










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.27 no.2 Bogotá Apr./June 2007
Papel de los receptores tipo toll en las infecciones virales: el VIH-1 como modelo
Juan Carlos Hernández, Carlos Julio Montoya, Silvio Urcuqui-Inchima
Grupo de Inmunovirología, Biogénesis, Universidad de Antioquia, Medellín, Colombia
Recibido: 23/11/06; aceptado: 12/03/07
Los receptores tipo toll son un componente esencial de la respuesta inmune innata y adaptativa, pues se encargan del reconocimiento de los diferentes agentes patógenos y desencadenan respuestas dirigidas a eliminarlos y a desarrollar memoria inmunológica. Durante las infecciones virales se activan diferentes receptores tipo toll que, generalmente, inducen una respuesta inmune protectora pero, también, pueden hacer parte de los mecanismos patogénicos del virus. Una de las infecciones virales en la que los receptores tipo toll participan de esta respuesta dual, es la infección por el VIH-1, en la cual varios de estos receptores se activan para desarrollar respuestas antivirales dirigidas por los interferones tipo 1; pero, la replicación y la diseminación del virus también se favorecen por las señales derivadas de la estimulación de dichos receptores, en particular, por las infecciones asociadas con microorganismos oportunistas, lo cual favorece la progresión de la infección por el VIH-1. Un entendimiento integral del comportamiento de estos receptores durante las infecciones virales, permitirá diseñar estrategias profilácticas o terapéuticas basadas en la modulación de su expresión y función, en particular, utilizando agonistas de estos receptores que sean eficaces en la lucha por el control de las infecciones virales.
Palabras clave: receptores toll-like /inmunología, virosis, VIH-1, infecciones oportunistas, inmunoterapia, inmunidad innata.
The role of toll-like receptors in viral infections: HIV-1 as a model
The toll-like receptors are an essential component of the innate and adaptive immune response. They are responsible for the recognition of different pathogens agents and trigger responses directed at eliminating the pathogens as well as the development of immunological long-term memory. During viral infections, several different toll-like receptors are activated. These generally induce a protective immune response, but at the same time, can also be part of the pathogenic mechanisms of the viral infection. One of the viral infections in which toll-like receptors participate is the HIV-1 infection. Here, several receptors are activated to develop antiviral responses mediated by interferon type I; however virus replication and spreading dissemination are also favoured by signals derived from stimulation of the toll-like receptors. Individuals co-infected with opportunistic microorganisms are particularly affected, promoting the progression of HIV-1 infection. An integral understanding of the behavior of toll-like receptors during viral infections will allow the design of prophylactic and/or therapeutic strategies, based on the modulation of the expression and function of these receptors. Agonists of these receptors can be used effectively to control these viral infections.
Key words: toll-like receptors/immunology, virus diseases, HIV-1, opportunistic infections, immunotherapy, immunity, natural.
Los receptores tipo toll (toll-like receptors, TLR) se identificaron en el humano con base en la homología con los receptores toll descritos en la mosca de la fruta, Drosophila melanogaster (1). Estructuralmente, los TLR presentan un dominio de unión a los ligandos constituido por repeticiones ricas en leucina (Leu), que interactúa directamente con los componentes microbianos (
figura 1 ), un dominio transmembrana y un dominio citoplas-mático muy conservado, responsable del inicio de la señalización intracelular (2).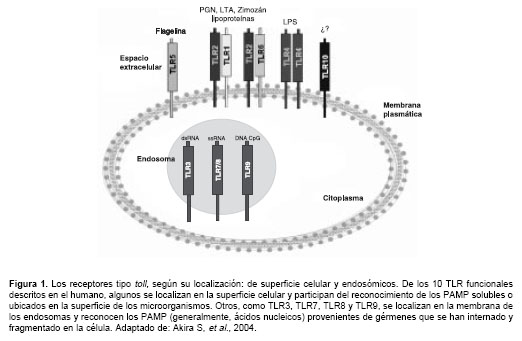
Los TLR hacen parte de los receptores para el reconocimiento de patrones moleculares asocia-dos a patógenos (pathogen recognition receptors, PRR), presentes tanto en células involucradas en la respuesta inmune como en células no inmunes; sus ligandos son los patrones moleculares asociados a patógenos (pathogen associated molecular patterns, PAMP), moléculas muy conservadas estructural y evolutivamente y que son vitales para la supervivencia de los microorganismos, tales como el lipopolisacárido (LPS), el zimosán, el peptidoglicano y los ácidos nucleicos (3).
