










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.31 no.1 Bogotá Jan./Mar. 2011
ARTÍCULO ORIGINAL
Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
Recibido: 28/01/10; aceptado:28/09/10
Introduction. Low infection rates in neonates born to HIV-1-seropositive mothers highlight the existence of natural defense mechanisms in the maternal-fetal interface. Human beta defensins (HBDs) inhibit HIV-1 replication in vitro and their variants are associated with HIV-1 resistance/susceptibility.
Objective. Levels of HBD mRNA expression in placentas were obtained from seropositive and healthy mothers to determine whether HIV-1 infection induces anti-viral factors.
Materials and methods. HBD-1, -2 and -3 transcripts were quantified by real time RT-PCR, and A692G/G1654A/A1836G variants in the DEFB1 gene were evaluated by sequencing.
Results. Transcript levels of HBD-1 were significantly higher, and those of HBD-3 were lower in placenta from seropositive mothers compared to controls. Additionally, simultaneous presence of the A692G A/G and A1836G G/G genotypes was associated with high expression of HBD-1 in all populations and the A692G variant in babies born to seropositive mothers was in Hardy-Weinberg disequilibrium.
Conclusion. Contrasting results in levels of HBDs were probably due to viral stimuli and suggest that HIV-1 induce a differential expression of HBDs in placenta and these proteins could be involved in protecting against HIV-1 at least early in pregnancy. However, it was not possible to associate these findings directly with protection against HIV-1 vertical transmission since none of the newborn infants became infected.
Key words: human beta-defensins, placenta, HIV-1; immunity, innate; infectious disease transmission, vertical.
Expresión diferencial en placenta de beta-defensinas humanas y detección de variantes alélicas en el gen DEFB1 de madres positivas para VIH-1
Introducción. Las bajas tasas de infección en neonatos nacidos de madres seropositivas para el VIH-1 resaltan la existencia de mecanismos de defensa natural en la interfase materno-fetal. Las beta-defensinas humanas inhiben la replicación del VIH-1 in vitro y sus polimorfismos están asociados con la resistencia o susceptibilidad al VIH-1.
Objetivo. Comparar los niveles de expresión de ARNm de beta-defensinas humanas en placentas de madres seropositivas y en seronegativas para determinar si la infección por VIH-1 induce factores antivirales que pudieran proteger a los bebés de la transmisión del VIH-1.
Materiales y métodos. Los transcritos de HBD-1, 2 y 3 se cuantificaron por PCR en tiempo real y las variantes A692G/G1654A/A1836G del gen DEFB1 se evaluaron por secuenciación.
Resultados. Los niveles de transcritos de HBD-1 fueron significativamente mayores, y los de HBD-3 fueron menores en placentas de madres seropositivas en comparación con los controles. Además, la presencia simultánea de los genotipos A692G A/G y A1836G G/G se asoció con alta expresión de HBD-1 en toda la población estudiada y la variante A692G estuvo en desequilibrio de Hardy-Weinberg en los bebés nacidos de madres seropositivas.
Conclusión. Los resultados contrastantes de los niveles de HBD se deben, probablemente, a estímulos virales y sugieren que el VIH-1 induce una expresión diferencial de beta-defensinas humanas en placenta y que estas proteínas podrían estar involucradas en la protección contra el VIH-1, al menos, en las etapas tempranas del embarazo. Sin embargo, no fue posible asociar estos hallazgos con la protección contra la transmisión vertical del VIH-1, puesto que ninguno de los bebés adquirió la infección.
Palabras clave: beta-defensinas; placenta; VIH-1, inmunidad innata, transmisión vertical de enfermedad infecciosa.
Similar to other infectious microorganisms, the type 1 human immunodeficiency virus (HIV-1) can be vertically transmitted to the fetus (1,2); in fact, most children acquire this infection through their mothers, in uterus, at delivery or during breast feeding (3-6). The rate of vertical transmission decreased markedly, from 15%-35% to 0.8%-3%, when pregnant women are given zidovudine (AZT) during pregnancy and their children are delivered by cesarean section (7-9).
The transplacental route of infection was the first identified mechanism of mother-to-fetus HIV-1 transmission, after detection of HIV in fetal tissue in 1985 (10,11). In fact, the fetus acquired the HIV-1 as early as eight weeks of gestation (12). Trophoblastic cells express CD4, CCR1, CCR3, CXCR4 and CCR5, all molecules involved in HIV-1 entry (13). In addition, reports have indicated that placental fibroblasts can act as viral reservoirs, capable of infecting other fetal cells (14). This relationship suggested avenues by which the fetus can acquire the infection by an alteration of the maternal-fetal barrier, by cell-to-cell contact from infected cells to trophoblastic cells or by HIV-1 transcytosis (15).
Cases have been documented of neonates born to seropositive (SP) mothers. These are known as HIV-1-exposed seronegatives (ESN), in whom a low rate of infection has been observed even in absence of preventive measures (16,17), a condition that indicates the existence of natural mechanisms for preventing infection during gestation.
The Δ32 mutation is the main genetic mechanism known to confer a high degree of resistance to HIV-1 infection (18,19). Homozygous individuals for the null allele are highly resistant to HIV-1 infection, and the heterozygous individuals exhibit a delay in AIDS progression, including children (18,20-22). However, the homozygous genotype is uncommon and explains only 3.6% of the resistant population to HIV-1 (23-25). In addition, the Δ32 mutation has not been found to confer protection against HIV-1 vertical transmission (22,26,27).
