










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkBiomédica
versión impresa ISSN 0120-4157versión On-line ISSN 2590-7379
Biomédica v.27 n.3 Bogotá jul./sep. 2007
Evaluación de la producción de
β -amiloide por la mutación E280A en el gen de la presenilina 1Andrés Villegas 1, Mónica M. Castañeda 1, Luis Fernando Arias 3,4, Beatriz Vieco 3,4, Francisco Lopera 1, Gabriel Bedoya 2
1 Grupo de Neurociencias de Antioquia, Universidad de Antioquia, Medellín, Colombia
2
Grupo de Genética Molecular, Universidad de Antioquia, Medellín, Colombia3
Departamento de Patología, Universidad de Antioquia, Medellín, Colombia4
Departamento de Patología, Hospital Universitario San Vicente de Paúl, Medellín, ColombiaRecibido: 03/10/06; aceptado: 14/05/07
Introducción.
La mutación E280A en el gen de presenilina 1 se encuentra asociada al grupo familiar más grande del mundo con enfermedad de Alzheimer. La presenilina 1 es un componente esencial del complejo g-secretasa, responsable de la producción del péptido β -amiloide, considerado clave en la fisiopatogenia de la enfermedad.Objetivo. Determinar si la mutación E280A en presenilina 1 incrementa la producción de b-amiloide, como reflejo de la ganancia de función
g -secretasa.Materiales y métodos. Se evaluaron, por la tinción con rojo congo e inmunohistoquímica, depósitos sistémicos de b-amiloide en tejidos de cadáveres afectados, portadores de la mutación E280A en la presenilina 1 y cadáveres de no afectados. Se cuantificó por la técnica de ELISA el
β -amiloide de 40 y 42 aminoácidos en cultivos de células CHO que expresan la proteína precursora de amiloide, transfectadas con los cADN de presenilina 1 portando las mutaciones M146L, E280A, D E9 y L392V.Resultados. Se encontraron depósitos proteicos en todos los tejidos estudiados y escasos depósitos de
β -amiloide. No se encontró diferencia en la cantidad de depósitos, ni en su localización. No se observó incremento en la producción de β -amiloide en las células transfectadas con los cADN de presenilina 1 con las mutaciones comparadas con los controles.Conclusiones. La mutación E280A en presenilina 1 no aumentó la producción de
β -amiloide en tejidos periféricos no neuronales o en el modelo in vitro, contrario a lo que sucede en una ganancia de función de g -secretasa.Palabras clave: beta amiloide, mutación E280A, presenilina-1, enfermedad de Alzheimer, rojo congo.
Evaluation of amyloid-b by the E280A mutation in presenilin gene
Introduction. The E280A mutation of the presenilin 1 gene has been found to be the most common associate in Alzheimers patients with a family history of this disease. Presenilin 1 is a critical component of the g-secretase complex and plays an essential role in the production of amyloid-
β peptide. This peptide has been strongly associated with the physiopathology of the disease.Objective. The E280A mutation in the presenilin 1 was investigated for increased production of amyloid-
β , as a response to gain in g-secretase function.Materials and methods. Levels of systemic amyloid-
β were measured with congo red staining and immuno-histochemistry of the tissues of affected cadavers, compared with non-affected cadavers. The 40 and 42 amino acid amyloid-β levels were quantified by ELISA assay in CHO cell cultures. The amyloid precursor protein expressed by the cultures was detected by transfection with the cDNAs of presenilin 1 carrying the M146L, E280A, DE9 y L392V mutations.Results. Protein deposits were found in all tissues investigatged, but only a few with
β -amyloid deposition. No differences were observed in the amount or location of amyloid-β between affected and unaffected cadavers. Not increase was noted in the production of amyloid-β from the CHO cells transfected with cDNA from any of the mutations of presenilin 1.Conclusions. The E280A mutation in the presenilin 1 gene was not associated with the increased production of amyloid-
β in non-neuronal peripheral tissues, or in the in vitro model. This is in contrast to the expectation in a g-secretase gain of function.Key words: amyloid beta-protein, mutation/genetics, presenilin-1, Alzheimer disease, congo red.
La enfermedad de Alzheimer fue descrita en 1907 por Alois Alzheimer como una amiloidosis cerebral (1) y años más tarde se describieron grupos familiares afectados por la enfermedad (2). El estudio de estos grupos familiares permitió la identificación de los genes que codifican para las proteínas precursora de amiloide, presenilina 1 y presenilina 2 (3). Una mutación en la presenilina 1 es la transversión de adenina por citosina en el codón 280, que provoca en la proteína un cambio de ácido glutámico por alanina. Esta mutación afecta al grupo familiar más grande del mundo con enfermedad de Alzheimer (4).
