










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.30 n.1 Bogotá jan./mar. 2010
1 Laboratorio de Micología y Fitopatología, Facultad de Ciencias, Universidad de los Andes, Bogotá, D.C., Colombia
2 Grupo de Genética Molecular, Corporación Corpogen, Bogotá, D.C., Colombia
Recibido: 30/01/09; aceptado:27/07/09
Introducción. El creciente número de genomas secuenciados pertenecientes al complejo Mycobacterium tuberculosis hace posible la comparación y el análisis genómico, que puede revelar importantes mecanismos de evolución y variación para entender la patogénesis de esta especie.
Objetivo. Mediante el uso de alineamientos múltiples se pretendió analizar las diferencias entre seis genomas del complejo M. tuberculosis, para encontrar regiones de variación que conduzcan a mejoras en la identificación de estas especies o en el tratamiento.
Materiales y métodos. Mediante el programa bioinformático Mauve, se realizaron alineamientos múltiples de seis genomas pertenecientes a especies del complejo M. tuberculosis. Las regiones genómicas exclusivas para cada genoma se anotaron usando la base de datos Tuberculosis Database.
Resultados. El porcentaje de similitud entre los seis genomas analizados estuvo entre 96,1% y 97,8%. La anotación de las regiones exclusivas reveló la presencia de elementos de transposición, familias de proteínas PPE y PE-PGRS, regiones asociadas a resistencia contra bacteriófagos y regiones intergénicas.
Conclusiones. A pesar de la gran similitud entre las cepas analizadas, existen variaciones entre ellas que pueden ser importantes para entender diferencias en comportamiento y virulencia, así como para mejorar los diagnósticos de cepas específicas. Regiones como aquéllas con genes para proteínas de membrana, posiblemente, relacionadas con la variación y la respuesta antigénica, son de particular interés para estudios futuros orientados a buscar tratamientos nuevos para el control de esta enfermedad
Palabras clave: Mycobacterium tuberculosis, genómica, tuberculosis.
Comparative analysis of six Mycobacterium tuberculosis complex genomes
Introduction. A growing number of sequenced genomes belonging to the Mycobacterium tuberculosis complex has enabled a comparison of strain traits and genomic constitution. These analyses may reveal mechanisms of evolution and genomic variation relevant to tuberculosis pathogenesis.
Objective. Multiple alignments were used to analyze the differences between six genomes of the M. tuberculosis complex and to locate regions of variation that may lead to improvements in species identification or in their treatment.
Materials and methods. The Mauve software package was used to perform a multiple alignment of 6 genomes belonging to the M. tuberculosis complex. Regions exclusive to each genome were annotated using the TB database.
Results. Percent similarity among the six genomes ranged between 96.1% and 97.8%. The annotation identified intergenic regions, regions associated with transposable elements of the PE-PGRS and PPE families, and regions associated with resistance against bacteriophage.
Conclusions. In spite of the high genetic similarity among the tuberculosis strains, genomic variations were elucidated that may be relevant to differences in behavior and virulence, as well as for improvement of strain diagnosis. Regions encoding membrane-associated proteins, possibly related with antigenic variation and immune response, are particularly interesting for studies aimed at seeking tuberculosis treatments.
Key words: Mycobacterium tuberculosis, genomics, tuberculosis.
Mycobacterium, el único género de la familia de las Mycobacteriaceae, incluye patógenos que causan graves enfermedades en los mamíferos, como lepra y tuberculosis (1). Mycobacterium tuberculosis es el agente causal de la tuberculosis, una de las enfermedades infectocontagiosas más prevalentes de la historia. En la actualidad, según cifras del informe de 2008 de la Organización Mundial de la Salud, se presentan 9,2 millones de casos nuevos al año (139 por cada 100.000 habitantes) y 1,7 millones de muertes por año (2). En 2006 se estimaron 0,5 millones de casos de tuberculosis multirresistente (2). En Colombia la incidencia de tuberculosis para el 2005 fue de 25,2 casos por cada 100.000 habitantes (3) y en el 2006 se reportaron 11.625 casos nuevos (4).
La gran mayoría de los casos de tuberculosis están relacionados con especies que forman el complejo Mycobacterium tuberculosis (M. tuberculosis, M. africanum, M. bovis, M. microti y M. cannetii) (5), Los miembros de este complejo comparten 99,95% de identidad a nivel de ADN (6,7), pero poseen una amplia variabilidad fenotípica en cuanto a sus huéspedes, tipo de enfermedad y gravedad de la enfermedad. En el momento se cuenta con varios genomas secuenciados, lo cual ofrece nuevas oportunidades para estudios de genómica comparativa.