Several reports have indicated that soluble factors, secreted by various types of cells during the immune response, have strong antiviral activity as a primary function or as a “collateral” effect. At least 5 factors have been identified in the maternal-fetus interface: stromal α-chemokine derived factor-1 (SDF-1) (28), leukemia inhibitory factor (LIF) (29), alloantigen-stimulated factor (ASF) (30), RNases associated with human chorionic gonadotropin (hCG) (31-33) and, most recently, the b and α-defensins (34-36). Defensins are small cysteine-rich cationic peptides that exhibit antimicrobial activity against a broad spectrum of microorganisms (37). To date, six human b-defensin (HBD) have been identified and characterized (38). HBD-1 is constitutively expressed by epithelial cells, while HBD-2 and 3 are induced by proinflammatory cytokines and microbes including viruses (39). Exposure of oral epithelial cells to HIV-1 induces mRNA expression for HBD-2 and -3. These defensins inhibit R5 and X4 HIV-1 infection and replication by a direct interaction with virions and by down-regulation oof the CXCR4 coreceptor expression (40,41). In addition, HBD-2 and -3 mRNA are increased in oral mucosa from ESN compared to healthy controls (HC) (42).
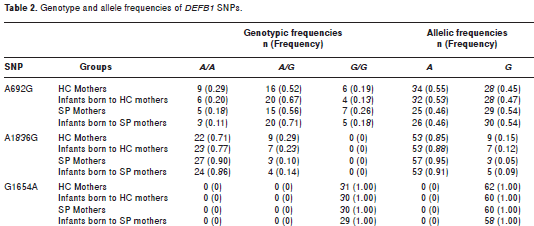
Thirty single-nucleotide polymorphisms (SNP) associated with different ethnic groups have been reported in HBD genes, particularly in DEFB1 and DEFB2 that code for HBD-1 and 2 respectively (43). Previous SNP studies have suggested that -44 C/G (site 668), -20 A/G (site 692) and -52 G/A (site 660) SNPs in DEFB1 are associated with differential risks of vertical transmission of HIV-1 (44-47). Furthermore, the frequency of the SNP genotype A692G G/G in the DEFB1 gene was significantly higher in ESN compared to SP individuals and suggested an association of this SNP with resistance to HIV-1 infection (42). Additional SNPs (692G, G1654A and A1836G) in the DEFB1 gene have characteristics that plausibly link them to differential gene expression or gene product function (43).
Expression of HBD-1, 2, and 3 mRNA in fetal membranes and placenta has been described previously (34,36). However, HBD expression levels in the placenta and other reproductive tissues have not been characterized as potential modifiers of the risk of HIV infection, nor has the influence of HIV-1 in the induction of these factors been explored.
To determine if the transcriptional expression of these proteins is modulated by the exposure to HIV-1, HBD-1, 2 and 3 mRNA levels in the placenta were quantified from HIV-1 positive and negative mothers. In addition, the presence of A692G, G1654A and A1836G SNPs were determined in the DEFB1 gene.
An increased expression of HBD-1 mRNA and a decreased expression of HBD-3 mRNA was found in placentas from HIV-1 positive mothers. In addition, when the SNP genotypes A692G A/G and A1836G G/G were present simultaneously, they were associated with high expression of HBD-1 but not HBD-3. A final observation was that the frequency of A692G SNP was in Hardy Weinberg disequilibrium in children born to HIV-1 positive mothers.
Materials and methods
Study subjects and samples
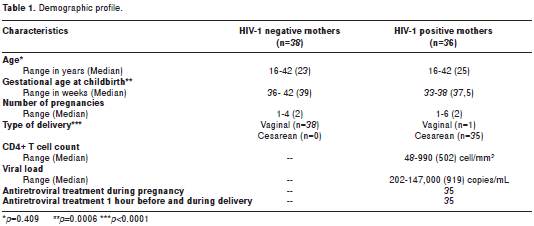
The study enrolled 74 mothers and their infants between 2004 and 2005 from three hospitals in Medellín, Colombia: Hospital Universitario San Vicente de Paul, Hospital General and Clínica Prado. Thirty six HIV-1 positive pregnant women were included. Healthy controls (HC) were 38 pregnant women volunteers with similar ethnic background to the seropositive (SP) individuals. The HC HIV-1 seronegative status was evaluated by an ELISA test, and presence of proviral DNA was ruled out in all HC samples by a negative nested PCR for env HIV-1 gene. Thirty five HIV-1-positive women received antiretroviral chemoprophylaxis during pregnancy, mainly zidovudine, (600 mg/day) plus lamivudine (300 mg/day) (Combivir®) and indinavir (800 mg/ every eight hours (q.8.h) or nelfinavir (750 mg q.8.h) or nevirapine (400 mg/day) according to physician criteria. These women also received zidovudine 2 mg per kilogram weight during first hour (mg/kg/h) before cesarean section and 1 mg/kg/h during labor (Table 1).
At the time of delivery, by labour or caesarean, a 20 ml sample of peripheral blood was collected from the mother. 35-40 ml of cord blood and placenta tissue were stored in RNA later, (QIAgen, Valencia, CA) at -70°C. Approximately 2-3 ml sample of peripheral blood was obtained from neonates during the first 24 hrs postpartum and at 3 and 6 months of age. By determination of viral load in these samples, the occurrence of perinatal infection was evaluated in newborns from HIV-1 positive women. At 18 months, an additional sample was taken to determine the presence of anti-HIV-1 antibodies by an ELISA test. A clear explanation of the objectives and the implications of the results were given to each participant. An institutional-approved informed consent was signed, according to Colombian government resolution 008430 of 1993 legislation.