La enfermedad de Alzheimer se caracteriza por depósitos amiloideos en el cerebro (placas seniles), cuyo componente principal es el péptido
β amiloide; este péptido es producto del procesamiento de la proteína precursora de amiloide por la g-secretasa (5). La g-secretasa es un complejo aspartil-proteasa conformado por heterodímeros de presenilina, nicastrina, aph-1 y pen-2 que corta proteínas integrales de membrana tipo 1 en la membrana citoplasmática (6-8). El procesamiento de la proteína precursora de amiloide se inicia con la proteólisis por las enzimas a-secretasa o b-secretasa, lo que produce la liberación al exterior de la membrana de fragmentos amino terminales de diferentes tamaños llamados: proteína precursora de amiloide soluble a o b, respectivamente, y deja anclados a la membrana los fragmentos carboxilo terminales llamados de acuerdo con su tamaño, C83 y C99, según si la proteólisis es realizada por a-secretasa o β -secretasa, respectivamente (9).Los polipéptidos C83 y C99 son sustratos para la
g-secretasa, que puede cortarlos en el sitio e (en el borde interno de la membrana citoplasmática) (10) o en el sitio g (porción media dentro de la membrana celular); cuando el sustrato es C83, el corte g genera un pequeño fragmento llamado p3 y, cuando lo hace en C99, se genera el b amiloide que posee un rango de longitud entre 37 a 43 aminoaácidos; en condiciones fisiológicas, el β amiloide 1-40 es el más abundante, seguido por el β amiloide 1-42 (11,12). La presenilina es el centro catalítico de la g-secretasa, que no sólo interviene en la proteólisis de la proteína precursora de amiloide, sino que interviene también en el procesamiento de otras proteínas involucradas en múltiples actividades celulares (13).Se han reportado depósitos de
β amiloide en piel e intestino, en individuos afectados con enfermedad de Alzheimer y en afectados con síndrome de Down (14). Las proteínas precursora de amiloide y presenilina 1 son proteínas de expresión ubicua (15,16) y el β amiloide es producido tanto por células neuronales como no neuronales como producto del metabolismo celular (17), por lo cual se ha pensado que el β amiloide generado en órganos no neuronales podría penetrar al cerebro por vía plasmática atravesando la barrera hematoencefálica; por ello se han realizado mediciones de b amiloide plasmático buscando una relación entre los niveles plasmáticos de b amiloide y los cambios patológicos en cerebros con enfermedad de Alzheimer. Esta relación aún no se ha encontrado, pero continúa siendo un importante foco de investigación en la enfermedad de Alzheimer (18).El incremento en la generación del péptido b amiloide 1-42 por mutaciones en presenilina 1, se ha demostrado, tanto en ratones como en cultivo de células neuroblastoides, y se ha encontrado que sólo unas pocas mutaciones en la presenilina 1 incrementan también el péptido b amiloide 1-40 (11,19,20). El incremento en la producción de
β amiloide 1-42 en los portadores de un alelo mutado de presenilina 1, lleva a pensar en una ganancia de función (o disfunción) del alelo mutante sobre el alelo silvestre, también llamado efecto dominante negativo (21).Estos hallazgos condujeron al grupo de Dennis J. Selkoe a proponer la hipótesis amiloidea (22). Esta hipótesis da un papel central al
β amiloide 1-42 en la génesis de la enfermedad de Alzheimer; también trata de explicar los casos que no son atribuidos a estas mutaciones por alteración en el proceso de degradación de β amiloide que llevan a que predomine el b amiloide 1-42 y, por medio de una cascada desencadenada por la toxicidad del mismo, se genera el fenotipo de demencia característico de la enfermedad (23). Con base en esta hipótesis, se ha propuesto reducir la producción del b amiloide como tratamiento para la enfermedad de Alzheimer (24).Un incremento en la función g-secretasa debe llevar a un aumento en la producción de b amiloide, por lo cual planteamos el objetivo de evaluar si la mutación E280A en la presenilina 1 incrementa la producción de b amiloide (ganancia de función). En el presente trabajo evaluamos la producción de b amiloide en tejidos no neuronales de afectados con enfermedad de Alzheimer familiar, portadores de la mutación E280A, y en cultivos de células CHO que expresan establemente la proteína precursora de amiloide y transfectadas con las mutaciones M146L, E280A, DE9 y L392V en presenilina 1.
Materiales y métodos
Población y tejidos estudiados
Los cadáveres de seis individuos afectados con enfermedad de Alzheimer familiar, portadores de la mutación E280A en presenilina 1, previa autorización de los familiares, fueron sometidos a autopsia y se extrajeron muestras de piel, músculo esquelético, pulmón, corazón, hígado, bazo, páncreas, intestino, riñón y glándula suprarrenal.
