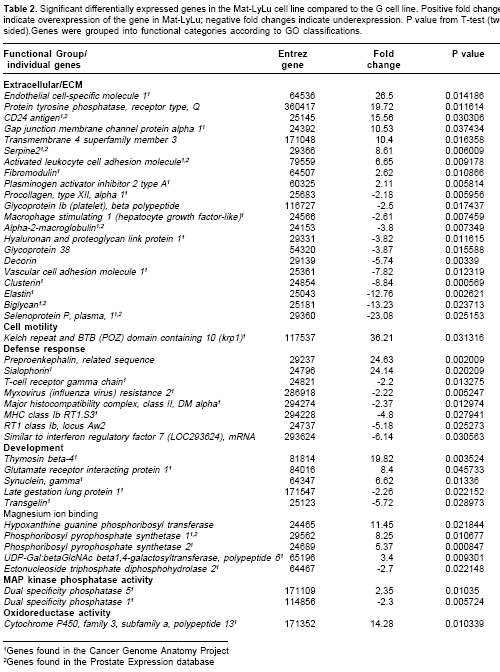
Many differentially expressed genes were identified from several signaling and metabolic pathways in MAT-LyLu ( Table 1). Among the signaling pathways, differential expression was found in the androgen receptor signaling pathway, focal adhesion, Wnt- signaling, hepatocyte growth factor receptor (HGF), epidermal growth factor (EGF) receptor 1, MAPK, integrin mediated cell adhesion, TGF beta, and Insulin signaling pathways (Table 1). In addition to signaling pathways, a number of genes were overexpressed in the cholesterol biosynthesis and nucleotide metabolic pathways (Table 1) Overexpression of these metabolic pathways may account for the high metabolic activity required for the MAT-LyLu cell line to maintain its characteristic high level of cell proliferation compared to the non-metastatic G line, which has a much lower doubling time (4,13). On the other hand, functional group analysis of the DNA microarray data revealed the significantly altered expression of 107 well-characterized genes (Table 2 ), 48 overexpressed and 59 underexpressed in MAT-LyLu compared to G cell lines, according to the criteria described in materials and methods.
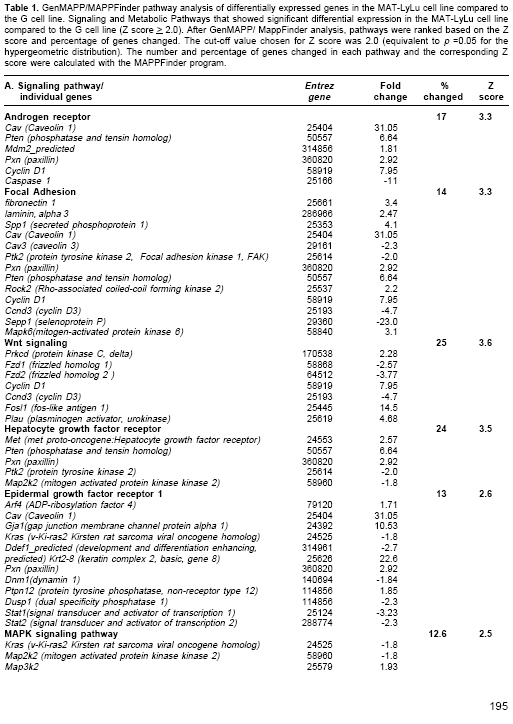
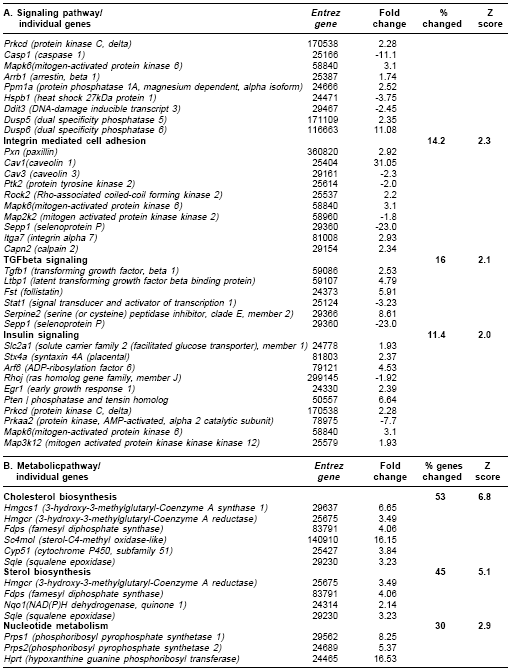
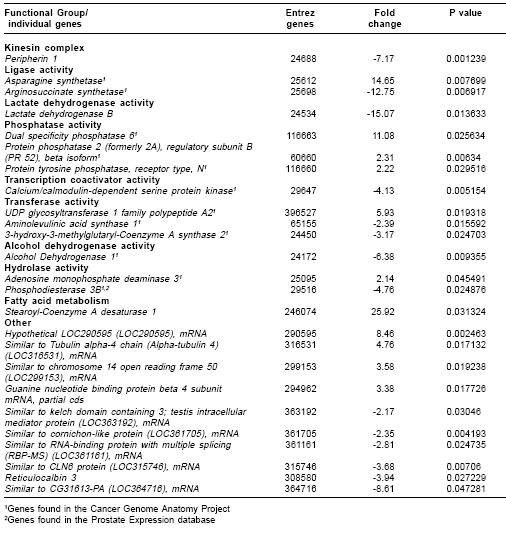
Hierarchical clustering of the significantly expressed genes clearly showed the gene expression differences between MAT-LyLu and G cell lines (
Figure 1 ). Significant functional clustering of the differentially expressed genes included the following GO categories: extracellular region/extracellular matrix (20% of the genes), development (5%), defense response (7%), proteolysis and peptidolysis (5%) and magnesium ion binding (5%), among others.