PCR amplification of CCR5 open reading frame (ORF)
The exon 4 of the CCR5 gene was amplified by PCR as previously described (23). The reactions were undertaken in a PTC-100 peltier Thermal Cycler (MJ research) with the following protocol: 95°C 3 min, 30 cycles of amplification at 95°C x 30 s, 56°C x 30 s and 72°C x 30 s, and finally 72°C x 5 min. The PCR product was separated by electrophoresis in a 2% agarose gel, stained with “SYBR green” and visualized by UV transillumination. For the standard genotype (CCR5/CCR5), a 225bp PCR product was obtained, whereas a product of 193 bp indicated a mutant homozygous genotype (D32/D32). Presence of both bands indicated a heterozygous genotype (CCR5/D32).
Restriction fragment length-polymorphisms (RFLPs) for SNPs A692G, G1654A and A1836G in the DEFB1 gene
Polymorphisms were determined by RFLP-PCR. The primer sequences and protocols were previously described (43). PCR products were digested in a final volume of 15µ containing 1X buffer, 0.2µ of the corresponding restriction enzyme and 4µ of PCR product. The mixture was incubated for 16 h at 37°C. The enzymes used were Bme1390I for the A692G SNP, Bst11071 for the A1836G SNP, and HpyCH4V for the G1654A SNP. The SNPs were verified by sequencing using a commercial service from Macrogen (Macrogen, Seoul, Korea). The sequences were analyzed with MEGA software version 3.1. DEFB1 exon 1 and 2 DNA sequences provided by Genebank NCBI (accession numbers U50930 and U50931, respectively) were used as reference.
Real time RT-PCR assay to quantify HBD mRNA
Total RNA was extracted from epithelial cells with TRizol®Reagent (INVITROGEN), following the manufacturer’s instructions. The amount and purity of the RNA were determined by spectrometry at 260/280 nm using the NanoDrop-1000 (Thermo Scientific). Isolated total RNA was treated with DNase I (Fermentas) to eliminate genomic DNA. cDNA was synthesized using the SuperScript III (Invitrogen) according to the manufacturer’s instructions. Each 25µ-real-time PCR mixture consisted of 3µ of cDNA, 1x reaction buffer, 5mM MgCl2, 0.2 mM dNTPs, 0.05U of platinum Taq DNA polymerase (Invitrogen), 0.2 µM of each primer and SYBR green dye diluted 1:2500 (Sigma, St. Louis. USA). The b-actin RNA was used to normalize the RNA content in each preparation. Thermal conditions and the oligonucleotides have been described previously (40,48,49).
The cycling profiles were: 95°C for 10 min followed by 45 cycles of 95°C for 15 s, 55°C for 30 s for HBD-1 (60°C for 30 s for HBD-2 and HBD-3, and 55°C for 30 s for b-Actin) and 72°C for 30 s. We included a melting curve to confirm the specificity of the PCR product. The PCR products were separated by electrophoresis in a 2% agarose gel, stained with “SYBR green” and visualized by UV transillumination. The band size was estimated with HyperLadder II (Bioline). All real-time RTâPCR amplifications were performed in duplicate and data acquisition was via the Chromo 4TM detector, software 2.03 (MJ Research). Quantity of HBD mRNA was determined by DCt method (50) normalized with the b-actin expression level.
Statistical analysis
Fisher’s-exact tests or chi-square tests were used to compare categorical variables. Differences in HBD mRNA expression levels were tested by Mann-Whitney statistic. Correlations between HBD mRNA expression level and immunological state of HIV-1 positive mothers were evaluated using Spearman’s rank correlation coefficient. A p<0.05 was considered statistically significant. Analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, CA, USA). Allele and genotype frequencies, as well as Hardy-Weinberg equilibrium (HWE) were calculated with GENEPOP software version 3.4. (51). Additionally, ARLEQUIN version 3.1 (52) was used to test for differences in haplotype frequencies.
Results
Demographic data
Thirty-eight HIV-1 negative mothers, 36 HIV-1 positive mothers and their infants were evaluated. One SP woman did not receive treatment during pregnancy or at delivery. Statistically significant differences were observed in gestational age; 33-38 weeks (median= 37.5) for HIV-1 positive mothers and 36-42 weeks (median= 39) for HIV-1 negative mothers, (p<0.001). None of the neonates born to HIV-1 SP mothers acquired HIV-1 infection perinatally. Demographic data are described in table 1.
Low frequency of CCR5 D32 mutation in the entire population
Since the presence of the D32 mutation can skew the results, this mutation was evaluated in all subjects. None exhibited the homozygous D32 genotype. Two infants born to SP mothers, two born to HC mothers and two SP mothers were delta-32/CCR5 heterozygous. No difference was observed between the evaluated groups. In addition, D32 was in Hardy-Weinberg equilibrium in the SP and HC groups, according to the expected frequencies (data not shown).
Hardy-Weinberg disequilibrium of A692G SNP in DEFB1 gene in babies born to SP mothers
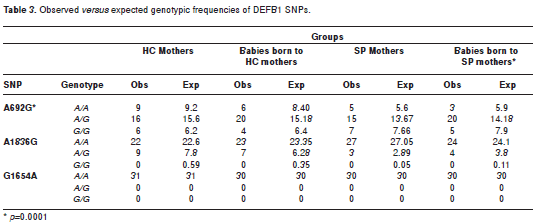
When the allele and genotype frequencies were analyzed for the A692G, G1654A and A1836G SNPs in SP and HC mothers and their infants, statistically significant differences were not observed (table 2). Similarly, the analysis of the observed vs expected frequencies under Hardy-Weinberg equilibrium model did not show statistically significant differences with one exception. The A692G SNP in the infants born to HIV-1 positive mothers was significantly deviant (p=0.0001) (table 3).
Increased expression of HBD-1 mRNA and decreased expression of HBD-3 mRNA in placenta from SP mothers
To determine if differences occurred in the expression of HBD-1, 2 or 3 mRNA among the studied groups, the number of mRNA copies was calculated in duplicate by RT-PCR from placenta tissue. The HBD-2 mRNA expression was examined in 20 placenta samples; it was detected in 2 samples from HC mothers and in 2 samples from SP mothers.