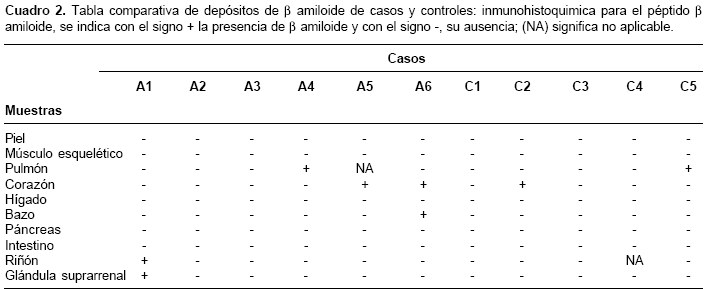
Como individuos control se usaron los tejidos de seis cadáveres provenientes de medicina legal, que carecían de historia familiar de demencia y que poseían edad y sexo homólogos a los de los cadáveres de los afectados con enfermedad de Alzheimer familiar. De los seis controles usados, uno debió ser excluido del estudio debido a problemas técnicos en el procesamiento de la muestra (
cuadros 1 y cuadro 2). Del grupo de los afectados, A2, A3 y A6 eran de sexo femenino, al igual que C2, C4 y C5, en el grupo control. La edad de la muerte fue 52, 58, 59, 60, 61 y 74 años para A1, A2, A3, A4, A5 y A6, respectivamente, y para el grupo control fue 51, 56, 60, 60 y 71 años para C1, C2, C3, C4 y C5, respectivamente.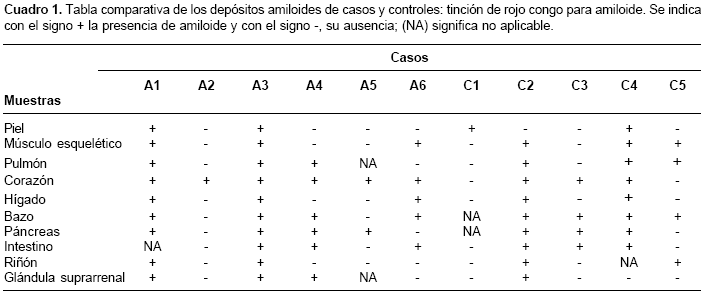
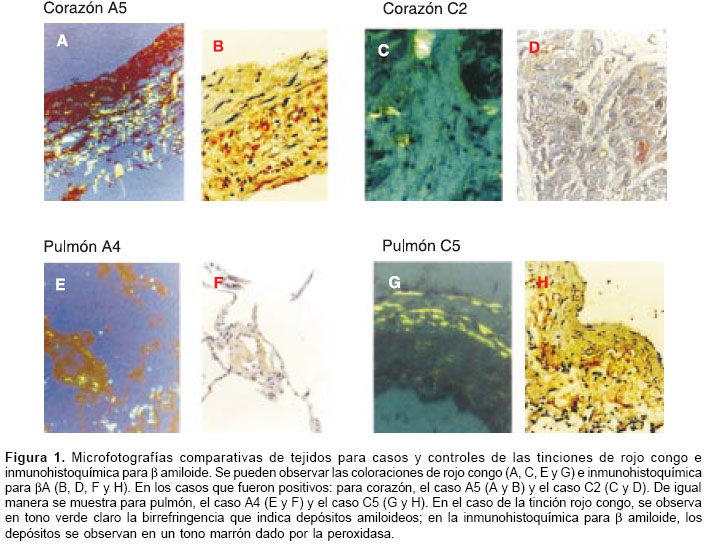
Rojo congo e inmunohistoquímica para
b amiloideLos tejidos se fijaron en formol al 10% con pH estabilizado en 7,4 y luego se incluyeron en bloques de parafina y se hicieron cortes de 4 µm con micrótomo de rotación convencional (Leica, 2035), los cuales se tiñeron mediante la técnica de rojo congo, para detectar depósitos amiloideos. Se reportaron como positivos cuando se observaba birrefringencia verde al examinar la muestra en el microscopio de luz polarizada (25).
Para identificar el componente de b amiloide en los cortes realizados y su distribución en los depósitos amiloideos identificados por el rojo congo, los cortes se sometieron a inmuno-histoquímica, para la detección del péptido b amiloide. Para la técnica de inmunohistoquímica, se usó la de biotina-avidina-peroxidasa, con el anticuerpo policlonal R1282 (dilución 1:1000, donado por D. Selkoe, Boston, MA), derivado de conejo y que reconoce bA 1-40 y 1-42 humanos (26). Para el proceso de tinción, se usó el estuche Dako (Glostrup, DK), de acuerdo con las instrucciones dadas por el fabricante; la presencia de b amiloide se evidenciaba por un precipitado café y en dicho caso se reportaba como positivo. Como controles positivos para la inmuno-histoquímica se emplearon cortes de cerebro de un caso de enfermedad de Alzheimer familiar portador de la mutación E280A, previamente estudiado.
Plásmidos y vectores
Para el efecto usamos los vectores pZeoPS1wt, pZeoPS1DE9, pZeoCMVPS1M146L y pZeoL392V, donados por el laboratorio dirigido por Weiming Xia (Center for Neurologic Diseases, Harvard Medical School); el pZeoE280A se fabricó a partir del vector pZeoPS1 wt, usando el estuche de mutagénesis dirigida Quik Change (Stratagene). Como control de transfección se usó el plásmido pZeo (Invitrogen, Carlsbad, CA).
Cultivos celulares
Las células CHO, que expresan en forma estable y en exceso la proteína precursora de amiloide humana (línea celular CHO-7W) (27), creciendo en platos de cultivo de seis pozos, fueron transfectadas transitoriamente mediante la técnica de lipofectamina (Invitrogen, Carlsbad, CA) con 1,0 µg de ADN de cada uno de los vectores: pZeoPS1 wt, pZeoPS1DE9, pZeoCMVPS1M146L, pZeoE280A, pZeoL392V y pZeo. Esos vectores portaban el cADN de presenilina 1 humana: silvestre, con la deleción del exón 9, presenilina 1 con la mutación M146L, presenilina 1 con la mutación E280A, presenilina 1 con la mutación L392V y el vector vacío, respectivamente. A las 36 horas de la transfección, las células fueron sometidas a ELISA y Western blot para evaluar la expresión de β amiloide (40 y 42) y presenilina 1, respectivamente.
Western blot
Después de la transfección, las células se cosecharon y lisaron en una solución tampón que contenía 50 mM trisHCl, pH 7,6, 150 mM NaCl, 2 mM EDTA, 0,2% SDS, 1% nonidet P-40 e inhibidor de proteasas en tabletas cóctel (Roche). La cuantificación de proteínas en el lisado se hizo con el estuche BCA (Pierce, Rockford, IL); 20 µg de cada una de las muestras se usaron para electroforesis en geles de glicina de 4%-20% SDS-PAGE, con la solución tampón de muestra (Invitrogen, Carlsbad, CA). Las proteínas fueron electrotransferidas a membranas PVDF (BioRad, Hercules, CA) y el blot se hizo con el anticuerpo monoclonal de antipresenilina 1 derivado de rata (dilución 1:750, Chemicon, Temecula, CA). Para evaluar la concentración de proteínas, se usó el anticuerpo policlonal anti-a-actinina derivado de conejo (dilución 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA). Los anticuerpos primarios se incubaron con las muestras durante 12 horas. Los anticuerpos secundarios conjugados con peroxidasa fueron: anti-IgG de rata derivado de conejo (1:3000, Sigma, Sant Louis, MI) y anti-IgG de conejo derivado de burro (1:5.000, Amersham Biosciences, Piscataway, NJ). Las membranas se sometieron a quimioluminiscencia con el estuche ECL (Santa Cruz Biotechnology, Santa Cruz, CA) y la reacción se detectó mediante autorradiografía (Kodak, Rochester, NY).