La caracterización morfológica y fisiológica ha sido la estrategia más usada por la comunidad científica para conocer los procesos de patogénesis de importantes microorganismos. Por otra parte, el incremento casi exponencial de la secuenciación de genomas, debido a los bajos costos, ha abierto la posibilidad de comparar genomas.
La genómica comparativa busca encontrar relaciones entre genomas, ya sean distantes o cercanos, a partir de los genomas secuenciados, y tiene el potencial de identificar componentes útiles para entender las relaciones bioquímicas, fisiológicas y patogénicas de los microorganismos (1). La genómica comparativa ha llevado a avances en los campos de la biología evolutiva (8,9), reconstrucción filogenética (1,10,11), programas de descubrimiento de medicamentos (12,13) y predicción de funciones en genes hipotéticos (14). A esto se le suma la identificación de regiones intrónicas (15), sitios de splicing y un mayor conocimiento de regiones no codificadoras (16,17).
La reconstrucción y la comparación de genomas de especies bacterianas han revelado que la organización cromosómica no se conserva a lo largo de la escala evolutiva (18). Un análisis más detallado puede ayudar a inferir relaciones entre las características de organización de los cromosomas y la fisiología celular. Uno de los principales objetivos en los análisis comparativos de los genomas secuenciados es identificar las secuencias que están conservadas entre las diferentes especies comparadas (19). Se han desarrollado poderosos algoritmos con el objetivo de reconstruir las circunstancias de los rearreglos cromosómicos (11,20,21).
Darling y colaboradores crearon en el 2004 un software llamado Mauve, para comparación múltiple de genomas, que identifica regiones genómicas conservadas, rearreglos e inversiones dentro de regiones conservadas y el punto exacto en donde ocurre el punto de quiebre de esos rearreglos genómicos a lo largo de múltiples genomas (22).
En este estudio se usaron alineamientos múltiples con el fin de analizar las diferencias entre genomas pertenecientes al complejo M. tuberculosis y encontrar regiones únicas que puedan servir para entender mejor las variaciones entre este grupo de micobacterias.
Materiales y métodos
Microorganismos y genomas
Para el análisis comparativo se usaron las secuencias genómicas ensambladas de las bacterias pertenecientes al complejo M. tuberculosis: M. tuberculosis CDC1551, M. tuberculosis F11, M. tuberculosis H37Ra, M. tuberculosis H37Rv, M. bovis BCG str. Pasteur 1173P2 y M. bovis AF2122/97 (cuadro 1). Además de estar completamente secuenciados, ensamblados y anotados, estos genomas se usan como referencia en la anotación de otros genomas del género Mycobacterium que están en proceso de secuenciación (1,9).
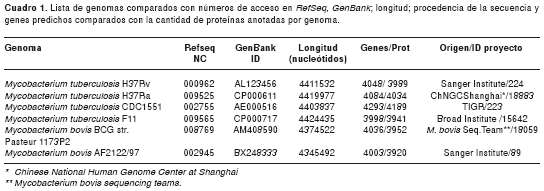
Alineamiento múltiple de genomas
Para el alineamiento múltiple de genomas se usó el programa Mauve, versión 2.2.0, desarrollado por Darling y colaboradores (22). Los alineamientos de los genomas pertenecientes al complejo M. tuberculosis, se hicieron usando el algoritmo progresivo de Mauve (progressive Mauve) que inicialmente identifica secuencias sucesivas con similitud exacta compartida por dos o más genomas y, junto a una matriz de distancia basada en la conservación genómica, construye un árbol guía. Posteriormente, selecciona subconjuntos de estas secuencias para hacer el anclaje y realizar un alineamiento usando el algoritmo de alineamientos locales MUSCLE (23). Estas regiones alineadas se denominan regiones locales de linealidad y representan una secuencia compartida por dos o más genomas incluidos dentro del alineamiento. Las regiones lineales vecinas se agrupan en bloques locales de linealidad (locally colinear blocks, LCB), que están separados por islas genómicas (22).