Some of these functional clusters reflected the changes believed to occur during the metastatic process, such as alteration of extracellular matrix components, neovascularization, changes in cell structure and increased motility (14). Thus, there is a strong concordance of our results with the scientific literature about the metastatic process.
We also compared our microarray data with other microarray analysis of prostate cancer metastasis (15,16). For example, in the study by LaTulippe et al, they reported the altered expression of several genes that correlate with the metastatic phenotype in prostate cancer and those genes were grouped into functional categories, such as cell cycle regulation, DNA repair, cell structure, and motility among others. Although we cannot correlate directly the results presented by LaTulippe et al (clinical samples) with our results (rat cell lines samples), those results by LaTulippe et al showed a similar trend of changes in the metastatic prostate cancer than our data (
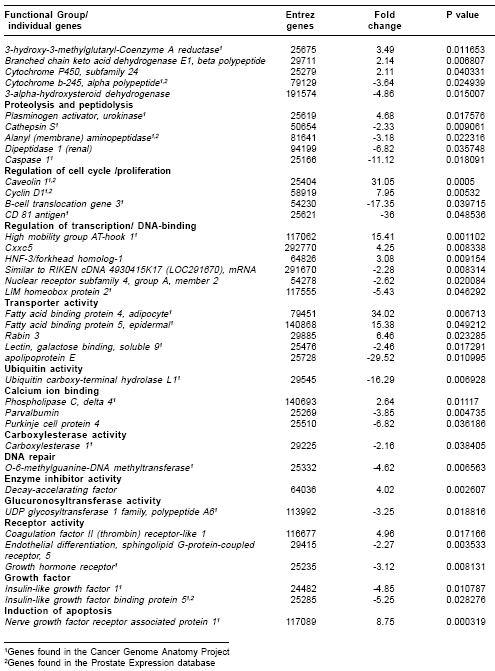
Table 2 ), demonstrating the validity of our approach and the significance of our results.For specific gene validation, two approaches were used-correlation with gene expression database results and with qRT-PCR analysis. In this way, the data were matched with the Cancer Genome Anatomy Project database (cgap.nci.nih.gov) and with the Prostate Expression database (17). Each gene was first matched with the corresponding human gene using the Entrez Gene identification number, since the databases are for Homo sapiens species.
Table 2 indicates the many genes were expressed in cancer and/or in prostate samples.For qRT-PCR analysis, we chose the krp1, ESM-1 and CAV-1 genes were chosen because they represented overexpressed genes relevant to the current study. These genes were validated by qRT-PCR with statistically significant p values (< 0.05) (
Table 3 ). The levels of gene expression determined by qRT-PCR analyses were consistent with those observed in the DNA microarray experiments. For example, the 36- fold increase in expression of krp1 determined by DNA microarray analysis was validated by the 48-fold increase observed by qRT-PCR. These results validated our DNA microarray analysis.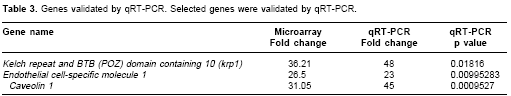
Discussion
Prostate cancer progression from a localized form to a metastatic state seems to be regulated by several oncogenic signals in the prostate epithelial cells and by changes in the stromal-epithelial interaction. These include the Hedgehog signaling and Wnt signaling pathways (18,19) as well as overexpression of several growth factors and their receptors, such as the epidermal growth factor, fibroblast growth factor, HGF, insulin-like growth factor-1 and vascular endothelial growth factor (VEGF) (20). In agreement with published observations, the metastatic phenotype of MAT-LyLu cell line was associated to overexpression of genes involved in several of these signaling pathways. Examples of the overexpressed genes included met proto-oncogene (HGF receptor), phosphatase and tensin homologue on chromosome 10 (PTEN), paxillin (Pxn), and caveolin (Cav)-all of which are important components of the HGF receptor, androgen receptor, epidermal growth factor receptor, integrin mediated cell adhesion, and focal adhesion signaling pathways ( Table 1 ).
