










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157On-line version ISSN 2590-7379
Biomédica vol.31 no.4 Bogotá Oct./Dec. 2011
COMUNICACIÓN BREVE
1Escuela de Medicina y Ciencias de la Salud, Universidad del Rosario, Bogotá, D.C, Colombia
2Instituto of Biotecnología, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
Author contributions: Ricardo A. Cifuentes: data analysis and manuscript writing. Emiliano Barreto: review and supervision of the whole process.
Recibido: 01/02/11; aceptado:27/07/11
Introduction: The different ways for selecting single nucleotide polymorphisms have been related to paradoxical conclusions about their usefulness in predicting chronic fatigue syndrome even when using the same dataset.
Objective: To evaluate the efficacy in predicting this syndrome by using polymorphisms selected by a supervised approach that is claimed to be a method that helps identifying their optimal profile.
Materials and methods: We eliminated those polymorphisms that did not meet the Hardy-Weinberg equilibrium. Next, the profile of polymorphisms was obtained through the supervised approach and three aspects were evaluated: comparison of prediction accuracy with the accuracy of a profile that was based on linkage disequilibrium, assessment of the efficacy in determining a higher risk stratum, and estimating the algorithm influence on accuracy.
Results: A valid profile (p<0.01) was obtained with a higher accuracy than the one based on linkage disequilibrium, 72.8 vs. 62.2% (p<0.01). This profile included two known polymorphisms associated with chronic fatigue syndrome, the NR3C1_11159943 major allele and the 5HTT_7911132 minor allele. Muscular pain or sinus nasal symptoms in the stratum with the profile predicted V with a higher accuracy than those symptoms in the entire dataset, 87.1 vs. 70.4% (p<0.01) and 92.5 vs. 71.8% (p<0.01) respectively. The profile led to similar accuracies with different algorithms.
Conclusions: The supervised approach made it possible to discover a reliable profile of polymorphisms associated with this syndrome. Using this profile, accuracy for this dataset was the highest reported and it increased when the profile was combined with clinical data.
Key words: genetic polymorphism, chronic fatigue syndrome, computational biology, artificial intelligence, systems biology, linkage disequilibrium.Selección supervisada de polimorfismos de nucleótido único en el síndrome de fatiga crónica
Introducción. Las diferentes formas de seleccionar polimorfismos de nucleótido único se han relacionado con conclusiones paradójicas respecto a su utilidad para predecir el síndrome de fatiga crónica, incluso utilizando los mismos datos.
Objetivo. Evaluar la eficacia para predecir este síndrome de los polimorfismos seleccionados mediante un enfoque supervisado, método que permite ayudar a identificar el perfil óptimo de los polimorfismos.
Materiales y métodos. Se eliminaron los polimorfismos que no estaban en equilibrio de Hardy- Weinberg. Luego obtuvimos el perfil de polimorfismos mediante el enfoque supervisado y evaluamos tres aspectos: comparación de la exactitud de predicción con la del perfil obtenido mediante una selección basada en el desequilibrio de ligamiento, evaluación de la eficacia para determinar un estrato con mayor riesgo y estimación de la influencia del algoritmo de clasificación sobre la exactitud de predicción.
Resultados. Se obtuvo un perfil válido (p<0,01) con mayor exactitud que el basado en el desequilibrio de ligamiento, 72,8 Vs. 62,2 % (p<0,01), que incluyó el alelo mayor de NR3C1_11159943 y el menor de 5HTT_7911132, conocidos polimorfismos asociados a este síndrome. El dolor muscular o los síntomas de los senos paranasales en el estrato con el perfil, predijeron la presencia del síndrome con mayor exactitud que estos síntomas en toda la población, 87,1 % Vs. 70,4 % (p<0,01) y 92,5 % Vs. 71,8 % (p<0,01) respectivamente. El perfil llevó a una exactitud similar con diferentes algoritmos.
Conclusiones. El enfoque supervisado permitió descubrir un perfil válido y confiable de polimorfismos asociado al síndrome de fatiga crónica. Se encontró la mayor exactitud reportada con estos datos que aumentó al combinarse con las variables clínicas.
Palabras clave: polimorfismo genético, síndrome de fatiga crónica, biología computacional, inteligencia artificial, biología de sistemas, desequilibrio de ligamiento
Groups of single nucleotide polymorphisms (SNP) could be markers for the genetic predisposition towards complex diseases given that a significant fraction of disease susceptibility may be explained by the relatively modest effects of a small number of common genetic variants (1). Thus, profiles of SNPs could be used as objective markers for diseases that are diagnosed by means of subjective symptoms such as the chronic fatigue syndrome (2).