ELISA para b amiloide
El β amiloide de 40 y 42 fue cuantificado en el medio recolectado y analizado por ELISA de sándwich doble, al igual que las células, las cuales fueron lisadas en una solución tampón enfriada en hielo que contenía 5,0 M guanidinaHCl y 50 mM trisHCl, pH 8,0; las muestras estuvieron en incubación por 3 a 4 horas y luego se diluyeron 1:10 en solución tampón enfriada en hielo con caseína al 0,25%, azida de sodio 0,05%, 20 µg/ml de aprotinina, 5 mM EDTA, pH 8,0, 10 µg/ml de leupeptina en PBS. Se centrifugaron a 16.000g por 20 minutos a 4oC.
La ELISA en sándwich específico para amiloide 1-42 utiliza el anticuerpo de captura 21F12, específico para los aminoácidos 33-42 y el anticuerpo reportero fue el biotinilado 3D6, específico para los aminoácidos 13-28 del b amiloide. Para la detección del β amiloide 1-40, el anticuerpo de captura fue el 2G3, específico para los aminoácidos 33-40 y el anticuerpo reportero fue igualmente el 3D6 biotinilado. Esta ELISA no detecta el péptido truncado en el N-terminal p3 (mayores detalles sobre la ELISA en Jonson-Wood K, et al. 1997 y Seubert P, et al. 1992) (28,29). Para la cuantificación del b amiloide plasmático se usó la técnica de ELISA, de acuerdo con el protocolo descrito por K. Johnson-Wood et al. (28).
Análisis estadístico
Para el análisis estadístico se empleó el programa SPSS y se aplicó ANOVA de una vía para buscar diferencias entre los grupos. Para analizar diferencias entre variables cualitativas de dos grupos, se aplicó el test exacto de Fisher.
Resultados
Depósitos amiloideos en portadores de la mutación E280A de presenilina 1
Con el rojo congo, 33 muestras fueron positivas para los afectados con enfermedad de Alzheimer, de las cuales, 10 correspondieron a un individuo; mientras que, en los individuos control, hubo 26 muestras positivas y 9 de éstas pertenecieron a un individuo (cuadro 1). Todas las muestras de corazón del grupo con enfermedad Alzheimer produjeron birrefringencia, mientras que para los controles, tres de las cinco muestras de corazón fueron positivas. En las muestras de piel, músculo esquelético, pulmón, bazo, intestino y riñón se presentaron igual número de casos positivos, tanto para portadores de la mutación, como para los controles. Mientras que en hígado la relación fue de tres a dos, en páncreas se presentaron cuatro a tres positivos de los individuos afectados frente a los controles, respectivamente. Para la glándula suprarrenal, la proporción de muestras positivas de los afectados con enfermedad de Alzheimer frente a los controles, fue de tres a uno. Al comparar los grupos (afectados con enfermedad de Alzheimer versus grupo control), el valor de p fue de 0,75; lo cual indica que no hubo diferencias en los resultados obtenidos.
b amiloide en portadores de la mutación E280A de presenilina 1
La reactividad contra b amiloide sólo se observó en seis muestras de los afectados con enfermedad de Alzheimer, frente a dos muestras en los controles. Los tejidos que mostraron depósitos de b amiloide en los portadores de la mutación fueron: corazón (dos casos), pulmón, bazo, riñón y glándula suprarrenal; con un caso positivo cada uno. Para los controles, las únicas muestras positivas fueron una de corazón y una de pulmón (cuadro 2). El análisis estadístico entre ambos grupos (afectados con enfermedad de Alzheimer versus grupo control) no indicó diferencias, con un valor de p de 0,23. El β amiloide, en todos los casos se encontró en depósitos difusos y perivasculares (figura 1); en ningún caso se encontraron depósitos en placas.
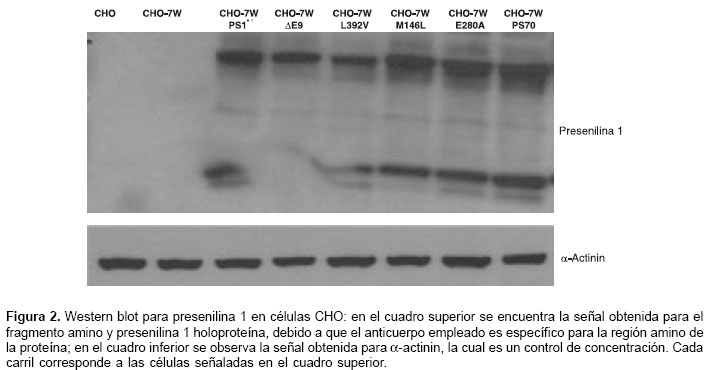
Expresión de presenilina 1 silvestre y presenilina 1 mutante en células CHO
Las células CHO-7W expresan establemente la proteína precursora de amiloide humana y fueron transfectadas con presenilina 1 silvestre y presenilina 1 portadora de las mutaciones M146L, E280A, DE9 y L392V. Se evaluó la expresión de presenilina 1 por medio del Western blot (figura 2) y la proteína a-actinina se usó como control de la concentración de proteínas. Se observó una adecuada expresión de presenilina 1 en las células transfectadas con los cDNA portadoras de presenilina 1 silvestre y con las mutaciones; en la mutación DE9, se observó ausencia del fragmento amino, fenómeno previamente reportado debido a que la proteína presenilina 1-DE9 no sufre endoproteólisis (30). No se observó inhibición de la proteólisis de presenilina 1 en las otras mutaciones evaluadas diferentes a DE9.