Aunque en otras especies se han encontrado múltiples TLR, en el ser humano se han descrito 10 TLR funcionales que reconocen una amplia gama de PAMP provenientes de diversos microorganismos (
figura 1 y cuadro 1 ). Estos TLR activan diferentes vías de señalización intracelular a partir de su dominio citoplásmico, el cual también se encuentra en la región intracelular del receptor de la interleucina 1 y se conoce como dominio TIR (dominio del receptor de interleucina 1/ toll); este dominio contiene múltiples repeticiones de Leu e interactúa con cuatro proteínas adaptadoras diferentes, según el TLR: proteína de diferen-ciación mieloide de 88 kd(MyD88), proteína adaptadora del dominio TIR (TIRAP), proteínas adaptadoras asociadas al dominio TIR inductoras de interferón-b (TRIF) y la molécula adaptadora relacionada con TRIF (TRAM) (4,5).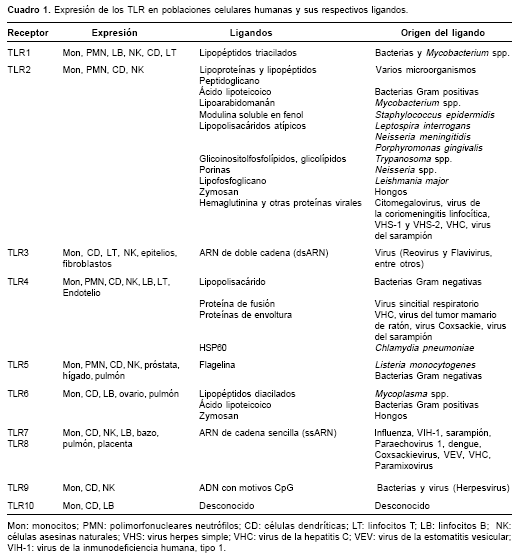
Las vías de señalización inducidas por los TLR activan las cinasas de la familia MAP y factores de transcripción, como NF-kappaB (NF-
kB), AP-1, CREB/ATF, NF-IL6 y IRF, que regulan la expresión de genes cuyos productos actúan tanto en la respuesta inmune innata (monocitos/macrófagos, células dendríticas y neutrófilos, principalmente), como en la inmunidad adaptativa (6,7). En el primer caso, los genes inducidos más importantes son los que codifican para las citocinas proinflamatorias, como el factor de necrosis tumoral a (FNT-a) y las interleucinas 1 (IL-1), 6, 8 y 18, los interferones a y b(IFN-a y b), las quimiocinas, los leucotrienos y las prosta-glandinas. En cuanto a la inmunidad adaptativa, las señales que dependen de los TLR inducen la expresión de IL-12, moléculas del HLA clase II y moléculas coestimuladoras como CD40, CD80 y CD86, proteínas indispensables para una adecuada presentación antigénica (4,7). La expresión de dichos genes depende de la célula que es estimulada, del tipo de estímulo, del tipo de TLR, de la proteína adaptadora y del micro-ambiente tisular.La activación por los TLR contribuye al recono-cimiento y eliminación de los microorganismos; además, constituye un puente entre la inmunidad innata y la adaptativa pues promueve la presentación antigénica y el establecimiento de un patrón específico de secreción de citocinas de una respuesta contra antígenos intracelulares (patrón Th1) o extracelulares (patrón Th2) (7). Por ejemplo, la estimulación del TLR9 expresado por células dendríticas plasmacitoides (pDC) induce la producción de IFNs
ay by de FNT-a, moléculas que activan las células iNKT para que produzcan otras citocinas, como el IFN-g, que contribuyen a madurar y activar las células dendríticas mieloides (mDC) para que realicen una adecuada presenta-ción de antígenos a los linfocitos T vírgenes (8).Aunque los TLR se describieron inicialmente como receptores extracelulares (1), los receptores TLR3, TLR7, TLR8 y TLR9 se expresan en comparti-mientos intracelulares (endosomas,
figura 1 ), donde reconocen ácidos nucleicos principalmente de origen viral, como ARN de doble cadena (dsARN), ARN de cadena sencilla (ssARN) y ADN no metilado con motivos CpG (9). Al alinear la secuencia de aminoácidos de los 10 TLR, se observa que tres de los cuatro TLR que reconocen ácidos nucleicos (TLR7, TLR8 y TRL9) presentan una mayor homología, que está entre el 33% y el 40%. Este alineamiento permitió definir que el TLR3 es muy diferente, excepto en una región rica en Leu que se encuentra en el extremo N-terminal de todos los TLR y que es muy conservada. Las regiones ricas en Leu están implicadas en interacciones proteína-proteína y unen proteínas relacionadas con la activación de las diferentes vías de señalización.El dsARN, un componente intermediario del ciclo replicativo de la mayoría de los virus, es uno de los responsables de activar la producción de IFN
ay b, proteínas que inducen la expresión de otros genes que median la respuesta innata ante las infecciones virales, como la proteína cinasa dependiente de dsARN (PKR) que inhibe la síntesis de proteínas virales. La expresión de los IFN ay binducida por el dsARN puede ser dependiente de señales mediadas por el TLR3 o independiente de ellas (10).El dominio del TLR3 que se une al dsARN está compuesto por 23 repeticiones ricas en Leu (dominio LRR, entre los aminoácidos 250 y 596) (11); como se puede apreciar en la
figura 2 , en la secuencia de aminoácidos del IFN-b (número de acceso: NP_002167) existe igualmente una secuencia de 28 Leu que se alinea perfectamente con el dominio LRR del TLR3 (número de acceso: NP_003256). La presencia de un dominio tipo LRR en IFNb sugiere que podría unir dsARN; sin embargo, los alcances de la homología entre el TLR3 y el IFN-b, y de la posible unión entre el dsARA y el IFN-b, en caso de existir, no se prevé por el momento.
El papel de los TLR en las infecciones virales
La relación entre los TLR y los virus se fundamenta en tres aspectos: i) los TLR son estimulados por moléculas derivadas de los virus (proteínas y ácidos nucleicos), activando vías de señalización que inducen la expresión de genes cuyos productos median la respuesta inmune antiviral, en particular, los IFN ay b; ii) las señales que dependen de los TLR median algunos efectos inmunopatogénicos, como la hiperactivación inmunológica, la apoptosis y un estado pro-inflamatorio crónico, entre otros; iii) algunas estrategias terapéuticas o profilácticas contra los virus se basan en la estimulación de los TLR mediante los respectivos agonistas.
De los 10 TLR descritos en el humano, 6 son activados por componentes virales; TLR2 y TLR4 por proteínas, mientras que TLR3, TLR7, TLR8 y TLR9 se activan por ácidos nucleicos ( cuadro 1 ) (12).
TLR2
Este receptor reconoce una amplia gama de productos microbianos. El TLR2, generalmente, funciona como un heterodímero, bien con TLR1 o con TLR6, para formar complejos funcionalmente activos con especificidad para lipopéptidos diacilados o triacilados provenientes de algunas bacterias Gram positivas (13,14).
Las señales que dependen del TLR2 pueden ser activadas en respuesta a infecciones virales (12); el virus de la coriomeningitis linfocítica, que causa desde síntomas gripales hasta cuadros graves de encefalomielitis, desencadena una respuesta inflamatoria que depende de TLR2 y MyD88, con secreción de citocinas y quimiocinas inflamatorias (IL-6, IL-8, MCP-1) antes del establecimiento de una respuesta inmune específica (15).
La hemaglutinina del virus del sarampión se une al TLR2 y tiene un efecto dual: por un lado, activa rápidamente la respuesta inmune e induce la expresión de citocinas inflamatorias como la IL-6, y, al mismo tiempo, induce la expresión de la molécula CD150 en los monocitos, molécula que es un receptor para todas las cepas del sarampión, con lo cual facilita la diseminación del virus y contribuye con la patogenia viral (16).