However, to discover clinically relevant profiles, the informative SNPs must be selected (3). In the case of high-dimensional data, the performance of most classification algorithms suffers as the number of features becomes excessively large (4). Moreover, in a small dataset, there is no single solution if a training set contains only a limited subset of all possible instances as each distinct training instance removes those possible SNP profiles where the guesses are wrong. Thus, the selection of SNPs is necessary since the number of possible instances increases when the number of variables does (5).
The algorithms for SNP selection typically use the genetic concept of linkage disequilibrium or, in other words, when the presence of one SNP predicts the presence of another SNP (6). Then, based on measurements of linkage disequilibrium such as r2 (7), a group of SNPs is selected to represent all the SNPs in a given dataset (8, 9). Other test statistic approaches such as feature ranking, feature subset selection constructive induction, induction scan statistics, score statistics, weighted-average statistics and the supervised recursive feature addition have been proposed for SNP selection in genetic association studies (10, 11). The supervised approach is claimed by its authors to be a method that helps to identify the optimal subset of SNPs necessary for discovering the variations associated with disease (10).
With regards to the chronic fatigue syndrome, different researchers reached contradictory results on the usefulness of SNPs for disease prediction in spite of the fact that they analyzed the same dataset of SNPs. However, there were differences in their methodologies.
A group of researchers from the United States that evaluated all the possible combinations of SNPs by using an enumerative search concluded that SNP profiles improve accuracy in predicting chronic fatigue syndrome (12). In contrast, a group from Trento, Italy made a selection with a supervised approach based on entropy and concluded that the SNPs in this dataset were not useful for prediction of the chronic fatigue syndrome (Bassetti M, Bernabe M, Borile M, Desilvestro C, Fedrizzi T, Giordani A, et al. Validation of CFS classification with different data sources. Critical Assessment of Microarray Data Analysis Conference, June 2006). However, these studies had differences in the SNP selection, the use of the Hardy-Weinberg equilibrium as a criterion for SNP exclusion, and the learning algorithm that was chosen for prediction. These differences in methodology showed a need for further research to evaluate the supervised approach.
As a consequence, the aim of this study was to evaluate the efficacy of the supervised approach based on entropy in selecting useful SNPs for predicting the chronic fatigue syndrome. That is the reason we evaluated three different aspects: comparison of the prediction accuracy of the supervised approach with the one that was based on linkage disequilibrium for SNP selection, assessment of the efficacy of the SNPs selected by the supervised approach in determining a population with a higher risk of chronic fatigue syndrome, and an estimate of the machine learning algorithm influence on the prediction accuracy.
Materials and methods
Database cleansing
From the SNP dataset, provided by the Critical Assessment of Microarray Data Analysis Conference, June 2006 (CAMDA 2006), available at http://www.camda.duke.edu/camda06/datasets/, we eliminated 9 of the 44 SNP that were not in the Hardy-Weinberg equilibrium. All of the excluded SNPs had fewer heterozygous genotypes than expected based on allele frequencies, something that was highly suggestive of technical error. Next, those subjects that did not have SNP data were excluded from the analysis thus leaving 43 cases of patients with chronic fatigue syndrome and 58 non-fatigued controls for analysis as previously described (12).
Comparison of SNP selection approaches
Two different approaches were used for selecting SNPs. One approach kept the SNPs that best represent the other SNPs in each one of the groups that were in LD (1). The first step was to determine the haplotype frequencies for each pair of SNPs by means of the expectation-maximization algorithm (8). Next, the r2 measure for each pair of SNPs in the same chromosome was calculated and the ones which were highly significant (p<0.01) were determined. This is supported by the fact that r2 times N (number of subjects in the study) is equal to the chi square value with 1 degree of freedom for the case of two loci with two alleles each. Finally, those SNPs for which the lowest highly significant r2 value was higher than the lowest highly significant r2 value of the other SNPs were kept.