Producción de b amiloide en células CHO
La actividad g-secretasa fue determinada por la detección del producto (b amiloide), el cual se discriminó entre b amiloide 1-40 y 1-42; además, se diferenció entre el excretado (detectado en el sobrenadante) y el que se encuentra en las células (proveniente de extractos celulares).
En las células CHO, CHO-7W y CHO-7W + pZeo, se detectó producción de b amiloide de 1-40 y 1-42; los niveles excretados de β amiloide de 1-40 fueron mayores con diferencia no significativa, en el grupo de células transfectadas con los cADN mutados en comparación con el de las células control (CHO, CHO-7W, CHO-7W + pZeo y CHO-7W + PS1wt). Todas las células produjeron β amiloide 1-40 excretado; pero, en los extractos celulares, las células CHO-7W y CHO-7W + pZeo no mostraron niveles detectables y las células CHO mostraron una mayor producción de β amiloide 1-40 intracelular que el excretado.
El b amiloide 1-42 extracelular de células CHO-7W mostró niveles muy significativos (p<0,001) al compararlos con los niveles de las otras células estudiadas. Las células CHO-7W fueron las únicas células control (CHO, CHO-7W y CHO-7W + pZeo) que mostraron producción de β amiloide 1-42 excretado. Todas las células control presentaron niveles intracelulares de b amiloide de 1-42, pero, fueron significativamente mayores en las células transfetadas con el cADN normal (presenilina 1 silvestre), al ser comparadas con respecto al de las células que portan las mutaciones L392V y E280A, con valores de p de 0,014 y 0,022, respectivamente. Las células CHO-7W y las transfectadas con el cDNA portadora de las mutaciones L392V y E280A tuvieron una producción de b amiloide 1-42 extracelular, mayor que la producción intracelular (figura 3).
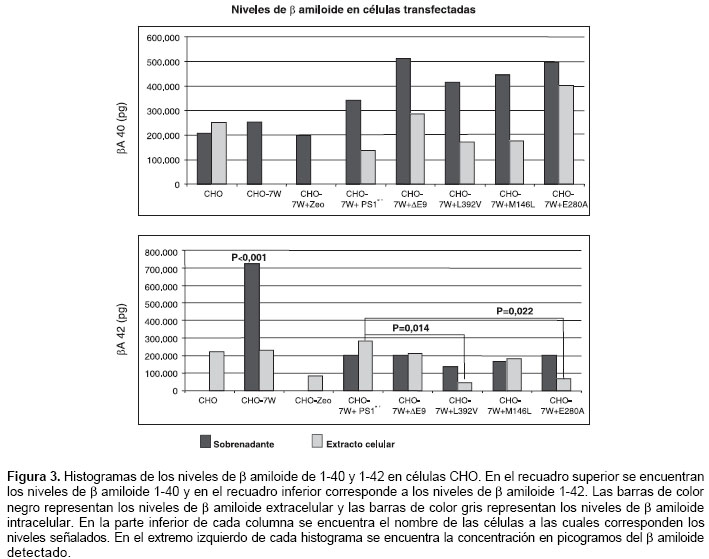
Las mediciones de
β amiloide 1-40 y 1-42 permitieron hacer un análisis de la relación entre el producido de uno con respecto al otro (b amiloide 1-42/1-40) y analizar la producción de cada uno de los dos β amiloides con respecto al espacio donde fueron detectados (extracelular o intracelular). Este análisis se obtiene al hacer la división de los promedios de los niveles obtenidos para cada una de las células en su respectivo espacio (intracelular o extracelular). De este análisis debieron excluirse en varias ocasiones las células CHO, CHO-7W y CHO-7W + pZeo, debido a niveles no detectables en los extractos celulares o en los sobrenadantes.Las células CHO-7W tuvieron una diferencia muy significativa (p<0,001), para la relación del
β amiloide 1-42/1-40 en el sobrenadante, con respecto a las otras células. Se pueden observar tendencias, como es el caso de una relación extracto/sobrenadante para el b amiloide 1-42, más baja para las células transfectadas con el cDNA mutante, comparadas con las CHO-7W y las transfectadas con el cDNA sin mutación (presenilina 1 silvestre), lo cual indica una proporción mayor de b amiloide 1-42 extracelular para las mutantes en presenilina 1 que las que no llevan la mutación (cuadro 3).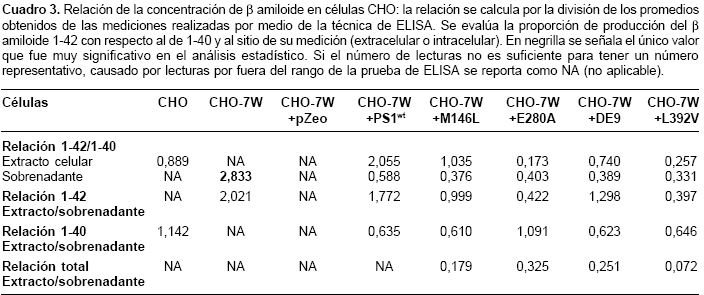
Discusión
En los portadores de la mutación E280A en la presenilina 1 no se encontraron depósitos de
β amiloide en los tejidos en los que previamente se habían reportado depósitos en personas afectadas con enfermedad de Alzheimer (piel e intestino) (14). Se observó un gran número de muestras positivas para rojo congo y pocas positivas para b amiloide, probablemente debido a la gran especificidad de la inmunohistoquímica para la detección del b amiloide. En corazón fue más evidente el fenómeno, puesto que este órgano es rico en colágeno, con el cual el rojo congo presenta reactividad cruzada (31). El b amiloide se encontró como depósitos difusos y perivasculares, lo que puede tener relación con el receptor de productos de glicosilasión avanzada, el cual se encuentra en el endotelio vascular y transporta b amiloide del plasma al parénquima cerebral (32).La inhibición de la endoproteólisis de presenilina 1 se ha descrito en mutaciones de presenilina 1 (33). Nuestros resultados no mostraron inhibición de endoproteólisis de presenilina 1, en las mutaciones M146L, E280A y L392V, a pesar de que E280A se encuentra muy cercana al sitio de corte y de la tirosina 288 que es considerada esencial para la endoproteólisis (34).