El alineamiento se realizó usando los siguientes parámetros: peso de la semilla de 15 nucleótidos, determinar LCB, asumir genomas lineales, alineamiento total, refinamiento iterativo y la opción del uso de familias de semilla en el anclaje se dejó desactivada. Se tomaron como archivo de entrada las secuencias organizadas filogenéticamente (1) dentro de un archivo "multifasta" (archivo de texto plano utilizado para representar las secuencias de ácidos nucleicos de cada uno de los genomas), de la siguiente manera: 1) M. tuberculosis H37Rv, 2) M. tuberculosis H37Ra, 3) M. tuberculosis CDC1551, 4) M. tuberculosis F11, 5) M. bovis BCG y 6) M. bovis AF2122/97. El análisis posterior se realizó utilizando el archivo de salida backbone, que relaciona los LCB presentes en dos o más genomas, identificando las coordenadas de nucleótidos de cada LCB.
Con el objetivo de extraer e identificar las regiones únicas por genoma, se desarrollaron herramientas bioinformáticas utilizando lenguaje de programación Perl y se utilizó la herramienta extractseq, disponible en el paquete de libre acceso EMBOSS, versión 5.0.0, (http://bioinfo.hku.hk/EMBOSS, European Molecular Biology Open software Suite, European Molecular Biology Open software Suite), para la extracción de secuencias.
La identificación de las regiones exclusivas o islas genómicas para cada uno de los genomas, se hizo empleando la anotación disponible en la base de datos de tuberculosis del Instituto Broad y la Escuela de Medicina de Stanford (www.tbdb.org).
En Tuberculosis Database se encuentran anotados algunos genomas de micobacterias; en el menú desplegable TB Genomes se accedió a la opción Download Sequence Data y se descargó por cada genoma analizado el archivo genome_summary_per_gene.txt que contiene la anotación completa de cada genoma. Se compararon las coordenadas de los LCB con las coordenadas de la anotación de cada genoma disponible en el archivo mencionado anteriormente, para identificar la posición de la región analizada.
Resultados
Matriz de distancias
El algoritmo progresivo usado para hacer las comparaciones genómicas inicialmente construye una matriz de conservación entre los genomas del complejo M. tuberculosis que refleja la similitud entre pares de genomas, en la que valores cercanos a 0 significan gran similitud (22).
A partir de dicha matriz se generó un gráfico en que se observa el grado de similitud de cada genoma estudiado en relación con los otros (figura 1). Cada vértice del hexágono representa un genoma y cada línea representa la similitud genética frente a los otros genomas estudiados. De esta manera, las líneas de M. tuberculosis H37Rv y M. tuberculosis H37Ra son comparables, lo que significa que genómicamente son similares.
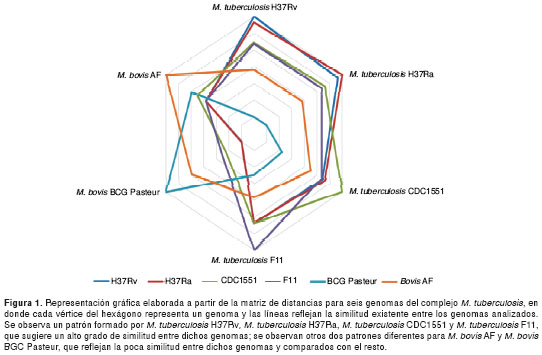
Por otra parte, los genomas de M. bovis BCG y M. bovis AF son los más divergentes con respecto a los demás genomas del complejo (figura 1). Con base en esta información, es posible definir dos grupos: uno formado por los genomas de M. tuberculosis H37Rv, M. tuberculosis H37Ra, M. tuberculosis CDC1551 y M. tuberculosis F11, y el otro formado por M. bovis BCG str. Pasteur 1173P2 y M. bovis AF2122/9.
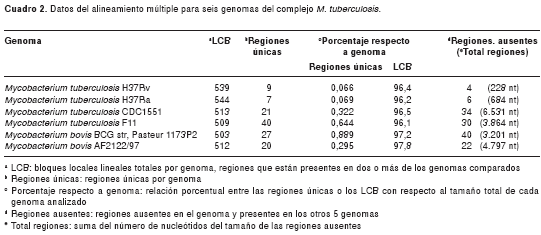
Bloques locales de linealidad
Se encontró un total de 684 LCB (cuadro 2), agrupados en un bloque de linealidad, de los cuales, 267 están presentes en todos los genomas. Al sumar la longitud de los LCB encontrados en cada genoma y comparar este valor con la longitud del genoma correspondiente, se encontró que estas regiones comunes cubrían entre 96,1% y 97,8% de cada genoma analizado (cuadro 2).