The presence of both defensins, HBD-1 and HBD-3, was observed in a significantly higher number of samples from SP mothers (15/36; 41.7%) compared to samples from HC mothers (4/38; 10.5%); (p=0.003; figure 1). Additionally, HBD-1 mRNA was detected in a significantly higher number of placenta samples from HIV-1 positive mothers (20/36; 55.6%) compared to samples from HC mothers (7/38; 18.4%) (p=0.002; figure 1). In addition, HIV-1 positive mothers had a significantly higher HBD-1 expression in placental tissue than HIV-1 negative mothers (p= 0.004; figure 2a).
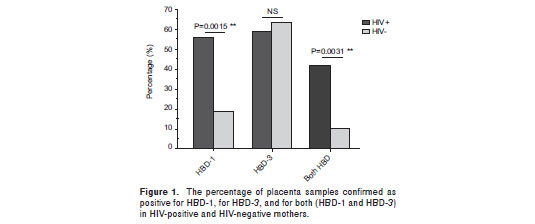
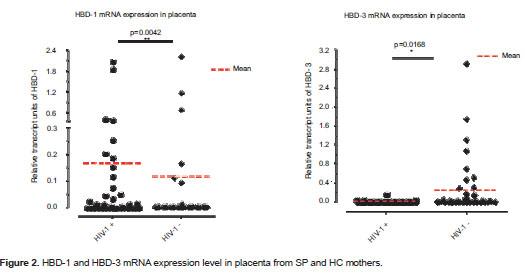
HBD-3 mRNA was detected in 21 of 36 (58.3%) placenta samples from HIV-1 positive mothers, and in 24 of 38 (63.2%) placenta samples from seronegative mothers; this difference was not statistically significant (p=0.81; figure 1). However, when HBD-3 mRNA levels were analyzed, a significantly lower expression was observed in SP than in HC mothers (p=0.017; figure 2b).
Correlation between HBD expression and SNP genotype
To verify if the presence of SNPs was associated with a differential antimicrobial peptide production, the presence of different genotypes in all groups was correlated with (1) the percentage of positive samples for HBD-1 or HBD-3, or both and (2) HBD mRNA expression level. Only one significant correlation was observed between the presence of both the A692G A/G and A1836G G/G SNPs genotypes and a high percentage of positive samples for HBD-1 (p=0.0391).
No correlation between HBD expression and mother-child HLA-I discordance
Class I MHC was characterized to learn if the HLA discordance was associated with a higher antimicrobial peptide production. The degree of allele concordance was correlated with percentage of samples positive for HBD-1/HBD-3, and with HBD mRNA expression level. No significant correlations were observed, suggesting that HLA discordance was not related with HBD expression. However, due to the small sample size, additional studies will be necessary to reenforce this finding.
No correlation between HBD expression level and immunological state of the HIV-1 positive mothers
Finally, no significant correlations were observed between HBD-1 and HBD-3 mRNA levels with viral load (r=0.199, r=0.141, respectively) and CD4+ T cell count (r=-0.299, r=0.288). This indicated that HBD mRNA levels were not influenced directly by the viral load or the immune status of the infected mother.
Discussion
The low rate of infection exhibited by ESN neonates born to HIV-1 SP mothers even in absence of preventive measures (16,17) has suggested the existence of natural mechanisms that prevent this infection during gestation. However, the mechanisms that avoid HIV-1 vertical transmission have not been fully described and represent an important research target. The rate of vertical transmission decreases markedly, from 15%-35% to 0.8%-3%, when pregnant women are given zidovudine (AZT) early in pregnancy and when their children are delivered by cesarean section (7-9). In the current study, all HIV-1 positive mothers but one were treated to reduce the possibility of vertical transmission. Here, in these cases, therefore, the lack of infection may have been due to combination of factors, making it difficult to specify the individual role in natural resistance to HIV-1.
The fetus can acquire the HIV-1 as early as eight weeks of gestation (12), and infection possibly result from an alteration of the maternal-fetal barrier or an interaction of infected cells with trophoblastic cells or by HIV-1 transcytosis (15). Antiretrovirals are administered to HIV-1 positive mothers late in pregnancy; therefore, the HBDs or other antiviral soluble factors must play their protective role early in pregnancy. However, in the current study, HBD expression was not associated with protection against HIV-1 infection.
Although the gestational age at delivery and the type of delivery (vaginal vs cesarean) were significantly different between HIV-1 negative and positive mothers (p=0.0006; p<0.0001, respectively; table 1), previous reports have not associated these variables with a differential expression of defensins. However, these differences are not the issue since defensins are expressed early in pregnancy, being promoted by the cytokne environment and other infectious agents as previosly reported (53). The proposal here is that those HIV-1 stimuli and the SNPs in the DEFB1 gene can differentially modulate the expression levels of HBDs mRNA, and these proteins serve as a defense mechanism involved in protecting against HIV-1 vertical transmission in early pregnancy.
The most important mechanism identified for natural resistance is the D32 mutation in the CCR5 gene (25). However, the homozygous genotype (Δ32/Δ32) was not encountered in the evaluated population; the D32/CCR5 heterozygotes were found in two newborns each of SP and HC mothers. These results agree with previous studies that have reported no association between the presence of the Δ32 allele and reduced risk of perinatally acquired HIV-1 infection (22,26,27). Other mechanisms proposed for the natural resistance to HIV-1 have included soluble factors with antiviral properties for preventing vertical transmission (28-30,32,33,54,55).