El citomegalovirus (CMV), un patógeno oportunista ubicuo de la familia Herpesviridae, interactúa con el TLR2induciendo la expresión de citocinas proinflamatorias mediante la activación del factor de transcripción NF-kB (17). En el modelo de ratón, las células NK son responsables de controlar la infección por el CMV de ratón (mCMV); Szomolanyi-Tsuda y colaboradores demostraron que el TLR2 juega un papel fundamental en dicho proceso (18).
También se ha descrito que el virus varicela-zóster (VVZ) activa la expresión de citocinas proinflamatorias en una vía dependiente NF-kB y TLR2 (19). Otros miembros de la familia Herpesviridae , así como el virus de la hepatitis C (VHC), inducen la expresión de IL-6, IL-8 y FNT-a mediante la estimulación del TLR2 (20,21).
TLR3
Este fue el primer TLR descrito con capacidad de reconocimiento de los virus (22). El TLR3 forma homodímeros activos que se unen al dsARN, activando vías de señalización mediadas por el NF-kB y el factor 3 regulador de interferón (IRF3), los cuales inducen una respuesta antiviral mediada por los IFN ay b; además, estimulan la producción de las citocinas IL-12, IL-6, y FNT-a (5,22).
Un análogo sintético del dsARN, el ácido poli-inosínico/policitidílico o poli(I:C), presenta una actividad inmunoestimuladora similar a la del dsARN en una vía que depende del TLR3. Otros estudios has demostrado que la viperina/cig5, inducida por la estimulación del TLR3, también contribuye en la respuesta antiviral en los astrocitos (23). Así, la respuesta antiviral mediada por el TLR3 luego de la unión al dsARN aparentemente depende del tipo celular; en fibroblastos y células dendríticas, la expresión de los IFN ay b es mediada por la activación de RIG-1, una ARN helicasa, mientras que en las pDC la estimulación del TLR3 induce la activación de IRF5 e IRF7 conduciendo a la producción de grandes cantidades de IFN-a (24,25).
Se han descrito varios virus con capacidad de activar las vías de señalización que dependen de la estimulación del TLR3, como el LMCV, mCMV, el virus de la estomatitis vesicular (VEV) y los reovirus (26). Las células epiteliales del intestino expresan el TLR3 y la estimulación con dsARN de rotavirus, induce no sólo la señalización intracelular (activación de IkB y MAPK ERK1/2), sino también, la producción de citocinas (IL-6, IFN-b y TNF-a) y la apoptosis (27); se observaron resultados similares en células mononucleares de sangre periférica de pacientes infectados con rotavirus (28); esto sugiere el papel protagónico del TLR3 en la patogénesis viral.
TLR4
A diferencia del TLR2, el TLR4 es estimulado por PAMP provenientes de bacterias Gram negativas, como el LPS; también es activado por ligandos endógenos como las b-defensinas, el fibrinógeno, la fibronectina y algunas proteínas de choque térmico (4). En el caso de los virus, el TLR4 es estimulado por la proteína de fusión del virus sincitial respiratorio (VSR) (29) y por lipopéptidos derivados del VHC (30); los neutrófilos de un grupo de pacientes con infección crónica por el VHC presentaron un incremento en la expresión de TLR2 y TLR4, lo que sugiere la existencia de una correlación entre el nivel de expresión de estos receptores, las características del daño de los hepatocitos y la intensidad de la necrosis (31).
En macrófagos humanos, se ha descrito que la proteína p30 del virus linfotrópico T humano tipo 1 (HTLV-1) interfiere con la vía de señalización inducida por TLR4, modulando la liberación de citocinas pro y antiinflamatorias (32). El virus del tumor mamario múrido (MMTV) estimula el TLR4, en forma independiente de la adherencia y fusión del virus con la célula (33), y se ha sugerido que la interacción entre MMTV y TLR4 es el mecanismo mediante el cual este virus activa los linfocitos B. Los Coxsackie virus también se unen y estimulan el TLR4 (34), interacción que incrementa la miocarditis aguda mediante la expresión de citocinas proinflamatorias en el corazón (35). La hemaglutinina del virus del sarampión se une a los receptores CD46 y SLAM para ingresar a las células blanco. Se ha reportado en células dendríticas de ratón transgénicas para el receptor SLAM humano, que el virus del sarampión inhibe selectivamente la síntesis de IL-12, pero no la de IL-6 y FNT-a (36), alterando la activación de las células presentadoras de antígeno y la inducción de una respuesta adaptativa; dicho efecto depende de la estimulación específica del TLR4 por las proteínas del virus del sarampión.
TLR7 y TLR8
Estos dos receptores están muy relacionados, pues ambos se localizan en el mismo compartimiento celular (vesículas intracelulares y endosomas) y son activados naturalmente por el mismo ligando, el ssARN (22,37,38). Estos TLR también se unen a algunos compuestos sintéticos de la familia de las imidazoquinolinas, que tienen actividad antiviral, antitumoral o ambas, como el R-848.
El TLR7 y el TLR8 son estimulados por ssARN proveniente de diferentes virus; recientemente se demostró que la activación en los endosomas del TLR7 por los virus del dengue (VD) y de influenza estaba relacionada con la fusión y con la pérdida de la cápside del genoma viral (39), lo cual indica la importancia de la liberación de ssARN para su interacción con los TLR.
En las pDC, el TLR7 es estimulado por el ssARN del VEV y de influenza, lo que conlleva a una expresión exagerada de moléculas coestimuladoras y de citocinas (40). Otros estudios han demostrado que el ssARN del virus de la inmunodeficiencia humano tipo 1 (VIH-1) activa TLR7 y TLR8 y que la activación de estos TLR con el agonista R-848 genera un potente efecto anti-VIH-1 (41). Además, se ha demostrado in vitro que el VIH-1 activa las pDC, bien sea por la interacción entre la proteína de envoltura viral gp120 y el CD4, o mediante el ssARN liberado en el citoplasma y que se une al TLR7 endosómico (42).