The other approach was to select SNPs through the supervised approach. This was done with the CfsSubsetEval function of the Weka software® (13) that chose the group of SNPs with the highest correlation to the diagnostic categories, namely, fatigued patients with chronic fatigue syndrome and non-fatigued controls. At the same time, this group also included only those SNPs with the lowest possible correlation to each other (14). This function used a best first search to determine the combinations of SNPs to be evaluated by the CfsSubsetEval function. A ten-fold cross validation was used to measure the generalization performance of the combinations in each of the ten different partitions of the dataset (15). The selected SNPs were those that were chosen by the CfsSubsetEval function from any of the ten partitions.
Next by using the SPSS® software, two predictive models, one for each group of SNPs selected by the two previously described approaches, were generated by logistic regression as described (Lee E, Cho S, Park T. Integration of expression data and genotype data: application of chronic fatigue syndrome data. Critical Assessment of Microarray Data Analysis Conference, June 2006). Observations with missing values were not taken into account and values were assigned to the SNPs according to the codominant inheritance as follows (12): two points for the homozygous genotype with two minor alleles, one point for the heterozygous genotype and zero points for the homozygous genotype with two major alleles.
In the case of sex linked SNPs, two points were assigned when the genotype was hemizygous for the minor allele and zero points when it was hemizygous for the major allele.
The SNP profiles that were kept in each model were chosen by forward selection and backward elimination based on the accuracy achieved with a leave-one-out cross validation (15). To compare the two models, the mean accuracies and their 95 percent confidence intervals were obtained in 30 different test sets. Each test set had 20% of the observations and these had the same percentage of both cases and controls that the original dataset did. This guaranteed that each class was properly represented in the test set which the leave-one-out cross validation does not do. The test sets were selected after setting a random seed by using the Bernoulli formula in the Compute Variable option in the Transform menu at SPSS®. Next, the validity of the model based on the supervised approach was evaluated by means of a permutation test. We created 200 shuffled versions of the data by randomly relabeling subjects as cases or controls and keeping an equal number of subjects in each category, as described (12).
Efficacy in determining a population with higher risk
The efficacy of the SNP profile from the model based on the supervised approach to determine a population with a higher risk of chronic fatigue syndrome was evaluated by a comparison between the prediction accuracy in the general population of the dataset and the prediction accuracy in the stratum that had the SNP profile associated with chronic fatigue syndrome, under nonspecific clinical conditions. Briefly, the accuracy values in both groups when there is one symptom, for instance sore throat was compared. The difference in the values of sensitivity, specificity and the positive predictive value (PPV) was also assessed (16). All these values were procured in 30 randomly generated test sets of 20% of the observations. Statistical significance of the difference was evaluated by T-test and comparison of their 95% confidence intervals. The same analysis was done with each one of the symptoms that were registered in the dataset: tender nodes, diarrhea, exertion fatigue, muscle pain, joint pain, fever, chills, unrefreshing sleep, sleep problems, headache, memory and concentration problems, nausea, abdominal pain, shortness of breath, photophobia, depression and sinus nasal problems.
Estimate of machine learning algorithm influence on prediction accuracy
The reliability of the SNP profile selected by the supervised approach was assessed by a comparison of the prediction accuracies obtained with different types of machine learning algorithms. To do this, all the algorithms included in the Weka® software were run after which they were divided into one of the five main types of algorithms: Functions, Bayesians, Instance Based Learners, Decision Trees and Rules. Then, ones that had the highest prediction accuracy in each of these groups were compared. The accuracies were procured by using a ten-fold cross-validation. The statistical significance of the comparison was evaluated with the Kruskal- Wallis test by using the SPSS® software. This nonparametric test was chosen as we did not assume a normal distribution of the data, which would not be the case with the analogous one-way analysis of variance, ANOVA (17). In this case, we used the data in each one of the ten partitions from the cross-validation to calculate each one of the mean accuracies.
Results
With the approach that was based on linkage disequilibrium, we kept 13 SNPs. In general, one SNP was selected from each one of the seven groups of SNPs that was in linkage disequilibrium. Each group corresponded to one of the following seven genes: Glucocorticoid receptor (NR3C1), corticotropin releasing hormone receptor-2 (CRHR2), tryptophan hidroxilase (TPH2), serotonin receptor 2A (HTR2A), corticotropin releasing hormone receptor-1 (CRHR1), serotonin transporter (5HTT) and catechol-O-methyltransferase (COMT). However, there were two exceptions: the two SNPs from the HTR2A gene and the SNPs in the TPH2 gene. The SNPs from the HTR2A gene were kept because there was only one r2 measure. In the case of the TPH2 gene, one SNP was not in linkage disequilibrium so we kept this single SNP and the one that best represented the group of SNPs that were in linkage disequilibrium. So, we kept 9 SNPs from the 7 groups in linkage disequilibrium and the 4 SNPs that were not in linkage disequilibrium: two from the tyrosine hidroxilase gene (TH) and two from genes that had only one SNP in the dataset: pro-opiomelanocortin (POMC) and monoamine oxidase B (MAOB) genes (Table 1).