The HGF receptor, also called c-Met, is a heterodimer with tyrosine kinase activity that associates with a multiprotein complex involved in downstream signal transduction (21).
c-Met is activated by the HGF and it has been shown to induce alterations in cell motility, cell shape, adhesion, resistance to apoptosis, and anchorage independent growth. These all contribute to the role of c-Met in cancer and makes this receptor a potential therapeutic target for cancer drugs (22-24). c-Met has also been found to target paxillin (overexpressed in the current study) and focal adhesion kinase (FAK), an activity that can lead to alterations in cell motility and tissue invasion by transformed cells (25,26). The overexpression of several genes in the HGF receptor pathway in the MAT-LyLu cell line ( Table1 ) was also notable.
The clinically aggressive phenotype distinctive of the metastatic cascade (14), has been studied at the gene expression level (15,16). In those studies, the overall gene expression alteration in the metastatic phenotype agrees with the data herein, with similar patterns of altered expression in genes associated with signaling transduction, ECM remodeling, motility, cell cycle regulation, and cell structure. An important validation of the cell line base model used herein are results from studies (15,16) done in human samples which fully agree with the MAT-LyLu data.
Genes relevant for the metastatic phenotype, found with altered expression in the current study, include ESM-1, CD24 antigen, biglycan (BGN), serpine2, fibromodulin, macrophage stimulating 1, alpha-2-macroglobulin, glycoprotein 38, decorin, vascular cell adhesion molecule 1, clusterin, and elastin. These 12 genes are grouped in the ECM interaction functional group. Several of these genes encode proteoglycan (PG) proteins (ESM-1, BGN, fibromodulin, and decorin) which are major components of the ECM that maintain the integrity of structural tissue. However, ESM-1, BGN and fibromodulin have not been associated previously with prostate cancer metastasis. Furthermore, the most overexpressed gene in MAT-LyLu, krp1- which encodes a protein associated with formation of elongated pseudopodia in transformed rat fibroblast (27)-has not been previously associated to prostate cancer metastasis. Additional studies are necessary to determine the association of these genes to prostate cancer metastasis in the human counterpart. Currently experiments are underway in our laboratory to determine the status of expression of ESM-1 gene and protein in human prostate cancer.
Overexpression of ESM-1 in the tumor tissue may constitute a negative prognostic factor and a marker for metastasis for cancer patients. This concept has been suggested by others in cases of breast cancer (28), renal cancer (29), and lung cancer (30). However, the current study is the first to link ESM-1 to metastatic prostate cancer.
Fibromodulin, overexpressed, and BGN and decorin, underexpressed, are genes that encode PG proteins that are members of the small leucine-rich PG family. They function, under physiological and pathophysiological conditions, as regulators of matrix assembly, cellular adhesion, migration and transforming growth factor beta (TGF-beta) activity (31,32). In the current study, differential expression of genes in the TGF-beta signaling pathway was found as well ( Table 1 ).
Other genes associated with cell-ECM interaction are CD24 and Serpine 2. CD24 gene, overexpressed in the current study, encodes a small, heavily glycosylated and mucin-like cell surface protein. CD24 has been found overexpres-sed previousely in prostate cancers (33), and this overexpression has been suggested as predictor of prostate cancer relapse and poor prognosis (34).
Serpine 2 ( protease nexin I), found over expressed in the current study, has been implicated in invasive potential of pancreatic cancer cells because it alters ECM production and organization within the tumors (35). Serpine 2 mRNA levels have been found overexpressed in prostate cancer cell lines and possibly interacts negatively with prostasin, an invasion suppressor protein (36).