Identificación de regiones
Se hallaron 417 regiones variables dentro del alineamiento. Éstas son regiones que se encuentran en un genoma o grupo de genomas determinados y no en otros.
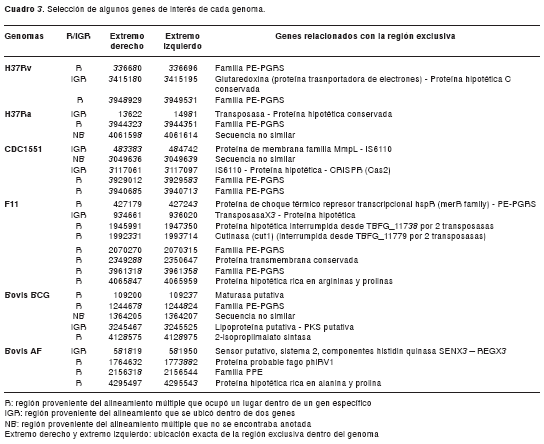
De la información del archivo de salida backbone se identificaron las regiones exclusivas de cada genoma: 9 regiones para M. tuberculosis H37Rv, 7 para M. tuberculosis H37Ra, 21 para M. tuberculosis CDC1551, 40 para M. tuberculosis F11, 27 para M. bovis BCG y 20 para M. bovis AF2122/97, que suman 124 regiones exclusivas.
El análisis de los genes presentes en estas regiones demostró la presencia de elementos de transposición, proteínas pertenecientes a las familias PPE, PE-PGRS y regiones intergénicas, como se observa en la figura 2. El cuadro 3 muestra algunas de las regiones exclusivas identificadas para cada genoma y su descripción. Las tablas con la información completa de los resultados, incluyendo LCB y regiones exclusivas, así como las herramientas bioinformáticas desarrolladas están disponibles por solicitud directa.

Discusión
La genómica comparativa permite acercarse de una manera global a la biología, fisiología, patogénesis y virulencia de especies genómicamente relacionadas (24-26). Esto se hace posible gracias al acceso a genomas de especies muy relacionadas que, a su vez, acelera la anotación funcional de nuevos genomas. Field y colaboradores agruparon en el 2005 los genomas secuenciados en dos grupos (26). El primero de ellos está conformado por los genomas anotados y disponibles en las bases de datos primarias y el segundo, por los genomas secundarios que se derivan de los primarios ya que están relacionados por semejanza taxonómica y afinidad de nicho ecológico.
Mediante aproximaciones genómicas, como hibridaciones ADN-ADN, arreglos en cromosomas bacterianos artificiales, hibridación sustractiva de genomas (6,27,28) y marcadores moleculares (8), se ha reportado que el grado de similitud entre especies del complejo M. tuberculosis es cercano a 99,95%.
Otros grupos han realizado comparaciones de genomas completos y concluyen que el grado de variación es mayor del que se creía (7,29); estas variaciones incluyen inserciones, deleciones, polimorfismos de secuencia larga y polimorfismos de nucleótido simple, entre otros. Sin embargo, en estos reportes no se propone un porcentaje de similitud específico.
En el presente trabajo se hizo una comparación entre múltiples genomas del complejo M. tuberculosis y fue posible observar que todos se agruparon en un bloque lineal general, lo que valida la información sobre la poca variabilidad genética dentro del complejo. Sin embargo, es importante anotar que aunque las aproximaciones genómicas in vitro arrojaron semejanzas mayores de 99%, la similitud entre las seis especies del complejo analizadas en este trabajo, con base en la identificación de LCB comunes, no supera el 98%.
Con base en los resultados de este análisis comparativo, encontramos que la variación entre los genomas del complejo M. tuberculosis se encuentra cerca del 2%; posiblemente, esto se deba a que se incluyen seis genomas en la comparación, a diferencia de estrategias empleadas por otros grupos en las que se hacen únicamente comparaciones entre dos genomas.
Las diferencias encontradas entre nuestros resultados y los resultados reportados para hibridaciones, cromosomas bacterianos artificiales y marcadores moleculares, se deben a las características propias de cada metodología. Técnicas moleculares como la hibridación sustractiva se han considerado de baja resolución (7), ya que dependen de variables como la correcta restricción y sustracción de los genomas. La ventaja de los alineamientos entre genomas es que se manejan pocas variables y, en nuestro caso específico, no sólo se comparan parejas de genomas sino que se comparan seis genomas a la vez, incluyendo así información valiosa que no se ha incluido en estudios de análisis pareados.