Recent studies have shown that HIV-1 induces expression of HBD-2 and -3 in normal human oral epithelial cells and that these peptides inhibit HIV-1 replication through direct binding to the virus and by CXCR4 down regulation (40,41). HBDs expressed by fetal membranes and the placenta (34,36) may create a key barrier to the establishment of different infections (34). However, HBD expression levels at placenta and other reproductive tissues have not been studied as potential modifiers of the risk of HIV infection. In addition, the role of HIV-1 in the induction of HBDs in these tissues has not been explored. The current study quantified the HBD-1, 2 and 3 mRNA from placenta samples of HIV-1 positive mothers and HC by real time RT-PCR. The percentage of placenta samples from SP mothers expressing HBD-1 mRNA was found in comparison higher that among HC mothers (figure 1). Similarly, relative mRNA units of HBD-1 were significantly increased in placenta samples from SP mothers compared to samples from HC (figure 2a).
Previous reports have indicated that HBD-1 is constitutively expressed in placenta (39). The current results may indicate that in SP individuals, the virus has an inducing effect on the expression of HBD-1, but the effect is modulated by the genetic background and gene interactions. This issue requires further assessment; however, in support of these observations, other studies have indicated an up-regulation of HBD-1 during the course of infection and the occurrence of inflammatory processes such as genital lichen sclerosus (56), inflamed dental pulps (57) and chronic sialadenitis (58).
The percentage of placentas positive for the HBD-3 defensin was similar in both groups (figure 1). However, the relative levels of its transcript units were increased in placenta samples from HC mothers compared to those from SP mothers (figure 2b). Possibly, in vivo exposure to HIV-1, under certain circumstances, down-regulates the expression of some soluble factors, including HBD-3. In this regard, HBD-3 promoter is known to contain a functional binding site for the activator protein 1 (AP-1) (59); AP-1-convergent signaling pathways [for example those of toll-like receptor (TLR), and particularly TLR-2] are upstream events in the regulation of HBD-3 expression (59). Furthermore, all TLRs are expressed in human placenta (60,61) and it is known that HIV-1 inhibits the normal activation of these receptors leading to a down-regulation of their downstream gene products (62,63). Therefore, the down-regulation of HBDs by HIV-1 probably occurs through the inhibition of normal activation of some receptors and different transcription factor signaling pathways such as AP-1.
Furthermore, the TH2 cytokines IL-4, IL-10, and IL-13 have shown to down-regulate the HBD-3 expression on keratinocytes (64), but are upregulated during pregnancy (65) and are also induced by HIV-1 (66,67). Therefore, HIV-1 may induce an over-expression of this cytokine profile, thereby decreasing the levels of HBD-3 in placenta tissue of seropositive mothers. These observations support the hypothesis of the differential expression of HBDs induced by HIV-1 stimuli in placenta.
The search for SNPs in DEFB1, DEFB4 and DEFB103 genes; which code for HBD-1, 2 and 3 respectively, represent a remarkable tool in their association with susceptibility to several infectious diseases and in particular with HIV-1. DEFB4 and DEFB103 genes exhibit normal copy number variation (68) and most likely their allelic distribution is in accordance with the Hardy-Weinberg equilibrium (69,70). However, the DEFB1 gene does not exhibit this phenomenon (69) and thereby becomes an excellent target for the search of SNPs associated with degree of susceptibility.
Although the anti-HIV-1 role of HBD-1 has not been previously reported, an association between SNPs in DEFB1 and the risk of acquiring HIV-1 infection has been suggested (44-47). In our group, studies have indicated that the A692G polymorphism is associated with resistance to HIV-1 infection among discordant couples (42). In asociation with the observation that placenta samples from SP mothers exhibited a significantly higher expression of HBD-1 mRNA compared to the placenta tissue from HC mothers (figure 1 and figure 2a), 3 SNPs were identified in the DEFB1 gene (A692G, G1654A, A1836G). These SNPs have been reported to be in high frequencies in several ethnic populations (43), and have characteristics that plausibly link them to differential gene expression or gene product function.
The current study examined the association of SNPs in DEFB1 gene with differential HBDs production, and compared the presence of several genotypes with the percentage of samples positive for HBD-1 and HBD-3, as well as with HBD mRNA expression level. Only the simultaneous presence of A692G A/G and A1836G G/G genotypes was correlated significantly with the high percentage of positive samples for HBD-1 (p=0.04). However significant difference was observed between SP and HC mothers, although this may be a consequence of the small sample size. The A1836G SNP has been positioned within a probable polyadenylation site this mutation possibly affects the transcription or translation of the gene (43). These authors also suggest that SNP A692G can be recognized by the transcription factor NFkB (43) and thereby regulates the HBD-1 production. Because the A allele in the A692G SNP has been previously associated with a reduction in the HBD-1 expression compared to the G allele (71), it is likely that the presence of A1836G G/G and not the A692G A/G SNP explains the up-regulation of the HBD-1 expression. The functional effect of the DEFB1 SNPs is still controversial (71,72) and requires more comprehensive studies. The G1654A SNP is adjacent to the first of six conserved cysteine residues and potentially has an effect on the folding of the peptide, thereby affecting peptide function (43). In the current study population, only the G allele in G1655A SNP occurred and therefore comparisons of its effects were not possible.
No differences in genotype and allele frequencies were detected between the experimental groups for any of the SNPs evaluated (table 2). The genotype frequencies were in Hardy-Weinberg equilibrium, except the SNP A692G in the newborn of SP-mothers (table 3). The deviation in allele frequency may be due to the following causes: (1) the small sample size leading to a perception of genetic drift, or (2) negative selection for the normal genotype. The probability that this pattern was due to chance is very low (p<0.0001). However, the relationship of the A692G SNP to HIV-1 susceptibility was not evaluated since none of the newborns had acquired a perinatal HIV-1 infection.