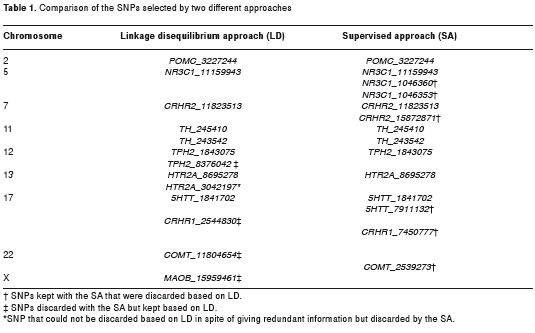
With the supervised approach, 14 SNPs were kept. In 8 cases, the SNPs were the same as those kept with the other approach. However, in some cases, this approach was more conservative because it held on to more than one SNP from some groups in linkage disequilibrium. At the same time, it eliminated some SNPs that had a low correlation with the diagnosis despite the fact that they represented other SNPs. The supervised approach also eliminated one of the two SNPs from the HTR2A gene that could not be differentiated by the approach based on linkage disequilibrium (Table 1).
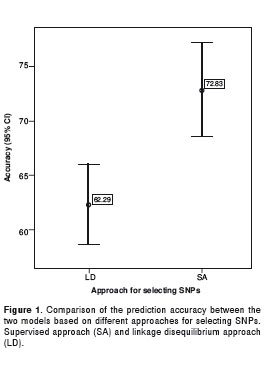
With regards to the purpose of comparing the two selection approaches, the models that were built showed different prediction accuracies. The model based on the supervised approach had a higher accuracy than the model based on linkage disequilibrium, 74.4% and 65.6% respectively, according to the result of the leave-one-out cross validation. Similar mean accuracies were obtained in the test sets, 72.8% and 62.2% respectively, p=4E-04 (Figure1).
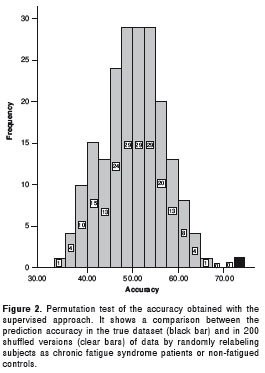
In detail, the model based on linkage disequilibrium showed an association of chronic fatigue syndrome with the presence of the major alleles of NR3C1_11159943 and TH_243542, and the minor allele of TPH_1843075. Only NR3C1_11159943 made a significant contribution to the model (p=0.015). In contrast, the model based on the supervised approach showed that the chronic fatigue syndrome was associated with the presence of the major alleles of NR3C1_11159943, CRHR1_7450777, and TH_245410, and minor alleles of 5HTT_7911132, TPH2_1843075, and HTR2A_8695278. Of the SNPs in the model only NR3C1_11159943 and 5HTT_7911132 made a significant contribution (p=0.01 and p=0.04 respectively). The permutation test showed that all 200 shuffled versions had lower accuracy than the actual version (Figure 2).
With regards to the efficacy of the SNPs selected through the supervised approach to determine a population with a higher risk, 37 subjects had the SNP profile associated with chronic fatigue syndrome. In this stratum, the presence of muscular pain led to a higher prediction accuracy than the presence of this symptom in the total population of the dataset, 87.1% vs. 70.4% respectively, p= 3E-07. In this stratum, the presence of sinus nasal symptoms also led to a higher prediction accuracy than the presence of this symptom in the general population, 92.5% vs. 71.8% respectively, p=2E-11 (Table 2).
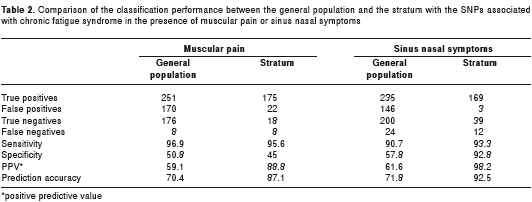
When there was muscular pain, the probability of having chronic fatigue syndrome was also higher (88.8%) in the stratum with the SNPs profile associated with chronic fatigue syndrome than in the general population (59.6%) based on the PPV values. However, the presence of muscular pain in chronic fatigue syndrome or its absence in controls were not higher in the stratum with the SNP profile than in the general population as indicated by the values of sensitivity and specificity, 95.6% vs. 96.9% and 45% vs. 50.8% respectively.