Los resultados esperados suponían un incremento sustancial del péptido
β amiloide 1-42, por las células transfectadas con las mutaciones en presenilina 1, con respecto a las controles (CHO, CHO-7W y CHO-7W + pZeo), al igual que dicho péptido debiera estar incrementado en sistemas diferentes al nervioso de los portadores de la mutación E280A.Sin embargo, los resultados se apartaron considerablemente de lo esperado. El incremento de producción de b amiloide excretado en líneas transfectadas con mutaciones en presenilina, se ha reportado como un incremento de b amiloide 1-42 y 1-40 (35), mientras que otros autores indican que el incremento sólo se encuentra en el b amiloide 1-42 (10) o que el incremento en la producción de b amiloide 1-42 se produce a expensas (reduciendo la frecuencia) de especies más cortas de
β amiloide (36).Un factor importante que incide en los diferentes reportes es el método que se utiliza para cuantificar el b amiloide; autores como Ikeuche et al. (35), usan técnicas cientos de veces más sensibles y específicas que la de ELISA, a tal punto que demuestran el incremento de b amiloide 1-40 y no sólo el incremento en la relación de b amiloide 1-42/1-40, como se reporta en la mayoría de las publicaciones (11,19,35,37-39).
En nuestro trabajo, la razón b amiloide 1-42/1-40 nos dio a la inversa de lo esperado, ya que fue mayor en células transfectadas con presenilina 1 silvestre que en las portadoras de las mutaciones, y fue más notable en extracto celular que en sobrenadante; estos resultados pueden ser explicados por factores como el tipo de célula utilizado, puesto que al utilizar células CHO es más difícil extrapolar los resultados a las células neuronales, mientras varios autores utilizan células neuronales obtenidas de ratones manipulados genéticamente.
Otro factor importante que influenció nuestros resultados es el sustrato utilizado para presenilina 1. Otros autores frecuentemente prueban las mutaciones de presenilina 1 con células que poseen la proteína precursora de amiloide con la mutación sueca (KM595/596NL) (11,35,40-42), que aceleran notablemente la producción de b amiloide 1-42 (43). En cambio, en nuestro trabajo, las células CHO-7W contienen la proteína precursora de amiloide silvestre.
Los altos niveles de
β amiloide 1-42 detectados en células CHO-7W demuestran que la presenilina 1 de ratón procesa la proteína precursora de amiloide humana en el sitio g y produce el mismo efecto que algunas mutaciones en la presenilina 1 humana.Esto puede deberse a que la homología entre la proteína precursora de amiloide humana y la de ratón no es del 100% (44,45). La variabilidad observada en la producción de
β amiloide se debió a que el complejo g-secretasa contiene solamente la presenilina 1 humana, ya que las otras tres proteínas que son cruciales para dicha función (nicastrina, aph-1 y pen-2) son de ratón, lo cual conlleva a que se conformen diferentes tipos de complejos g-secretasa (46); las células de ratón pueden generar ocho combinaciones de heterodímero (presenilina, nicastrina, aph-1 y pen-2) y no seis combinaciones, como en las células humanas (47,48).Además, las células CHO conservan la presenilina 1 y presenilina 2 de ratón, las transfectadas con los cADN de presenilina 1 (silvestre o mutado), pueden formar el complejo con un fragmento de ratón y el otro humano, lo cual aumenta el número de combinaciones; y, si se tiene en cuenta que en humanos se forman complejos con heterodímeros que contienen fragmentos de presenilina 1 y presenilina 2, incluso dímeros de holoproteínas, se puede comprender el comportamiento tan variable en la producción del b amiloide (49-52).
El panorama se puede hacer aún más complejo si consideramos que, para que la g-secretasa sea funcional, deben unirse, al menos, dos complejos tetraméricos (presenilina, nicastrina, aph-1 y pen-2) y estos complejos pueden estar conformados por las combinaciones ya mencionadas (53,54). Además, el fragmento C99 humano se ancla en microdominios lipídicos de una membrana de ratón, cuya composición en colesterol y esfingomielinas debe ser diferente a los microdominios lipídicos humanos, no sólo por el aspecto filogenético, sino por el tipo de célula.
En las células CHO-7W + pZeo, los niveles de b amiloide 1-40 y 1-42 (tanto intracelular como extracelular) fueron menores que los producidos por CHO-7W, lo cual se explica por un fenómeno de competencia entre los vectores empleados (55), puesto que ambas líneas celulares portan la proteína precursora de amiloide expresada bajo un promotor de CMV; pero, las CHO-7W + pZeo portan, además, el vector pZeo que también posee un promotor de CMV, el cual compite por los elementos que intervienen en la transcripción; como resultado lógico, producirá menores niveles de expresión de la proteína precursora de amiloide, que se reflejan en los resultados del b amiloide producido.