In sum, these results suggested that the increased levels of HBD-1 found in placenta from HIV-1 positive mothers was due to viral stimuli and potentially represent one of the mechanisms for avoidance of vertical transmission of HIV-1. In contrast, the virus appears to down-regulate HBD-3. Finally, the analysis of three SNPs in the DEFB1 gene did not elucidate the role of these genetic variants in the HBD-1 function, although the A692G A/G and A1836G G/G genotypes may provide a partial explanation for the increased expression of HBD-1 in placental tissue.
In conclusion, the HIV-1 stimuli and the SNPs in the DEFB1 gene are proposed to differentially modulate the expression levels of HBDs, and these proteins are a potential defense mechanism protecting against HIV-1 vertical transmission in early pregnancy.
Acknowledgments
We thank to the staff from Hospital Universitario San Vicente de Paúl, Hospital General and Clínica Prado from Medellín, Colombia for providing the patients.
Conflict of interest
The authors report no competing financial interests exist.
Financing
This study was supported by The Committee for Research Development from the University of Antioquia; the Colombian government institute created to support scientific research (COLCIENCIAS 111534319143).
Corresponding author: María Teresa Rugeles, Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia, Calle 62 No. 52-59, laboratorio 532, Medellín, Colombia. Phone: (574) 219 6551; fax:(574) 219 6482 mtrugel@udea.edu.co
References
1. Lamptey P, Wigley M, Carr D, Collymore Y. De frente a la pandemia del VIH/SIDA. Washington, D.C.: Population Reference Bureau; 2002. [ Links ]
2. Ahumada C, González A, Ribadeneira N, Russo M, Villa L. Making the linkages: HIV/AIDS and sexual and reproductive rights. Ottawa: The Youth Coalition; 2006. [ Links ]
3. UNAIDS. WHO. AIDS epidemeic update. Geneva: UNAIDS; 2007. [ Links ]
4. Chouquet C, Burgard M, Richardson S, Rouzioux C, Costagliola D. Timing of mother-to-child HIV-1 transmission and diagnosis of infection based on polymerase chain reaction in the neonatal period by a non-parametric method. AIDS. 1997;11:1183-4. [ Links ]
5. Rouzioux C, Costagliola D, Burgard M, Blanche S, Mayaux M, Griscelli C, et al. Estimated timing of mother-to-child human immunodeficiency virus type 1 (HIV-1) transmission by use of a Markov model. The HIV Infection in Newborns French Collaborative Study Group. Am J Epidemiol. 1995;142:1330-7. [ Links ]
6. The International Perinatal HIV Group. The mode of delivery and the risk of vertical transmission of human immunodeficiency virus type 1, a meta-analysis of 15 prospective cohort studies. N Engl J Med. 1999;340:977-87. [ Links ]
7. Kind C, Rudin C, Siegrist C, Wyler C, Biedermann K, Lauper U, et al. Prevention of vertical HIV transmission: Additive protective effect of elective cesarean section and zidovudine prophylaxis. AIDS. 1998;12:205-10. [ Links ]
8. Lansky A, Jones J, Wan P, Lindegren M, Wortley P. Trends in zidovudine prescription for pregnant women infected with HIV. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:289-92. [ Links ]
9. Sperling R, Shapiro D, Coombs R, Todd J, Herman S, McSherry G, et al. Maternal viral load, zidovudine treatment, and the risk of transmission of human immunodeficiency virus type 1 from mother to infant. Pediatric AIDS Clinical Trials Group Protocol 076 Study Group. N Engl J Med. 1996;335:1621-9. [ Links ]
10. Jovaisas E, Koch MA, Schafer A, Stauber M, Lowenthal D. LAV/HTLV-III in 20-week fetus. Lancet. 1985;2:1129. [ Links ]
11. Lapointe N, Michaud J, Pekovic D, Chausseau JP, Dupuy JM. Transplacental transmission of HTLV-III virus. N Engl J Med. 1985;312:1325-6. [ Links ]
12. Lewis SH, Reynolds-Kohler C, Fox HE, Nelson JA. HIV-1 in trophoblastic and villous Hofbauer cells, and haematological precursors in eight-week fetuses. Lancet. 1990;335:565-8. [ Links ]
13. Mognetti B, Moussa M, Croitoru J, Menu E, Dormont D, Roques P, et al. HIV-1 co-receptor expression on trophoblastic cells from early placentas and permittivity to infection by several HIV-1 primary isolates. Clin Exp Immunol. 2000;119:486-92. [ Links ]
14. Fazeley F, Hu J, Thirkill TL, Douglas GC. Infection of primary human placental fibroblasts with HIV-1, HIV-2, and SIV. Arch Virol. 1997;142:2237-48. [ Links ]
15. Lagaye S, Derrien M, Menu E, Coito C, Tresoldi E, Mauclere P, et al. Cell-to-cell contact results in a selective translocation of maternal human immunodeficiency virus type 1 quasispecies across a trophoblastic barrier by both transcytosis and infection. J Virol. 2001;75:4780-91. [ Links ]
16. Shearer GM, Clerici M. Protective immunity against HIV infection: Has nature done the experiment for us? Immunol Today. 1996;17:21-4. [ Links ]
17. Shacklett BL. Understanding the "lucky few": The conundrum of HIV-exposed, seronegative individuals. Curr HIV/AIDS Rep. 2006; 3: 26-31. [ Links ]
18. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367-77. [ Links ]
19. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722-5. [ Links ]