Regarding the probability of having chronic fatigue syndrome when there were sinus nasal symptoms, this was also much higher (98.2%) in the stratum with the SNPs profile associated with chronic fatigue syndrome than in the general population (61.6%) according to the PPV values. The presence of sinus nasal symptoms in chronic fatigue syndrome was a little higher in the stratum with the SNP profile than in the general population as indicated by the values of sensitivity, 93.3% vs. 90.7% respectively. But furthermore, the absence of sinus nasal symptoms in controls was much higher in the stratum with the SNP profile than in the general population as indicated by the values of specificity, 92.8% vs.57.8% respectively.
With regards to the influence on the accuracy of data mining techniques, the algorithms that achieved the highest accuracies among all of the algorithms already included in the Weka® software were: Decorate-Simple Logistic and Decorate-Logistic (76% and 74% respectively) in the group of Functions, Complement Naive Bayes (72%) in the group of Bayesian Learners, One-R Multiboost (73%) in the group of Rules, Kstar (68.9%) in the group of Instance Based Learners, and ADTree (66.6%) in the group of Decision Trees. However, in spite of the fact that the Functions tended to get higher accuracies, the differences in prediction accuracy among these algorithms were not significant (Figure 3).
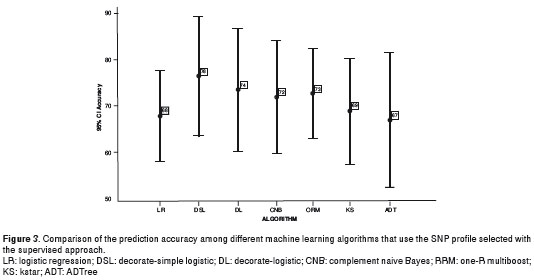
Discussion
The supervised approach based on entropy made it possible to find a SNP profile useful for prediction of chronic fatigue syndrome in the individuals included in the study. This approach, which takes into account the correlation with diagnostic categories, selected a SNP profile that predicted the chronic fatigue syndrome with the highest accuracy reported so far. It also determined a stratum of the population with a higher risk of having this disease in presence of nonspecific symptoms. Furthermore, the supervised approach enabled us to find a reliable SNP profile since similar results were produced while using different machine learning algorithms.
The classification accuracy with the supervised approach was not only 10 points above the accuracy of the approach based on linkage disequilibrium, but also higher than the 64.5% based on crossvalidation results reported by another research group that also used the same data and an approach based on LD (Lim S, Le W, Hu P, Xing B, Greenwood CMT, Bayene J. Integration of clinical, SNP, and microarray gene expression measurements in prediction of Chronic Fatigue Syndrome. Critical Assessment of Microarray Data Analysis Conference, June 2006). Moreover, the accuracy with the supervised approach was not significantly different from the highest reported of 76% (12) that was only achieved in a training set of observations. In other words, this accuracy was not obtained by the accepted methods for determining the prediction such as cross-validation or the estimate in an independent test set.
In addition, the model based on selection by a supervised approach demonstrated statistical and biological validity. The SNPs in the model predicted the dummy classifications with lower accuracies than the actual one. Furthermore, the two significant SNPs in the model were genetic variants with an already established association with chronic fatigue syndrome. NR3C1 has been implicated as regulator of the hypothalamic-pituitary-adrenal axis (18) and of the immune function: the increase in its ligand affinity has led to a reduction in T cell numbers and to a change in susceptibility to autoimmune diseases (19). A higher frequency of the NR3C1_11159943 major allele has also been reported in patients with chronic fatigue syndrome (20). Moreover, the model associated the syndrome with the minor allele of 5HTT_7911132, a SNP that belongs to a gene that decreases the level of active serotonin when the allelic variants with increased transcriptional activity are present (21). Taken together, these findings agree with an additive effect that leads to a lower level of cortisol (22, 23) and to alterations in the neurotransmission and in the immune function (24, 25) already described in the chronic fatigue syndrome (Figure 4).