Estas evidencias indican que las señales que dependen de TLR7 y TLR8 pueden contribuir al control de la infección por el VIH-1. El virus de influenza y el parechovirus 1 humano, un picornavirus ssARN productor de miocarditis y encefalitis, también estimulan las señales mediadas por TLR7 y TLR8 (38,40,43). En ratas, la administración de agonistas de TLR7 y TLR8 protege de los efectos nocivos derivados de la infección con el virus de influenza (44).
TLR9
Este receptor reconoce ADN bacteriano o viral hipometilado y rico en motivos CpG; en los virus, este PAMP se encuentra preferentemente en miembros de la familia Herpesviridae, principal-mente virus Herpes simplex 1 y 2 (VHS-1 y –2) (20,45). El TLR9 también se une a compuestos sintéticos constituidos por oligodeoxinucleótidos no metilados con motivos CpG (CpG ODN) (9). En las células dendríticas, los VHS son reconocidos tanto por el TLR2 como por el TLR9, y entre estos dos receptores dirigen una respuesta inmune mediada por la síntesis de IL-6 e IL-12 (46).
En el modelo de ratón, la infección corneal por VHS-1 incrementa la expresión de las quimiocinas CXCL9 y CXCL10 mediante la estimulación del TLR9 (47). Durante la infección primaria por el VHS-2, la expresión de los IFN ay b que depende de la estimulación del TLR9 es fundamental para controlar la infección (48). También en el modelo de ratón, la administración intranasal de baculovirus indujo una respuesta inmune mediada por el TLR9 y dependiente de IFN-a, la cual protegió de la infección letal por influenza (49).
Los TLR en la infección por el VIH-1
El VIH-1 es el agente causal del síndrome de inmunodeficiencia adquirida (sida), una de las principales causa de muerte en el mundo contemporáneo: aproximadamente, 40 millones de personas están actualmente infectadas con el VIH-1 y, desde su aparición, esta infección ha causado más de 25 millones de defunciones (50). Aunque la introducción de la terapia HAART (highly active anti-retroviral therapy) ha disminuido significa-tivamente la morbimortalidad asociada a la infección por el VIH-1 y el sida, esta terapia se asocia con la aparición de cepas virales resistentes y con reacciones adversas frecuentes, lo que hace necesario desarrollar nuevas estra-tegias de tratamiento, como la inmunoterapia.
Muy pocos estudios realizados in vivo han evaluado la expresión de los TLR en células de los individuos infectados con el VIH-1. Los monocitos de estos pacientes presentan mayores niveles de expresión del TLR2 que los de controles sanos (51); además, la secreción de citocinas como el FNT-a es mayor por los monocitos de pacientes positivos para VIH-1 y aumenta cuando son estimulados con la proteína gp120 de VIH-1. Estos resultados sugieren que puede existir una regulación de la expresión o la función (inducción de la secreción de citocinas proinflamatorias) de los TLR en las células de los individuos infectados por VIH-1 que, posiblemente, depende de las proteínas virales, como gp120, y que el aumento en la expresión/función de los TLR tenga un papel potencial en la activación crónica del sistema inmune y la replicación de VIH-1.
En una evaluación preliminar sobre el nivel de expresión de TLR2 y TLR4 en diferentes subpoblaciones de leucocitos provenientes de pacientes infectados con el VIH-1, hemos observado una disminución en el porcentaje de mDC y pDC que expresan estos TLR. Sin embargo, la intensidad de expresión de esos TLR en las mDC y pDC positivas (número de moléculas por célula), es mucho mayor en los individuos infectados con VIH-1 (resultados sin publicar). Ambos hallazgos son más notorios en los pacientes con replicación viral activa (carga viral plasmática >400 copias de ARN viral/ml) o en ausencia de tratamiento antirretroviral.
Estos resultados, aunque preliminares, sugieren que el VIH-1 o sus productos podrían modular la expresión de los TLR en las células de los sujetos infectados, lo que puede constituirse en otro elemento fisiopatológico pues, ante una mayor expresión de los TLR, tanto las coinfecciones como las infecciones oportunistas tendrían una mayor probabilidad de estimular la replicación del genoma del VIH-1, al activar la señalización de los TLR y el factor de trascripción NF-kB.
En la infección por el VIH-1 y el CMV, la estimulación de las células dendríticas con los ligandos de TLR3, TLR7, TLR8 y TLR9 favoreció la respuesta inmune celular innata y adaptativa contra estos virus (52). Sin embargo, la activación del sistema inmune por medio de estos receptores también puede conducir, directa o indirectamente, a un aumento de la replicación viral y agravamiento de la evolución de estas infecciones.
Al respecto, se ha demostrado que tanto las mDC como las pDC son blanco de la infección por el VIH-1; gracias a su patrón particular de expresión de TLR (las pDC expresan TLR7 y TLR9, mientras que las mDC expresan TLR1, TLR2, TLR3, TLR4, TLR8); ellas reconocen tanto ácidos nucleicos como proteínas de origen viral (53,54) y la activación desencadenada de ese reconocimiento puede explicar el papel fisiopatológico que cumplen las coinfecciones virales durante la infección por el VIH-1. En particular, el genoma del VIH-1 posee en sus extremos 5 y 3 las secuencias LTR (long terminal repeats ), que contienen múltiples secuencias promotoras de la transcripción que son unidas y estimuladas por diversos factores de transcripción normales en las células, tales como NF-kB y Sp1. Se ha demostrado que la activación de los TLR favorece un aumento en la transcripción del VIH-1 que depende del factor NF-kB (55,56).
La activación de la respuesta inmune por diferentes agentes infecciosos se ha propuesto como uno de los mecanismos que influyen en la progresión de la enfermedad en los individuos infectados con el VIH-1 (55). Esto se ha evidenciado en algunas coinfecciones con bacterias, como Mycobacterium tuberculosis (57)y Neisseria gonorrhoeae (58), que inducen la producción de citocinas proinflamatorias por medio de la interacción del factor NF-B con el LTR del VIH-1 (55, 56).
Equils y colaboradores describieron la inducción de la replicación del VIH-1 por la estimulación con LPS en una forma dependiente de TLR4 y NF-kB (59). La activación del LTR del VIH-1, dependiente de NF-kB, también se observó estimulando simultáneamente varios TLR con sus respectivos agonistas (LPS, CpG ODN, factores solubles del M. tuberculosis y modulina de Staphylococcus epidermidis) (60). También, las proteínas asociadas a los lípidos de la membrana de Mycoplasma spp. activan la replicación de VIH-1 a través de la estimulación de TLR1, TLR2 y TLR6 (61).