20. Buseyne F, Janvier G, Teglas JP, Ivanoff S, Burgard M, Bui E, et al. Impact of heterozygosity for the chemokine receptor CCR5 32-bp-deleted allele on plasma virus load and CD4 T lymphocytes in perinatally human immunodeficiency virus-infected children at 8 years of age. J Infect Dis. 1998;178:1019-23. [ Links ]
21. Marmor M, Sheppard HW, Donnell D, Bozeman S, Celum C, Buchbinder S, et al. Homozygous and heterozygous CCR5-Delta32 genotypes are associated with resistance to HIV infection. J Acquir Immune Defic Syndr. 2001; 27:472-81. [ Links ]
22. Villalba N, Pérez-Olmeda M, de José M, Hernández M, Sirera R, Espanol T, et al. CCR5 genotype and human immunodeficiency virus type 1 infection in perinatally exposed infants. Eur J Clin Microbiol Infect Dis. 1999;18:389-91. [ Links ]
23. Díaz FJ, Vega JA, Patiño PJ, Bedoya G, Nagles J, Villegas C, et al. Frequency of CCR5 delta-32 mutation in human immunodeficiency virus (HIV)-seropositive and HIV-exposed seronegative individuals and in general population of Medellin, Colombia. Mem Inst Oswaldo Cruz. 2000;95:237-42. [ Links ]
24. Rugeles MT, Solano F, Díaz FJ, Bedoya VI, Patiño PJ. Molecular characterization of the CCR 5 gene in seronegative individuals exposed to human immunodeficiency virus (HIV). J Clin Virol. 2002;23:161-9. [ Links ]
25. Huang Y, Paxton W, Wolinsky S, Zhang L, He T, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996; 2: 1240-3. [ Links ]
26. Edelstein RE, Arcuino LA, Hughes JP, Melvin AJ, Mohan KM, King PD, et al. Risk of mother-to-infant transmission of HIV-1 is not reduced in CCR5/delta32ccr5 heterozygotes. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:243-6. [ Links ]
27. Rousseau CM, Just JJ, Abrams EJ, Casabona J, Stein Z, King MC. CCR5del32 in perinatal HIV-1 infection. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:239-42. [ Links ]
28. Coulomb-L´Hermine A, Emilie D, Durand-Gasselin I, Galanaud P, Chaouat G. SDF-1 production by placental cells: A potential mechanism of inhibition of mother-to-fetus HIV transmission. AIDS Res Hum Retroviruses. 2000;16:1097-8. [ Links ]
29. Patterson BK, Behbahani H, Kabat WJ, Sullivan Y, O'Gorman MR, Landay A, et al. Leukemia inhibitory factor inhibits HIV-1 replication and is upregulated in placentae from nontransmitting women. J Clin Invest. 2001; 107: 287-94. [ Links ]
30. Bedoya VI, Jaimes FA, Delgado JC, Rugeles C, Usuga X, Zapata W, et al. Fetal-maternal HLA-A and -B discordance is associated with placental RNase expression and anti-HIV-1 activity. Curr HIV Res. 2008;6:380-7. [ Links ]
31. Lee-Huang S, Huang PL, Sun Y, Kung HF, Blithe DL, Chen HC. Lysozyme and RNases as anti-HIV components in beta-core preparations of human chorionic gonadotropin. Proc Natl Acad Sci USA. 1999;96:2678-81. [ Links ]
32. Bourinbaiar AS, Nagorny R. Inhibitory effect of human chorionic gonadotropin (hCG) on HIV-1 transmission from lymphocytes to trophoblasts. FEBS Lett. 1992;309:82-4. [ Links ]
33. Polliotti BM, Gnall-Sazenski S, Laughlin TS, Miller RK. Inhibitory effects of human chorionic gonadotropin (hCG) preparations on HIV infection of human placenta in vitro. Placenta. 2002;23(Suppl.A):S102-6. [ Links ]
34. King AE, Paltoo A, Kelly RW, Sallenave JM, Bocking AD, Challis JR. Expression of natural antimicrobials by human placenta and fetal membranes. Placenta. 2007;28:161-9. [ Links ]
35. Svinarich DM, Gómez R, Romero R. Detection of human defensins in the placenta. Am J Reprod Immunol. 1997;38:252-5. [ Links ]
36. Zhao C, Wang I, Lehrer RI. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett. 1996;396:319-22. [ Links ]
37. Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: More than just microbicidal. Trends Immunol. 2002;23:291-6. [ Links ]
38. Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol. 2004;22:181-215. [ Links ]
39. Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nat Rev Immunol. 2006;6:447-56. [ Links ]
40. Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS. 2003;17:F39-48. [ Links ]
41. Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra GM, et al. Human beta-defensins suppress human immunodeficiency virus infection: Potential role in mucosal protection. J Virol. 2005;79:14318-29. [ Links ]
42. Zapata W, Rodríguez B, Weber J, Estrada H, Quiñones-Mateu M, Zimermman P, et al. Increased levels of human beta-defensins mRNA in sexually HIV-1 exposed but uninfected individuals. Curr HIV Res. 2008;6:531-8. [ Links ]
43. Jurevic RJ, Chrisman P, Mancl L, Livingston R, Dale BA. Single-nucleotide polymorphisms and haplotype analysis in beta-defensin genes in different ethnic populations. Genet Test. 2002;6:261-9. [ Links ]
44. Braida L, Boniotto M, Pontillo A, Tovo PA, Amoroso A, Crovella S. A single-nucleotide polymorphism in the human beta-defensin 1 gene is associated with HIV-1 infection in Italian children. AIDS. 2004;18:1598-600. [ Links ]
45. Milanese M, Segat L, Pontillo A, Arraes LC, de Lima Filho JL, Crovella S. DEFB1 gene polymorphisms and increased risk of HIV-1 infection in Brazilian children. AIDS. 2006;20:1673-5. [ Links ]