Another overexpressed gene in the highly metastatic MAT-LyLu cell line, was the Cav-1 gene. Cav-1 was grouped in the regulation of cell proliferation group and encodes a protein that is a component of caveolae membrane coats. In human and animal studies, Cav-1 was overexpressed in metastatic prostate cancer; antisense-mediated down regulation of Cav-1 in prostate cancer cells in vitro seems to reduce the metastatic phenotype (37,38). Therefore, Cav-1 is considered a metastasis-related gene in prostate cancers.
Finally, the most underexpressed gene was the CD81 gene that encodes a cell-surface protein and belongs to the tetraspanin superfamily (TM4SF). Loss of CD81 has been found in poorly differentiated hepatocellular carcinoma and extrahepatic metastasis patients (39). CD81 has not been associated with prostate cancer previously; however, another member of the tetraspanin family, KAI1/CD82, has been suggested as a prostate cancer metastasis suppressor gene. The underexpression of the CD81 gene in the current study warrants further investigation to determine its exact function and its potential as a suppressant of metastasis in prostate cancer.
In summary, overexpressed and underexpressed genes were successfully identified in a highly metastatic rat prostate cancer cell line, when compared to a non metastatic rat prostate cancer cell line. The cell lines had been previously established as in vitro models for studying prostate cancer. To extrapolate results obtained from this work to the human context, further studies are needed with human samples and detailed clinical characterization of the patients for appropriate clinical correlations. Nonetheless, the identification of genes differentially expressed in these two rat cell lines by DNA microarray analysis is a first logical step in the search for dependable biomarkers for human prostate cancer metastasis.
Acknowledgements
The authors wish to thank the Microarray Facility of the Center for Molecular Medicine at the SUNY at Stony Brook School of Medicine. The authors also thankJohn Isaacs, M.D., for kindly providing the rat cell lines.
Conflict of interests
The authors declare that they have no competing interests.
Financial support
This study was supported by the US National Institute of Health (NIH grant # 1R21CA088982 awarded to Jan Geliebter, PhD).
"The data discussed in this publication have been deposited in NCBls Gene Expresion Omnibus (Geo, http://www.ncbi.nlm.nih.gov/geo/ ) and are accessible through Geo Series accession number GSE7703".
Corresponding author:
Niradiz Reyes, Universidad de Cartagena, School of Medicine, Department of Basic Sciences, Cartagena, Colombia.
Telefax: 654486/654772
References
1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al . Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106-30. [ Links ]
2. Dong JT, Rinker-Schaeffer CW, Ichikawa T, Barrett JC, Isaacs JT. Prostate cancer biology of metastasis and its clinical implications. World J Urol. 1996; 14:182-9. [ Links ]
3. Dunning WF. Prostate cancer in the rat. Natl Cancer Inst Monogr. 1963;12:351-69. [ Links ]
4. Isaacs JT, Isaacs WB, Feitz WF, Scheres J. Establishment and characterization of seven Dunning rat prostatic cancer cell lines and their use in developing methods for predicting metastatic abilities of prostatic cancers. Prostate. 1986;9:261-81. [ Links ]
5. Isaacs JT, Yu GW, Coffey DS. The characterization of a newly identified, highly metastatic variety of Dunning R 3327 rat prostatic adenocarcinoma system: the MAT LyLu tumor. Invest Urol. 1981;19:20-3. [ Links ]
6. Luo J, Sharma N, Seftor EA, De Larco J, Heidger PM, Hendrix MJ, et al. Heterogeneous expression of invasive and metastatic properties in a prostate tumor model. Pathol Oncol Res 1997;3:264-71. [ Links ]
7. Sharma N, Luo J, Kirschmann DA, OMalley Y, Robbins ME, Akporiaye ET et al. A novel immunological model for the study of prostate cancer. Cancer Res. 1999;59:2271-6. [ Links ]
8. Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. [ Links ]
9. Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 2001;98:31-6. [ Links ]
10. Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19-20. [ Links ]
11. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-9. [ Links ]
12. Rajeevan MS, Ranamukhaarachchi DG, Vernon SD, Unger ER. Use of real-time quantitative PCR to validate the results of cDNA array and differential display PCR technologies. Methods. 2001;25:443-51. [ Links ]