La inducción de la replicación viral mediada por la estimulación de los TLR, también se ha descrito en mastocitos infectados latentemente por el VIH-1 (62). Algunos factores endógenos se han relacionado con este proceso, como el factor estimulante de colonias de granulocitos y monocitos (GM-CSF) que, combinado con el LPS, aumenta 100 veces la producción de partículas virales del VIH-1 en monocitos, en comparación con células estimuladas sólo con LPS (63). Esto se puede explicar porque el GM-CSF regula positivamente la expresión del TLR4, receptor de LPS.
También se ha descrito que el ssARN del VIH-1, rico en guanosina y uridina, estimula los macrófagos y las células dendríticas vía TLR7 y TLR8, induciendo la producción de IFN-a y otras citocinas proinflamatorias (43); estas citocinas pueden promover la replicación de VIH-1, como se ha sido descrito antes (56,57). En contraste, la estimulación del TLR3 con poli(I:C) induce la expresión de la proteína Viperin/Cig5 en los astrocitos infectados con el VIH-1 e inhibe la replicación del genoma viral (23); la Viperin/Cig5 también ha presentado actividad antiviral en los modelos de infección por el VHC y el CMV.
Existe evidencia que indica que la inducción de la tolerancia con exposición repetida a los agonistas de TLR2, TLR4 y TLR9, se acompaña de una disminución en la transactivación del LTR del VIH-1.
Lo anterior se demostró en monocitos humanos que expresaban el gen de la luciferasa bajo el control del promotor LTR del VIH-1 y macrófagos humanos infectados con VIH-1 (64). Sin embargo, se observó el fenómeno contrario en macrófagos de un modelo de ratón transgénico para el genoma completo de VIH-1, en el cual se presentó una disminución en la intensidad de la respuesta inmune, con menor activación del factor NF-kB y menor producción de las citocinas proinflamatorias, pero, con un aumento en la replicación del VIH-1 evidenciada por una mayor producción del antígeno p24 (65); esto se puede explicar por las limitaciones del modelo de ratón, para estudiar los efectos de la activación de los TLR sobre la replicación del VIH-1 y su patogénesis, ya que no es un hospedero natural de este virus; por esto, se requieren alternativas experimentales que permitan una mejor comprensión de la relación entre la infección por el VIH-1 y los TLR.
Los TLR en la patogénesis de otros virus
Varias evidencias sugieren que la estimulación de la respuesta inmune mediada por los TLR puede generar una activación inmunológica "peligrosa" que, en lugar de proteger al individuo, contribuye con la patogénesis de las enfermedades infecciosas.
La barrera hematoencefálica representa un obstáculo fisiológico para la entrada de microor-ganismos al sistema nervioso central (SNC). Se ha demostrado en ratones que el virus del Nilo occidental induce, mediante las señales derivadas del TLR3, un estado inflamatorio sistémico caracterizado por la hipersecreción de FNT-a e IL-6 o "tormenta de citocinas", lo que altera la permeabilidad y la integridad de la BHE y permite la infección del SNC por el virus del Nilo occidental (66); en consecuencia, los ratones deficientes en TLR3 son resistentes a la infección letal por el virus del Nilo occidental. Este fenómeno favorece el ingreso y la infección del tejido cerebral por el virus del Nilo occidental, ocasionando encefalitis letal, e incrementa la patogénesis de esta infección (figura 3 ).
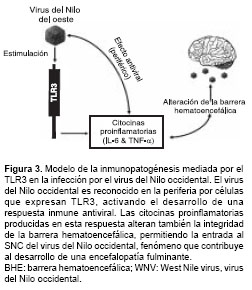
Durante la infección del SNC por el virus de encefalomielitis múrida de Theiler, también se ha observado que los astrocitos producen citocinas (IL-8) y quimiocinas (MCP-1, CCL-2, CXCL-8) luego de la activación mediada por el TLR3 (67); esto contribuye al desarrollo en ratones de una enfermedad desmielinizante similar a la esclerosis múltiple de los humanos. En otro modelo de ratón, la estimulación del TLR2 por el VHS-1 activa la secreción de citocinas, aumentando la seriedad de la encefalitis y la mortalidad (20); por el contrario, los ratones que no expresan TLR2 presentan una baja mortalidad.
Es bien conocido que algunos virus modulan la respuesta inmune como un mecanismo de evasión y de supervivencia (68). Algunos filovirus causantes de fiebres hemorrágicas, como los virus del Ébola y Marburg, disminuyen la expresión de genes relacionados con el sistema inmune, incluso los TLR, fenómeno que les permite escapar de la respuesta inmune y persistir en el organismo (68). Sin embargo, otros virus como el VSR, regulan positivamente la expresión de TLR3 y TLR4, además de que alteran la localización del TLR3 (69,70); este proceso sensibiliza la vía respiratoria del individuo infectado por el VSR, de manera que la exposición adicional a otros patógenos promueve una respuesta inmune exagerada que favorece el daño tisular.
Triantafilou y colaboradores demostraron el papel protagónico del TLR7 y el TLR8 en la respuesta inflamatoria inducida por el Coxsackie virus del grupo B (CVB), agente causal de una cardiomio-patía inflamatoria crónica (37); estos receptores se unen con el ssARN viral en los endosomas e inducen la producción de FNT-
a, IL-6 e IFN-b.El papel de los TLR en la inmunoterapia de las infecciones virales
Considerando el papel central que tienen los TLR en la inducción de la respuesta innata e inflamatoria, y en la generación de la competencia de las células presentadoras de antígenos para el desarrollo de la respuesta inmune adaptativa, la comprensión de los mecanismos moleculares implicados en la activación y la regulación de la expresión de los TLR podría permitir el diseño de estrategias terapéuticas con un gran potencial en enfermedades infecciosas, autoinmunes y neoplásicas.