46. Segat L, Milanese M, Boniotto M, Crovella S, Bernardon M, Costantini M, et al. DEFB-1 genetic polymorphism screening in HIV-1 positive pregnant women and their children. J Matern Fetal Neonatal Med. 2006;19:13-6. [ Links ]
47. Baroncelli S, Ricci E, Andreotti M, Guidotti G, Germano P, Marazzi MC, et al. Single-nucleotide polymorphisms in human beta-defensin-1 gene in Mozambican HIV-1-infected women and correlation with virologic parameters. AIDS. 2008;22:1515-7. [ Links ]
48. Trabattoni D, Caputo SL, Maffeis G, Vichi F, Biasin M, Pierotti P, et al. Human alpha defensin in HIV-exposed but uninfected individuals. J Acquir Immune Defic Syndr. 2004;35:455-63. [ Links ]
49. Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863-9. [ Links ]
50. Walker NJ. Tech.Sight. A technique whose time has come. Science. 2002;296:557-9. [ Links ]
51. Raymond M, Rousset F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248-9. [ Links ]
52. Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47-50. [ Links ]
53. Soto E, Espinoza J, Nien JK, Kusanovic JP, Erez O, Richani K, et al. Human beta-defensin-2: A natural antimicrobial peptide present in amniotic fluid participates in the host response to microbial invasion of the amniotic cavity. J Matern Fetal Neonatal Med. 2007;20:15-22. [ Links ]
54. Farquhar C, Mbori-Ngacha DA, Redman MW, Bosire RK, Lohman BL, Piantadosi AL, et al. CC and CXC chemokines in breastmilk are associated with mother-to-child HIV-1 transmission. Curr HIV Res. 2005;3:361-9. [ Links ]
55. Farquhar C, van Cott TC, Mbori-Ngacha DA, Horani L, Bosire RK, Kreiss JK, et al. Salivary secretory leukocyte protease inhibitor is associated with reduced transmission of human immunodeficiency virus type 1 through breast milk. J Infect Dis. 2002;186:1173-6. [ Links ]
56. Gambichler T, Skrygan M, Tigges C, Kobus S, Glaser R, Kreuter A. Significant upregulation of antimicrobial peptides and proteins in lichen sclerosus. Br J Dermatol. 2009;161:1136-42. [ Links ]
57. Paris S, Wolgin M, Kielbassa AM, Pries A, Zakrzewicz A. Gene expression of human beta-defensins in healthy and inflamed human dental pulps. J Endod. 2009;35:520-3. [ Links ]
58. Pantelis A, Wenghoefe M, Haas S, Merkelbach-Bruse S, Pantelis D, Jepsen S, et al. Down regulation and nuclear localization of human beta-defensin-1 in pleomorphic adenomas of salivary glands. Oral Oncol. 2009; 45: 526-30. [ Links ]
59. Menzies BE, Kenoyer A. Signal transduction and nuclear responses in Staphylococcus aureus-induced expression of human beta-defensin 3 in skin keratinocytes. Infect Immun. 2006;74:6847-54. [ Links ]
60. Ma Y, Krikun G, Abrahams VM, Mor G, Guller S. Cell type-specific expression and function of toll-like receptors 2 and 4 in human placenta: Implications in fetal infection. Placenta. 2007;28:1024-31. [ Links ]
61. Patni S, Wynen LP, Seager AL, Morgan G, White JO, Thornton CA. Expression and activity of Toll-like receptors 1-9 in the human term placenta and changes associated with labor at term. Biol Reprod. 2009;80:243-8. [ Links ]
62. Martinelli E, Cicala C, van Ryk D, Goode DJ, Macleod K, Arthos J, et al. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci USA. 2007;104:3396-401. [ Links ]
63. Pathak S, De Souza GA, Salte T, Wiker HG, Asjo B. HIV induces both a down-regulation of IRAK-4 that impairs TLR signalling and an up-regulation of the antibiotic peptide dermcidin in monocytic cells. Scand J Immunol. 2009;70:264-76. [ Links ]
64. Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006;121:332-8. [ Links ]
65. Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, et al. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin Exp Immunol. 1996;106:127-33. [ Links ]
66. Becker Y. The spreading of HIV-1 infection in the human organism is caused by fractalkine trafficking of the infected lymphocytes âa review, hypothesis and implications for treatment. Virus Genes. 2007;34:93-109. [ Links ]
67. Buonaguro L, Tornesello ML, Gallo RC, Marincola FM, Lewis GK, Buonaguro FM. Th2 polarization in peripheral blood mononuclear cells from human immunodeficiency virus (HIV)-infected subjects, as activated by HIV virus-like particles. J Virol. 2009;83:304-13. [ Links ]
68. Hollox EJ, Armour JA, Barber JC. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am J Hum Genet. 2003;73:591-600. [ Links ]
69. Boniotto M, Ventura M, Eskdale J, Crovella S, Gallagher G. Evidence for duplication of the human defensin gene DEFB4 in chromosomal region 8p22-23 and implications for the analysis of SNP allele distribution. Genet Test. 2004;8:325-7. [ Links ]
70. Linzmeier RM, Ganz T. Human defensin gene copy number polymorphisms: Comprehensive analysis of independent variation in alpha- and beta-defensin regions at 8p22-p23. Genomics. 2005;86:423-30. [ Links ]
71. Milanese M, Segat L, Crovella S. Transcriptional effect of DEFB1 gene 5´ untranslated region polymorphisms. Cancer Res. 2007; 67: 5997. [ Links ]
72. Sun CQ, Arnold R, Fernández-Golarz C, Parrish AB, Almekinder T, He J, et al. Human beta-defensin-1, a potential chromosome 8p tumor suppressor: Control of transcription and induction of apoptosis in renal cell carcinoma. Cancer Res. 2006;66:8542-9. [ Links ]