A pesar de la poca variación entre las especies del complejo M. tuberculosis, existe variabilidad representada por mutaciones puntuales, polimorfismos largos y eventos de inserción y transposición (1,8,17). Estas diferencias genotípicas, a su vez, pueden resultar en cambios fenotípicos grandes. La representación gráfica (figura 1) de las diferencias entre los genomas analizados revela la presencia de dos grupos.
La agrupación M. tuberculosis H37Rv, M. tuberculosis H37Ra, M. tuberculosis CDC1551 y M. tuberculosis F11, concuerda con estudios filogenéticos basados en 16S ADNr que agrupan a los tres primeros microorganismos dentro del mismo conglomerado y, asociado a ellos, al último de los genomas anteriormente mencionado (1).
Este análisis ubicó a M. tuberculosis H37Rv y M. tuberculosis CDC1551 en posiciones cercanas y separó a M. bovis BCG str. Pasteur 1173P2 y M. bovis AF2122/9, cepas con un patrón de distancias diferente al ser comparado con los demás genomas.
Estas agrupaciones son coherentes con análisis anteriores que encontraron diferencias entre los genomas de M. tuberculosis H37Rv y M. bovis BCG debido a rearreglos en la región de repeticiones directas, que es la base para genotipificar las bacterias pertenecientes al complejo M. tuberculosis (1), y con el análisis filogenético de las secuencias codificadoras interrumpidas para cuatro genomas pertenecientes al complejo (10).
La comparación entre genomas demostró la gran similitud existente entre M. tuberculosis H37Ra y H37Rv (figura 1), genomas que, además, contienen pocas regiones exclusivas, 9 y 7, respectivamente, también llamadas islas genómicas por algunos autores (30,31). La anotación de estas islas genómicas demostró la presencia de elementos de transposición, familias de proteínas PPE y PE-PGRS, y regiones intergénicas (cuadro 3).
El alto grado de similitud entre estas dos cepas ha sido reportado por Zeng y colaboradores, quienes también hallaron variaciones en regiones PE/PPE/PE-PGRS y demostraron que las diferencias radican en 53 inserciones y 21 deleciones (9). Las proteínas de las familias PPE y PE-PGRS se han encontrado en la pared celular de las micobacterias (32) y se han implicado en variación antigénica y evasión de la respuesta inmune (33). A su vez, las regiones intergénicas también pueden alterar la regulación de la expresión de genes adyacentes, generando cambios fenotípicos (9).
El análisis detallado de algunas de las regiones únicas para cada genoma reveló aspectos interesantes para futuras investigaciones. Una de las regiones exclusivas para M. tuberculosis CDC1551 contenía el elemento de inserción IS6110 y una región CRISPR, región de la cual no se sabe mucho en el complejo M. tuberculosis, pero que ha despertado gran interés recientemente puesto que se ha encontrado asociada a resistencia contra bacteriófagos en diversas especies bacterianas (34-36).
El análisis de M. tuberculosis F11 reveló, a su vez, una gran cantidad de regiones asociadas con elementos de inserción. Estos elementos están asociados con la generación de variabilidad genómica (37) y, en ocasiones, como cuando interrumpen un marco de lectura determinado o alteran expresión de genes adyacentes, pueden ocasionar profundos efectos sobre el fenotipo (37,38).
Algunas de las regiones exclusivas para M. bovis BCG y AF2122 se encontraron dentro de un marco abierto de lectura, lo que podría verse reflejado en cambios de las proteínas resultantes. En M. bovis AF2122 se encuentra una región exclusiva situada en regiones intergénicas entre dos proteínas asociadas a un sistema de dos componentes SenX3 y RegX3 (cuadro 3); esta región puede ser regulada por ARN no codificadores.
La comparación de múltiples genomas también reveló la existencia de extensas regiones conservadas, que sugieren la presencia de secuencias sujetas a fuertes presiones de selección. Así mismo, se detectaron rearreglos grandes debidos a la presencia del fago phiRv1, lo cual coincide con estudios anteriores (39).