En este sentido, ya se ha empezado a acumular evidencia sobre la manipulación terapéutica de la estimulación de los TLR con sus agonistas o antagonistas en diferentes modelos de enfermedad; como ejemplo, la apoptosis o muerte celular programada es uno de los mecanismos de regulación de la respuesta inmune, del control de los tumores y de la respuesta antiviral. Recientemente se observó que las células cancerígenas estimuladas con dsARN presentaban aumento en la apoptosis en un mecanismo mediado por la estimulación del TLR3, e independiente de las cinasas activadas por el dsARN (71).
La familia de las imidazoquinolinas comprende varios compuestos agonistas de TLR7 y TLR8 con actividad antiviral; la aplicación tópica de imiquimod y resiquimod se ha utilizado con éxito en el tratamiento del carcinoma de células basales (un tipo de cáncer de piel), de verrugas genitales y otras lesiones epiteliales, generalmente asociadas con infección crónica por el virus del papiloma humano (VPH); estos compuestos inducen, entre otras cosas, apoptosis de las células infectadas con el VPH y de otras células epiteliales con cambios displásicos o neoplásicos (72,73).
También se ha sugerido la inmunoterapia con agonistas de los TLR en la infección causada por el VHC; se ha demostrado que la administración de isatoribine (otro compuesto que estimula selectivamente el TLR7) genera actividad antiviral con pocos efectos secundarios (74). In vivo, se demostró modulación de la replicación del genoma del HBV luego de la administración intravenosa de los ligandos específicos para los receptores TLR3, TLR4, TLR5, TLR7 y TLR9 (75); en todos los casos, el efecto fue mediado por la producción de IFN
a y b.En la mucosa cérvico-vaginal se ha observado inducción de una respuesta antiviral innata tras la administración de agonistas de los TLR y antes de la exposición a los virus. La estimulación desencadenada por los CpG ODN en la mucosa vaginal promueve un estado antiviral que protege contra la infección por el VHS-2, aunque indujo inflamación local (76). Ashkar y colaboradores demostraron que los CpG ODN inhibían la replicación del genoma del VHS-2, pero no la entrada de este virus a la célula; sin embargo, cuando reemplazaron el CpG ODN por dsARN, se observó un incremento significativo en la actividad antiviral, con disminución de la respuesta inflamatoria local (77).
El potencial protector que tiene la aplicación de CpG ODN o de imiquimod en la infección vaginal por el virus de la inmunodeficiencia simiana (SIV) se ha evaluado en un modelo animal; el tratamiento con estos agonistas de los TLR indujo la producción de citocinas proinflamatorias (IL-1 e IL-6) y de moléculas con efecto antiviral (IFN
ay b), pero no protegió contra esta infección. Al contrario, esta estrategia potenció la infección por el SIV al reclutar en la mucosa vaginal diferentes células mononucleares, como los linfocitos T CD4+ y las células dendríticas, lo que puede facilitar la transmisión de la infección (78).En estudios clínicos de fase II, Cooper y colaboradores evaluaron el efecto inmuno-modulador de los CpG ODN como adyuvantes para vacunas contra el HBV o el virus de la influenza, tanto en individuos sanos como en pacientes infectados con el VIH-1. Los resultados demostraron que los CpG ODN son seguros en humanos; en los pacientes positivos para VIH-1 la administración de esos productos no se asoció con aumento en la carga viral ni con otros efectos secundarios potenciales (79-81).
Recientemente, se demostró en un modelo de ratón que la administración intranasal de la proteína de matriz del VIH-1 (p17) con un agonista de los receptores TLR2 y TLR6 como adyuvante (un lipopéptido derivado de Mycoplasma spp.), estimuló una fuerte respuesta inmune humoral (evidenciada por anticuerpos IgG e IgA anti-p17), así como una buena respuesta celular (determinada por la respuesta proliferativa de los linfocitos T ante el reto con p17 y por la producción de IFN-
g), tanto a nivel sistémico como de mucosas (82).Estos resultados sugieren que las vacunas basadas en la coadministración de agonistas de los TLR como adyuvantes, en combinación con proteínas estructurales o reguladoras del VIH-1, pueden ser candidatas atractivas para el diseño de una intervención que active el sistema inmune, para el uso profiláctico o terapéutico en la infección por el VIH-1.
Conclusiones
La relación entre los TLR y las infecciones virales apenas se está empezando a comprender; sin embargo, ya existe un número importante de publicaciones que permiten entender la magnitud del papel de los TLR en la respuesta inmune de defensa contra muchas infecciones virales, pero también su participación en la inmunopatogénesis de otras infecciones virales.
La infección por el VIH-1 constituye un buen ejemplo de la función dual que cumplen los TLR en varias infecciones virales: por un lado, contribuyen con el reconocimiento y la eliminación de estos microorganismos pero, al mismo tiempo, inducen la secreción de citocinas que favorecen un estado proinflamatorio crónico, la replicación viral y la diseminación de los viriones. Esas evidencias permiten postular que la modulación de la actividad de los TLR, positiva o negativa, puede utilizarse como una nueva estrategia de inmunoterapia con fines profilácticos o terapéuticos.
Agradecimientos
Los autores agradecen a A. Kumar, G. St. Laurent ( George Washington University), D. Hernández-Verduny y Anne-Lise Haenni (Institut Jacques Monod), por la lectura crítica del texto y por sus comentarios y sugerencias; a la Facultad de Medicina de la Universidad de Antioquia, a la Sede de Investigación Universitaria-SIU y a Colciencias, por todo el apoyo brindado.
Conflictos de interés
Los autores manifiestan no incurrir en ningún conflicto de interés mediante la realización de esta revisión.
Financiación
Este trabajo fue financiado por Colciencias a través del proyecto 1115-04-16402/RC 269-2004.
Correspondencia:
Silvio Urcuqui-Inchima, Sede de Investigación Universitaria, Universidad de Antioquia, Calle 62 # 52 - 59, Laboratorio 532, AA 1226. Medellín, Colombia.