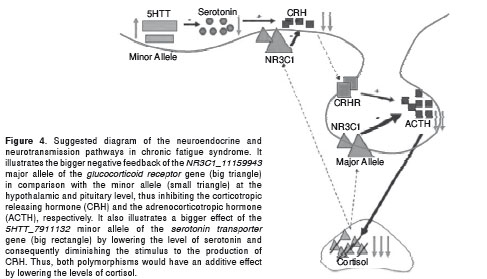
With regards to the finding that there is a higher probability of the group of people with the SNP profile associated with chronic fatigue syndrome having the disease when there were muscular pain or sinus nasal symptoms, it is remarkable that this was discovered through the leave-one-out cross validation. This offers the chance to squeeze the maximum out of a small dataset and to procure as accurate an estimate as possible (15) such as in the cases of using molecular markers to predict the response cancer patients will have to a treatment (26) or of the genes involved in the pluripotency of stem cells (27).
Furthermore, the results of the leave-one-out crossvalidation were confirmed in stratified test sets of 20% of the observations (five-fold cross validation), thus improving the reliability of the prediction accuracy (28). These results also suggest that muscular pain and the sinus nasal symptoms seem to represent other pathways related to chronic fatigue syndrome in addition to the described alterations in the immune or neuroendocrine system that have been related to the SNPs successfully selected by the supervised approach. Thus, the reported disturbance of excitability and increase in oxidative response in the muscle (29, 30), the hyperalgesia to pressure at paranasal sinuses, the rhinosinusitis complaints (31), and the increased prevalence of non-allergic rhinitis in patients with CFS (32) agree with our results.
With regards to the data mining techniques, the similar accuracies obtained with different learning algorithms makes the use of more than one algorithm to get strong evidence about the validity and reliability of a SNP profile feasible and even advisable. The most significant SNP in our study, NR3C1_11159943, had also been found to be highly relevant for distinguishing cases of chronic fatigue syndrome from non-fatigued individuals by a Bayesian approach (33). Moreover, the finding that the Function type of algorithms tended to get higher prediction accuracies seems to be interesting for further research. This could be explained by the fact that these algorithms allow the use of SNP values based on the type of inheritance and that the output gives different weights to the SNPs according to their contribution to the model. But, what is even more interesting is that these algorithms can be complemented by meta-algorithms especially useful for small samples, such as Decorate, that improve the classification task by using several artificial training sets (34) and by the selection of kernels that allow better discrimination between the diagnostic categories (35, 36).
In conclusion, the supervised approach helped to find a reliable and valid SNP profile for prediction of chronic fatigue syndrome in this dataset that deserves to be tested as a possible marker for chronic fatigue syndrome. Perhaps, having four or more symptoms, as described in chronic fatigue syndrome, it will not be necessary to make the diagnosis (37) and what is even better, people with the SNP profile could be closely observed given that the chronic fatigue syndrome could be hidden for periods of time (38). With regards to research, new approaches such as systems biology could integrate these genetic marker data with gene coexpression network analysis to identify diseaserelated pathways (39). As a final point, the supervised approach seems to be useful for identifying the optimal SNP profiles for prediction of other complex diseases after an appropriate database cleansing based on the genetics problem.