La genómica comparativa es una herramienta poderosa que permite hacer un análisis detallado de las diferencias genómicas entre cepas cercanamente relacionadas, como las del complejo M. tuberculosis. Este tipo de análisis in silico puede, por consiguiente, contribuir a la identificación y clasificación filogenética de especies, al conocimiento de los mecanismos de evolución de las micobacterias del complejo M. tuberculosis y a la posible identificación de regiones únicas que puedan estar asociadas con diferencias fenotípicas.
La evolución del complejo es de gran interés desde el punto de vista de su adaptación al huésped y el desarrollo de su patogénesis, mientras que un mayor conocimiento acerca de las regiones y proteínas exclusivas podría conducir a un mejor entendimiento de la patogénesis de este microorganismo y a la posible identificación de nuevos blancos para tratamientos.
En este trabajo, con la genómica comparativa se encontró que la similitud entre los seis genomas analizados no supera el 98% (cuadro 2) y que las variaciones identificadas se deben en gran parte a la presencia de regiones asociadas a elementos de transposición, familias de proteínas PPE y PE-PGRS y regiones intergénicas (figura 2 y cuadro 3). También se encontraron regiones exclusivas para cada genoma, que pueden servir de base para análisis futuros más detallados que puedan conducir a la posible identificación de secuencias o proteínas interesantes para entender diferencias fenotípicas, como virulencia de cepas del complejo M. tuberculosis, y relacionar cambios genómicos con la biología del microorganismo.
Agradecimientos
Los autores agradecen a Alan Durham, del Instituto de Matemáticas y Estadística de la Universidad de Sâo Paulo, por su asesoría en el desarrollo bioinformático, y a Andrés Cubillos de la Corporación Corpogen, por sus ideas para el desarrollo de este estudio y la revisión y corrección de este artículo.
Conflicto de intereses
Declaramos que la investigación a partir de la cual se originó el presente manuscrito no presenta conflictos de intereses.
Financiación
Este trabajo fue financiado por la Facultad de Ciencias de Universidad de los Andes (convocatoria 2008-2), Corporación Corpogen y Colciencias (Proyecto No. 6570-408-20410).
Correspondencia: María Mercedes Zambrano, Corporación Corpogen, Carrera 5 Nº 66A-34, Bogotá, D.C., Colombia. Teléfono: (571) 805 0106; fax (571) 348 4607 mzambrano@corpogen.org
Referencias
1. Brosch R, Pym A, Gordon S, Cole S. The evolution of mycobacterial pathogenicity: clues from comparative genomics. Trends Microbiol. 2001;9:454-8. [ Links ]
2. World Health Organization. WHO report 2008. Global tuberculosis control. 2008. Fecha de consulta: 2 de octubre 2008. Disponible en: http://www.who.int/tb/publications/global_report/2008/en/ [ Links ]
3. Garzón M, Angée D, Llerena C, Orjuela D, Victoria J. Vigilancia de la resistencia de Mycobacterium tuberculosis a los fármacos antituberculosos, Colombia 2004-2005. Biomédica. 2008;28:319-6. [ Links ]
4. Ministerio de la Protección Social, Organización Panamericana de la Salud. Situación de salud en Colombia. Indicadores básicos. Bogotá, D.C.: Ministerio de la Protección Social; 2006. [ Links ]
5. Tiruviluamala P, Reichman LB. Tuberculosis. Annu Rev Public Health. 2002;23:403-26. [ Links ]
6. Imaeda T. Deoxyribonucleic acid relatedness among selected strains of Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium micoti and Mycobacterium africanum. Int J Syst Bacteriol. 1985;35:147-50. [ Links ]
7. Fleischmann R, Alland D, Eisen J, Carpenter L, White O, Peterson J, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479-90. [ Links ]
8. Sreevatsan S, Pan X, Stockbauer K, Connell N, Kreiswirth B, Whittam T, et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA. 1997;94:9869-74. [ Links ]
9. Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, et al. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS One. 2008;3:e2375. [ Links ]
10. Deshayes C, Perrodou E, Euphrasie D, Frapy E, Poch O, Bifani P, et al. Detecting the molecular scars of evolution in the Mycobacterium tuberculosis complex by analyzing interrupted coding sequences. BMC Evol Biol. 2008;8:78. [ Links ]
11. Tang J, Moret BM. Scaling up accurate phylogenetic reconstruction from gene-order data. Bioinformatics. 2003;19(Suppl.1):i305-12. [ Links ]