Se observó una disociación entre el b amiloide intracelular y el extracelular. Pocos trabajos han evaluado la concentración de b amiloide intracelular, puesto que la mayoría se basan en la producción de b amiloide excretado; algunos autores reportan poco efecto de las mutaciones en la proteína precursora de amiloide (56), presenilina 1 y presenilina 2, sobre la concentración de
β amiloide intracelular (42); las mutaciones en la presenilina 1 son las que exhiben el menor incremento en la producción intracelular de β amiloide. Llama la atención que varios autores hayan identificado al β amiloide intracelular como el primer indicador de disfunción neuronal, sináptica y cognitiva, y el primer paso en la formación de placas seniles (57-59).Agradecimientos
Al Departamento de Patología, así como a los Laboratorios de Inmunovirología y Genética Molecular de la Facultad de Medicina de la Universidad de Antioquia. Al Instituto de Medicina Legal y Ciencias Forenses. A Dennis Selkoe de la Universidad de Harvard por la donación del anticuerpo policlonal R1282 contra b amiloide empleado en este trabajo. A. Weiming Xia de la Universidad de Harvard, por donar los vectores pZeoPS1 wt, pZeoPS1DE9, pZeoCMVPS1M146L y pZeoL392V empleados en este trabajo.
Conflictos de interés
Los investigadores declaramos que durante la realización del proyecto de investigación no existió conflicto de intereses que pudieran haber afectado los resultados del mismo.
Financiación
Este trabajo fue financiado por el Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnologia "Colciencias", bajo el proyecto No. 1115-04-404-98.
Correspondencia:
Andrés Villegas, Laboratorio de Neurociencias, Universidad de Antioquia, calle 62 # 52-59, torre 2, piso 4, laboratorios 411-412, Medellín, Antioquia, Colombia. Telefax: (574) 210 6444
andres.villegas@neurociencias.udea.edu.coReferencias
1. Graeber MB, Mehraein P. Reanalysis of the first case of Alzheimers disease. Eur Arch Psychiatry Clin Neurosci. 1999;249 (Suppl.3):10-3. [ Links ]
2. Bird TD. Clinical genetics of familial Alzheimer disease. En: Terry RD, Katzman R, Bick KL, eds. Alzheimer Disease. First ed. New York: Raven Press; 1994. p.65-74. [ Links ]
3. Bertoli-Avella AM, Oostra BA, Heutink P. Chasing genes in Alzheimers and Parkinsons disease. Hum Genet. 2004;114:413-38. [ Links ]
4. Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793-9. [ Links ]
5. Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimers disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50:397-409. [ Links ]
6. Pietrzik C, Behl C. Concepts for the treatment of Alzheimers disease: molecular mechanisms and clinical application. Int J Exp Pathol. 2005;86:173-85. [ Links ]
7. De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9-12. [ Links ]
8. Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486-8. [ Links ]
9. Vardy ER, Catto AJ, Hooper NM. Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimers disease. Trends Mol Med. 2005;11:464-72. [ Links ]
10. Brunkan AL, Goate AM. Presenilin function and gamma-secretase activity. J Neurochem. 2005;93:769-92. [ Links ]
11. Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, et al. Familial Alzheimers disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17: 1005-13. [ Links ]
12. Vetrivel KS, Thinakaran G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology. 2006;66 (Suppl. 1):S69-73. [ Links ]
13. Chan SL, Culmsee C, Haughey N, Klapper W, Mattson MP. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism invol-ving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol Dis. 2002;11:2-19. [ Links ]
14. Joachim CL, Mori H, Selkoe DJ. Amyloid beta-protein deposition in tissues other than brain in Alzheimers disease. Nature. 1989;341:226-30. [ Links ]
15. Govoni S, Gasparini L, Racchi M, Trabucchi M. Peripheral cells as an investigational tool for Alzheimers disease. Life Sci. 1996;59:461-8. [ Links ]
16. Suzuki T, Nishiyama K, Murayama S, Yamamoto A, Sato S, Kanazawa I, et al. Regional and cellular presenilin 1 gene expression in human and rat tissues. Biochem Biophys Res Commun. 1996;219:708-13. [ Links ]
17. Selkoe DJ. Physiological production of the beta-amyloid protein and the mechanism of Alzheimers disease. Trends Neurosci. 1993;16:403-9. [ Links ]
18. Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP. Amyloid beta protein 1-40 and 1-42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett. 2001;304:102-6. [ Links ]
19. Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proc Natl Acad Sci USA. 1996;93:13170-5. [ Links ]
20. Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. Familial Alzheimers disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J Neurochem. 2005;94:1189-201. [ Links ]
21. Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimers disease. Hum Mutat. 1998;11:183-90. [ Links ]
22. Hardy J. Testing times for the "amyloid cascade hypothesis". Neurobiol Aging. 2002;23:1073-4. [ Links ]
23. Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627-38. [ Links ]
24. Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Arch Neurol. 2005;62:192-5. [ Links ]
25. Instituto de Patología de las Fuerzas Armadas de los Estados Unidos de América (AFIP). Hidratos de carbono. En: Prophet EB, Mills B, Arrington JB, Sobin LH, Heffess CS, Mullick FG, eds. Métodos histotecnológicos. Washington, D.C.: Instituto de Patología de los Estados Unidos de América (ARP); 1995. p.151-73. [ Links ]
26. Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322-5. [ Links ]
27. Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, et al. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977-82. [ Links ]
28. Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci. USA 1997;94:1550-5. [ Links ]
29. Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimers beta-peptide from biological fluids. Nature. 1992;359:325-7. [ Links ]
30. Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181-90. [ Links ]
31. Westermark GT, Johnson KH, Westermark P. Staining methods for identification of amyloid in tissue. Methods Enzymol. 1999;309:3-25. [ Links ]
32. Deane R, Du YS, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907-13. [ Links ]
33. Mercken M, Takahashi H, Honda T, Sato K, Murayama M, Nakazato Y, et al. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodies: Evidence that Alzheimer mutations affect proteolytic processing. FEBS Lett. 1996;389:297-303. [ Links ]
34. Laudon H, Karlstrom H, Mathews PM, Farmery MR, Gandy SE, Lundkvist J, et al. Functional domains in presenilin 1: the Tyr-288 residue controls gamma-secretase activity and endoproteolysis. J Biol Chem. 2004;279:23925-32. [ Links ]
35. Ikeuchi T, Dolios G, Kim SH, Wang R, Sisodia SS. Familial Alzheimer disease-linked presenilin 1 variants enhance production of both Abeta 1-40 and Abeta 1-42 peptides that are only partially sensitive to a potent aspartyl protease transition state inhibitor of "gamma-secretase". J Biol Chem. 2003;278:7010-8. [ Links ]
36. Murphy MP, Uljon SN, Golde TE, Wang R. FAD-linked mutations in presenilin 1 alter the length of Abeta peptides derived from betaAPP transmembrane domain mutants. Biochim Biophys Acta. 2002;1586:199-209. [ Links ]
37. Durkin JT, Murthy S, Husten EJ, Trusko SP, Savage MJ, Rotella DP, et al. Rank-order of potencies for inhibition of the secretion of abeta40 and abeta42 suggests that both are generated by a single gamma-secretase. J Biol Chem. 1999;274:20499-504. [ Links ]
38. Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, et al. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem. 2002;277:36521-6. [ Links ]
39. Nakaya Y, Yamane T, Shiraishi H, Wang HQ, Matsubara E, Sato T, et al. Random mutagenesis of presenilin-1 identifies novel mutants exclusively generating long amyloid beta-peptides. J Biol Chem. 2005;280:19070-7. [ Links ]
40. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159-70. [ Links ]
41. Walker ES, Martínez M, Brunkan AL, Goate A. Presenilin 2 familial Alzheimers disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J Neurochem. 2005;92:294-301. [ Links ]
42. Takeda K, Araki W, Tabira T. Enhanced generation of intracellular Abeta42 amyloid peptide by mutation of presenilins PS1 and PS2. Eur J Neurosci. 2004;19:258-64. [ Links ]
43. Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939-45. [ Links ]
44. De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14:4932-8. [ Links ]
45. Sarasa M. Experimental models for Alzheimers disease research. Rev Neurol. 2006;42:297-301. [ Links ]
46. Ma G, Li T, Price DL, Wong PC. APH-1a is the principal mammalian APH-1 isoform present in gamma-secretase complexes during embryonic development. J Neurosci. 2005;25:192-8. [ Links ]
47. Hebert SS, Serneels L, Dejaegere T, Horre K, Dabrowski M, Baert V, et al. Coordinated and widespread expression of gamma-secretase in vivo: evidence for size and molecular heterogeneity. Neurobiol Dis. 2004;17:260-72. [ Links ]
48. Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H. Identification of distinct gamma-secretase complexes with different APH-1 variants. J Biol Chem. 2004;279:41340-5. [ Links ]
49. Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, et al. The proteolytic fragments of the Alzheimers disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem. 1998;273:3205-11. [ Links ]
50. Herl L, Lleo A, Thomas AV, Nyborg AC, Jansen K, Golde TE, et al. Detection of presenilin-1 homodimer formation in intact cells using fluorescent lifetime imaging microscopy. Biochem Biophys Res Commun. 2006;340:668-74. [ Links ]
51. Stromberg K, Hansson EM, Laudon H, Bergstedt S, Naslund J, Lundkvist J, et al. Gamma-secretase complexes containing N- and C-terminal fragments of different presenilin origin retain normal gamma-secretase activity. J Neurochem. 2005;95:880-90. [ Links ]
52. Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, et al. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415-22. [ Links ]
53. Evin G, Canterford LD, Hoke DE, Sharples RA, Culvenor JG, Masters CL. Transition-state analogue gamma-secretase inhibitors stabilize a 900 kDa presenilin/nicastrin complex. Biochemistry. 2005;44:4332-41. [ Links ]
54. Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, et al. A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci USA. 2003;100:13075-80. [ Links ]
55. Chen PH, Tseng WB, Chu Y, Hsu MT. Interference of the simian virus 40 origin of replication by the cytomegalovirus immediate early gene enhancer: evidence for competition of active regulatory chromatin conformation in a single domain. Mol Cell Biol. 2000;20:4062-74. [ Links ]
56. Grimm HS, Beher D, Lichtenthaler SF, Shearman MS, Beyreuther K, Hartmann T. gamma-Secretase cleavage site specificity differs for intracellular and secretory amyloid beta. J Biol Chem. 2003;278: 13077-85. [ Links ]
57. Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15-20. [ Links ]
58. Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimers disease. Neurobiol Aging. 2005;26: 1235-44. [ Links ]
59. Turner RS, Suzuki N, Chyung AS, Younkin SG, Lee VM. Amyloids beta40 and beta42 are generated intracellularly in cultured human neurons and their secretion increases with maturation. J Biol Chem. 1996;271: 8966-70. [ Links ]