Teléfono: (4) 210 64 83, fax: (4) 210 64 81
silviourcuqui@gmail.com
Referencias
1. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394-7. [ Links ]
2. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197-216. [ Links ]
3. Takeda K, Akira S. Microbial recognition by toll-like receptors. J Dermatol Sci. 2004;34:73-82. [ Links ]
4. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499-511. [ Links ]
5. West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409-37. [ Links ]
6. Zhang G, Ghosh S. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107:13-9. [ Links ]
7. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335-76. [ Links ]
8. Montoya CJ, Jie HB, Al-Harthi L, Mulder C, Patino PJ, Rugeles MT, et al. Activation of plasmacytoid dendritic cells with TLR9 agonists initiates invariant NKT cell-mediated cross-talk with myeloid dendritic cells. J Immunol. 2006;177:1028-39. [ Links ]
9. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740-5. [ Links ]
10. Matsumoto M, Funami K, Oshiumi H, Seya T. Toll-like receptor 3: a link between toll-like receptor, interferon and viruses. Microbiol Immunol. 2004;48:147-54. [ Links ]
11. Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, et al. The molecular structure of the toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10976-80. [ Links ]
12. Finberg RW, Kurt-Jones EA. Viruses and toll-like receptors. Microbes Infect. 2004;6:1356-60. [ Links ]
13. Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766-71. [ Links ]
14. Wyllie DH, Kiss-Toth E, Visintin A, Smith SC, Boussouf S, Segal DM, et al. Evidence for an accessory protein function for toll-like receptor 1 in anti-bacterial responses. J Immunol. 2000;165:7125-32. [ Links ]
15. Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, et al. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822-30. [ Links ]
16. Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. 2002;76:8729-36. [ Links ]
17. Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and toll-like receptor 2. J Virol. 2003;77:4588-96. [ Links ]
18. Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. 2006;80:4286-91. [ Links ]
19. Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via toll-like receptor 2. J Virol. 2005;79:12658-66. [ Links ]
20. Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, et al. Herpes simplex virus 1 interaction with toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315-20. [ Links ]
21. Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513-24. [ Links ]
22. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732-8. [ Links ]
23. Rivieccio MA, Suh HS, Zhao Y, Zhao ML, Chin KC, Lee SC, et al. TLR3 ligation activates an antiviral response in human fetal astrocytes: a role for viperin/cig5. J Immunol. 2006;177:4735-41. [ Links ]
24. Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19-28. [ Links ]
25. Meylan E, Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell. 2006;22:561-9. [ Links ]
26. Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231-8. [ Links ]
27. Sato A, Lizuka M, Nakagomi O, Suzuki M, Horie Y, Konno S, et al. Rotavirus double-stranded RNA induces apoptosis and diminishes wound repair in rat intestinal epithelial cells. J Gastroenterol Hepatol. 2006;21:521-30. [ Links ]
28. Xu J, Yang Y, Sun J, Ding Y, Su L, Shao C, et al. Expression of toll-like receptors and their association with cytokine responses in peripheral blood mononuclear cells of children with acute rotavirus diarrhoea. Clin Exp Immunol, 2006;144:376-81. [ Links ]
29. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398-401. [ Links ]
30. Duesberg U, von dem Bussche A, Kirschning C, Miyake K, Sauerbruch T, Spengler U. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)-core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol Lett. 2002;84:89-95. [ Links ]
31. Wisniewska-Ligier M, Wozniakowska-Gesicka T, Glowacka E, Lewkowicz P, Banasik M, Tchorzewski H. Involvement of innate immunity in the pathogenesis of chronic hepatitis C in children. Scand J Immunol. 2006;64:425-32. [ Links ]
32. Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J Biol Chem. 2006;281:23414-24. [ Links ]
33. Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci USA. 2002;99:2281-6. [ Links ]
34. Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003;170:4731-7. [ Links ]
35. Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15:17-27. [ Links ]
36. Hahm B, Cho JH, Oldstone MB. Measles virus-dendritic cell interaction via SLAM inhibits innate immunity: Selective signaling through TLR4 but not other TLRs mediates suppression of IL-12 synthesis. Virology. 2007;358:251-7. [ Links ]
37. Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005;7:1117-26. [ Links ]
38. Triantafilou K, Vakakis E, Orthopoulos G, Ahmed MA, Schumann C, Lepper PM, et al. TLR8 and TLR7 are involved in the hosts immune response to human parechovirus 1. Eur J Immunol. 2005;35:2416-23. [ Links ]
39. Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J Immunol. 2006;177:7114-21. [ Links ]
40. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598-603. [ Links ]
41. Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888-95. [ Links ]
42. Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265-75. [ Links ]
43. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526-9. [ Links ]
44. Hammerbeck DM, Burleson GR, Schuller CJ, Vasilakos JP, Tomai M, Egging E, et al. Administration of a dual toll-like receptor 7 and toll-like receptor 8 agonist protects against influenza in rats. Antiviral Res. 2007;73:1-11. [ Links ]
45. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513-20. [ Links ]
46. Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci USA. 2006;103:17343-8. [ Links ]
47. Wuest T, Austin BA, Uematsu S, Thapa M, Akira S, Carr DJ. Intact TRL 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J Neuroimmunol. 2006;179:46-52. [ Links ]
48. Svensson A, Bellner L, Magnusson M, Eriksson K. Role of IFN-alpha/beta signaling in the prevention of genital herpes virus type 2 infection. J Reprod Immunol. 2006. [Epub ahead of print]. [ Links ]
49. Abe T, Hemmi H, Miyamoto H, Moriishi K, Tamura S, Takaku H, et al. Involvement of the toll-like receptor 9 signaling pathway in the induction of innate immunity by baculovirus. J Virol. 2005;79:2847-58. [ Links ]