13. Wenger AS, Mickey DD, Hall M, Silverman LM, Mickey GH, Fried FA. In vitro characterization of MAT LyLu: a Dunning rat prostate adenocarcinoma tumor subline. J Urol. 1984;131:1232-6. [ Links ]
14. Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature. 1980;283:139-46. [ Links ]
15. LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499-506. [ Links ]
16. Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822-6. [ Links ]
17. Nelson PS, Clegg N, Eroglu B, Hawkins V, Bumgarner R, Smith T, et al. The prostate expression database (PEDB): status and enhancements in 2000. Nucleic Acids Res. 2000;28:212-3. [ Links ]
18. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signaling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707-12. [ Links ]
19. Yang X, Chen MW, Terry S, Vacherot F, Bemis DL, Capodice J, et al.Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene. 2006;25:3436-44. [ Links ]
20. Hellawell GO, Brewster SF. Growth factors and their receptors in prostate cancer. BJU Int. 2002;89:230-40. [ Links ]
21. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309-25. [ Links ]
22. Hurle RA, Davies G, Parr C, Mason MD, Jenkins SA, Kynaston HG et al. Hepatocyte growth factor/scatter factor and prostate cancer: a review. Histol Histopathol. 2005;20:1339-49. [ Links ]
23. Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41-59. [ Links ]
24. Kim SJ, Johnson M, Koterba K, Herynk MH, Uehara H, Gallick GE. Reduced c-Met expression by an adenovirus expressing a c-Met ribozyme inhibits tumorigenic growth and lymph node metastases of PC3-LN4 prostate tumor cells in an orthotopic nude mouse model. Clin Cancer Res. 2003;9:5161-70. [ Links ]
25. Parr C, Davies G, Nakamura T, Matsumoto K, Mason MD, Jiang WG. The HGF/SF-induced phosphorylation of paxillin, matrix adhesion, and invasion of prostate cancer cells were suppressed by NK4, an HGF/SF variant. Biochem Biophys Res Commun. 2001;285:1330-7. [ Links ]
26. Beviglia L, Kramer RH. HGF induces FAK activation and integrin-mediated adhesion in MTLn3 breast carcinoma cells. Int J Cancer. 1999;83:640-9. [ Links ]
27. Spence HJ, Johnston I, Ewart K, Buchanan SJ, Fitzgerald U, Ozanne BW. Krp1, a novel kelch related protein that is involved in pseudopod elongation in transformed cells. Oncogene. 2000;19:1266-76. [ Links ]
28. van t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530-6. [ Links ]
29. Amatschek S, Koenig U, Auer H, Steinlein P, Pacher M, Gruenfelder A, et al.Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res 2004;64:844-56. [ Links ]
30. Borczuk AC, Shah L, Pearson GD, Walter KL, Wang L, Austin JH, et al. Molecular signatures in biopsy specimens of lung cancer. Am J Respir Crit Care Med. 2004;170:167-74. [ Links ]
31. Burton-Wurster N, Liu W, Matthews GL, Lust G, Roughley PJ, Glant TT, et al. TGF beta 1 and biglycan, decorin, and fibromodulin metabolism in canine cartilage. Osteoarthr Cartil. 2003;11:167-76. [ Links ]
32. Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, et al. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302:527-34. [ Links ]
33. Schostak M, Krause H, Miller K, Schrader M, Weikert S, Christoph F, et al.Quantitative real-time RT-PCR of CD24 mRNA in the detection of prostate cancer. BMC Urol. 2006;6:7. [ Links ]
34. Kristiansen G, Pilarsky C, Pervan J, Sturzebecher B, Stephan C, Jung K, et al.CD24 expression is a significant predictor of PSA relapse and poor prognosis in low grade or organ confined prostate cancer. Prostate. 2004;58:183-92. [ Links ]
35. Buchholz M, Biebl A, Neesse A, Wagner M, Iwamura T, Leder G, et al. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 2003;63:4945-51. [ Links ]
36. Chen LM, Zhang X, Chai KX. Regulation of prostasin expression and function in the prostate. Prostate. 2004;59:1-12. [ Links ]
37. Tahir SA, Yang G, Ebara S, Timme TL, Satoh T, Li L, et al. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res. 2001;61:3882-5. [ Links ]
38. Thompson TC, Timme TL, Li L, Goltsov A. Caveolin-1, a metastasis-related gene that promotes cell survival in prostate cancer. Apoptosis 1999;4:233-7. [ Links ]
39. Inoue G, Horiike N, Onji M. The CD81 expression in liver in hepatocellular carcinoma. Int J Mol Med. 2001;7:67-71. [ Links ]
40. Bienstock RJ, Barrett JC. KAI1, a prostate metastasis suppressor: prediction of solvated structure and interactions with binding partners; integrins, cadherins, and cell-surface receptor proteins. Mol Carcinog. 2001;32:139-53. [ Links ]