12. De Groot A, Bosma A, Chinai N, Frost J, Jesdale B, Gonzalez M, et al. From genome to vaccine: in silico predictions, ex vivo verification. Vaccine. 2001;19:4385-95. [ Links ]
13. Mustafa A. Development of new vaccines and diagnostic reagents against tuberculosis. Mol Immunol. 2002:39:113-9. [ Links ]
14. Raman K, Yetura K, Chandra N. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol. 2008;2:109. [ Links ]
15. Morris R, Drouin G. Similar ectopic gene conversion frequencies in the backbone genome of pathogenic and nonpathogenic Escherichia coli Strains. Genomics. 2008;92:168-72. [ Links ]
16. Azhikina T, Gvozdevsky N, Botvinnik A, Fushan A, Shemyakin I, Shemyakin V, et al. A genome-wide sequence-independent comparative analysis of insertion-deletion polymorphisms in multiple Mycobacterium tuberculosis strains. Res Microbiol. 2006;157:282-90. [ Links ]
17. Wang X, Galamba A, Warner D, Soetaert K, Merkel J, Kalai M, et al. IS1096-mediated DNA rearrangements play a key role in genome evolution of Mycobacterium smegmatis. Tuberculosis (Edinb). 2008:88:399-409. [ Links ]
18. Fremez R, Faraut T, Fichant T, Gouzy J, Quentin Y. Phylogenetic exploration of bacterial genomic rearrangements. Bioinformatics. 2007:23:72-4. [ Links ]
19. Vishnoi A, Roy R, Bhattacharya A. Comparative analysis of bacterial genomes: identification of divergent regions in mycobacterial strains using an anchor-based approach. Nucleic Acids Res. 2007;35:3654-67. [ Links ]
20. Bourque G, Pevzner PA. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res. 2002;12:26-36. [ Links ]
21. Moret BM, Wyman S, Bader DA, Warnow T, Yan M. A new implementation and detailed study of breakpoint analysis. Pac Symp Biocomput. 2001:583-94. [ Links ]
22. Darling A, Mau B, Blatter F, Perna N. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394-403. [ Links ]
23. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. [ Links ]
24. Domenech P, Barry C, Cole S. Mycobacterium tuberculosis in the post-genomic age. Curr Opin Microbiol. 2001;4:28-34. [ Links ]
25. Malik A, Godfrey-Faussett P. Effects of genetic variability of Mycobacterium tuberculosis strains on the presentation of disease. Lancet Infect Dis. 2005:5:174-83. [ Links ]
26. Field D, Feil E, Wilson G. Databases and software for the comparison of prokaryotic genomes. Microbiology. 2005;151:2125-32. [ Links ]
27. Gordon S, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole S. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol. 1999;32:643-56. [ Links ]
28. Mahairas G. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178:1274-82. [ Links ]
29. Garnier T, Eiglmeier K, Camus J, Medina N, Mansoor H, Pryor M, et al. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci USA. 2003;100:7877-82. [ Links ]
30. Ou H, Chen L, Lonnen J, Chaudhuri R, Thani A, Smith R, et al. A novel strategy for the identification of genomic islands by comparative analysis of the contents and contexts of tRNA sites in closely related bacteria. Nucleic Acids Res. 2006;34:1e3. [ Links ]
31. Jang J, Becq J, Gicquel B, Deschavanne P, Neyrolles O. Horizontally acquired genomic islands in the tubercle bacilli. Trends Microbiol. 2008;16:303-8. [ Links ]
32. Delogu G, Pusceddu C, Bua A, Fadda G, Brennan M, Zanetti S. Rv1818c-encoded PE-PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol. 2004;52:725-33. [ Links ]
33. Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol Microbiol. 2002;44:9-19. [ Links ]
34. Brouns S, Jore M. Lundgren M, Westra E, Slijkhuis R, Snijders A, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960-3. [ Links ]
35. Barrangou R, Fremaux, C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709-12. [ Links ]
36. Sorek R, Kunin V, Hugenholtz P. CRISPR- a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181-6. [ Links ]
37. Darling A, Miklos I, Ragan M. Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 2008;4:e1000128. [ Links ]
38. Soto C, Menéndez M, Pérez E, Samper S, Gómez S, García M, et al. IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J Clin Microbiol. 2004;42:212-9. [ Links ]
39. Cubillos-Ruíz A, Morales JP, Zambrano MM. Analysis of the genetic variation in Mycobacterium tuberculosis strains by multiple genome alignments. BMC Res Notes. 2008;1:110. [ Links ]