50. UNAIDS U, WHO. AIDS epidemic update December 2006. Geneva: WHO; 2006. [ Links ]
51. Heggelund L, Muller F, Lien E, Yndestad A, Ueland T, Kristiansen KI, et al. Increased expression of toll-like receptor 2 on monocytes in HIV infection: possible roles in inflammation and viral replication. Clin Infect Dis. 2004;39:264-9. [ Links ]
52. Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, et al. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J Immunol. 2003;171:4320-8. [ Links ]
53. Ito T, Wang YH, Liu YJ. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by toll-like receptor (TLR) 7 and TLR9. Springer Semin Immunopathol. 2005;26:221-9. [ Links ]
54. Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863-9. [ Links ]
55. Pomerantz RJ, Feinberg MB, Trono D, Baltimore D. Lipopolysaccharide is a potent monocyte/macrophage-specific stimulator of human immunodeficiency virus type 1 expression. J Exp Med. 1990;172:253-61. [ Links ]
56. Lu YC, Touzjian N, Stenzel M, Dorfman T, Sodroski JG, Haseltine WA. The NF kappa B independent cis-acting sequences in HIV-1 LTR responsive to T-cell activation. J Acquir Immune Defic Syndr. 1991;4:173-7. [ Links ]
57. Poli G, Fauci AS. The effect of cytokines and pharmacologic agents on chronic HIV infection. AIDS Res Hum Retroviruses. 1992;8:191-7. [ Links ]
58. Zhang J, Li G, Bafica A, Pantelic M, Zhang P, Broxmeyer H, et al. Neisseria gonorrhoeae enhances infection of dendritic cells by HIV type 1. J Immunol. 2005;174:7995-8002. [ Links ]
59. Equils O, Faure E, Thomas L, Bulut Y, Trushin S, Arditi M. Bacterial lipopolysaccharide activates HIV long terminal repeat through toll-like receptor 4. J Immunol. 2001;166:2342-7. [ Links ]
60. Equils O, Schito ML, Karahashi H, Madak Z, Yarali A, Michelsen KS, et al. Toll-like receptor 2 (TLR2) and TLR9 signaling results in HIV-long terminal repeat trans-activation and HIV replication in HIV-1 transgenic mouse spleen cells: implications of simultaneous activation of TLRs on HIV replication. J Immunol. 2003;170:5159-64. [ Links ]
61. Shimizu T, Kida Y, Kuwano K. Lipid-associated membrane proteins of Mycoplasma fermentans and M. penetrans activate human immunodeficiency virus long-terminal repeats through toll-like receptors. Immunology. 2004;113:121-9. [ Links ]
62. Sundstrom JB, Little DM, Villinger F, Ellis JE, Ansari AA. Signaling through toll-like receptors triggers HIV-1 replication in latently infected mast cells. J Immunol. 2004;172:4391-401. [ Links ]
63. Osiecki K, Xie L, Zheng JH, Squires R, Pettoello-Mantovani M, Goldstein H. Identification of granulocyte-macrophage colony-stimulating factor and lipopolysaccharide-induced signal transduction pathways that synergize to stimulate HIV type 1 production by monocytes from HIV type 1 transgenic mice. AIDS Res Hum Retroviruses. 2005;21:125-39. [ Links ]
64. Equils O, Salehi KK, Cornataeanu R, Lu D, Singh S, Whittaker K, et al. Repeated lipopolysaccharide (LPS) exposure inhibits HIV replication in primary human macrophages. Microbes Infect. 2006;8:2469-76. [ Links ]
65. Bafica A, Scanga CA, Equils O, Sher A. The induction of toll-like receptor tolerance enhances rather than suppresses HIV-1 gene expression in transgenic mice. J Leukoc Biol. 2004;75:460-6. [ Links ]
66. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366-73. [ Links ]
67. So EY, Kang MH, Kim BS. Induction of chemokine and cytokine genes in astrocytes following infection with Theilers murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia. 2006;53:858-67. [ Links ]
68. Kash JC, Muhlberger E, Carter V, Grosch M, Perwitasari O, Proll SC, et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol. 2006;80:3009-20. [ Links ]
69. Monick MM, Yarovinsky TO, Powers LS, Butler NS, Carter AB, Gudmundsson G, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem. 2003;278:53035-44. [ Links ]
70. Groskreutz DJ, Monick MM, Powers LS, Yarovinsky TO, Look DC, Hunninghake GW. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J Immunol. 2006;176:1733-40. [ Links ]
71. Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894-901. [ Links ]
72. Stockfleth E, Trefzer U, Garcia-Bartels C, Wegner T, Schmook T, Sterry W. The use of toll-like receptor-7 agonist in the treatment of basal cell carcinoma: an overview. Br J Dermatol. 2003;149(Suppl.66):53-6. [ Links ]
73. Meyer T, Nindl I, Schmook T, Ulrich C, Sterry W, Stockfleth E. Induction of apoptosis by toll-like receptor-7 agonist in tissue cultures. Br J Dermatol. 2003;149(Suppl. 66):9-14. [ Links ]
74. Horsmans Y, Berg T, Desager JP, Mueller T, Schott E, Fletcher SP, et al. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology. 2005;42:724-31. [ Links ]
75. Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005;79:7269-72. [ Links ]
76. Ashkar AA, Bauer S, Mitchell WJ, Vieira J, Rosenthal KL. Local delivery of CpG oligodeoxynucleotides induces rapid changes in the genital mucosa and inhibits replication, but not entry, of herpes simplex virus type 2. J Virol. 2003;77:8948-56. [ Links ]
77. Ashkar AA, Yao XD, Gill N, Sajic D, Patrick AJ, Rosenthal KL. Toll-like receptor (TLR)-3, but not TLR4, agonist protects against genital herpes infection in the absence of inflammation seen with CpG DNA. J Infect Dis. 2004;190:1841-9. [ Links ]
78. Wang Y, Abel K, Lantz K, Krieg AM, McChesney MB, Miller CJ. The toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J Virol. 2005;79:14355-70. [ Links ]
79. Cooper CL, Davis HL, Morris ML, Efler SM, Krieg AM, Li Y, et al. Safety and immunogenicity of CPG 7909 injection as an adjuvant to Fluarix influenza vaccine. Vaccine. 2004;22:3136-43. [ Links ]
80. Cooper CL, Davis HL, Morris ML, Efler SM, Adhami MA, Krieg AM, et al. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J Clin Immunol. 2004;24:693-701. [ Links ]
81. Cooper CL, Davis HL, Angel JB, Morris ML, Elfer SM, Seguin I, et al. CPG 7909 adjuvant improves hepatitis B virus vaccine seroprotection in antiretroviral-treated HIV-infected adults. Aids. 2005;19:1473-9. [ Links ]
82. Becker PD, Fiorentini S, Link C, Tosti G, Ebensen T, Caruso A, et al.The HIV-1 matrix protein p17 can be efficiently delivered by intranasal route in mice using the TLR 2/6 agonist MALP-2 as mucosal adjuvant. Vaccine. 2006;24:5269-76. [ Links ]