Financial support
This work was supported by the Instituto de Biotecnología, Universidad National de Colombia
Conflict of interest The authors declare no conflict of interest
Corresponding author: Emiliano Barreto, Avenida carrera 30 N° 45-03, Edificio Manuel Ancízar, Bogotá, D.C., Colombia Telephone: (571)316 5000, extensión 16956; fax: (571) 316 5415 ebarretoh@unal.edu.co
References
1. Stram DO. Tag SNP selection for association studies. Genet Epidemiol. 2004;27:365-74. [ Links ]
2. Whistler T, Unger ER, Nisenbaum R, Vernon SD. Integration of gene expression, clinical, and epidemiologic data to characterize Chronic Fatigue Syndrome. J Transl Med. 2003;1:10 [ Links ]
3. He J, Zelikovsky A. Informative SNP selection methods based on SNP prediction. IEEE Trans Nanobioscience. 2007;6:60-7. [ Links ]
4. Sun Y, Todorovic S, Goodison S. Local-learning-based feature selection for high-dimensional data analysis. IEEE Trans Pattern Anal Mach Intell. 2010;32:1610-26. [ Links ]
5. Alpaydin E. Introduction to Machine Learning. Cambridge, Massachusetts: Massachussetts Institute of Technology Press; 2004. [ Links ]
6. Lalouel J, White R. Emery and Rimoin´s Principles and Practice of Medical Genetics. New York, NY: Churchill and Livingston; 1996. [ Links ]
7. VanLiere JM, Rosenberg NA. Mathematical properties of the r2 measure of linkage disequilibrium. Theor Popul Biol. 2008;74:130-7. [ Links ]
8. Stram DO, Haiman CA, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, et al. Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered. 2003;55:27-36. [ Links ]
9. Halperin E, Kimmel G, Shamir R. Tag SNP selection in genotype data for maximizing SNP prediction accuracy. Bioinformatics. 2005;21:i195-203. [ Links ]
10. Liu Q, Yang J, Chen Z, Yang MQ, Sung AH, Huang X. Supervised learning-based tagSNP selection for genomewide disease classifications. BMC Genomics. 2008;9:S6. [ Links ]
11. Bellazzi R, Zupan B. Predictive data mining in clinical medicine: current issues and guidelines. Int J Med Inform. 2008;77:81-97. [ Links ]
12. Goertzel BN, Pennachin C, de Souza Coelho L, Gurbaxani B, Maloney EM, Jones JF. Combinations of single nucleotide polymorphisms in neuroendocrine effector and receptor genes predict chronic fatigue syndrome. Pharmacogenomics. 2006;7:475-83. [ Links ]
13. Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479-81. [ Links ]
14. Hall M, Smith LA. Feature Subset Selection: A Correlation Based Filter Approach. New Zealand: University of Waikato;1998. [ Links ]
15. Witten IH, Frank E. Data Mining Practical Machine Learning Tools and Techniques. Second ed. San Francisco, CA: Elsevier; 2005. [ Links ]
16. Shortliffe E, Perreault L, Wiederhold G, Fagan L. Medical Informatics Computer Applications in Health Care and Biomedicine. New York, NY: Springer-Verlag; 2001. 17. Norman GR, Streiner DL. Biostadística. Madrid, España: Mosby/Doyma; 1996. [ Links ] [ Links ]
18. Lee E, Cho S, Kim K, Park T. An integrated approach to infer causal associations among gene expression, genotype variation, and disease. Genomics. 2009;94:269-77. [ Links ]
19. van den Brandt J, Luhder F, McPherson KG, de Graaf KL, Tischner D, Wiehr S, et al. Enhanced glucocorticoid receptor signaling in T cells impacts thymocyte apoptosis and adaptive immune responses. Am J Pathol. 2007;170:1041-53. [ Links ]
20. Rajeevan MS, Smith AK, Dimulescu I, Unger ER, Vernon SD, Heim C, et al. Glucocorticoid receptor polymorphisms and haplotypes associated with chronic fatigue syndrome. Genes Brain Behav. 2007;6:167-76. [ Links ]
21. Narita M, Narita N. Genetic background of chronic fatigue syndrome. Nippon Rinsho. 2007;65:997-1002. [ Links ]
22. Klimas NG, Koneru AO. Chronic fatigue syndrome: inflammation, immune function, and neuroendocrine interactions. Curr Rheumatol Rep. 2007;9:482-7. [ Links ]
23. van Den Eede F, Moorkens G, Van Houdenhove B, Cosyns P, Claes SJ. Hypothalamic-pituitary-adrenal axis function in chronic fatigue syndrome. Neuropsychobiology. 2007;55:112-20. [ Links ]
24. Miwa S, Takikawa O. Chronic fatigue syndrome and neurotransmitters. Nippon Rinsho. 2007;65:1005-10. [ Links ]
25. Parker AJ, Wessely S, Cleare AJ. The neuroendocrinology of chronic fatigue syndrome and fibromyalgia. Psychol Med. 2001;31:1331-45. 26. Barros Filho MC, Katayama ML, Brentani H, Abreu AP, Barbosa EM, Oliveira CT, et al. Gene trio signatures as molecular markers to predict response to doxorubicin cyclophosphamide neoadjuvant chemotherapy in breast cancer patients. Braz J Med Biol Res. 2010;43:1225-31. [ Links ] [ Links ]
27. Xu H, Lemischka IR, Ma´ayan A. SVM classifier to predict genes important for self-renewal and pluripotency of mouse embryonic stem cells. BMC Syst Biol. 2010;4:173. [ Links ]
28. Subramanian J, Simon R. An evaluation of resampling methods for assessment of survival risk prediction in highdimensional settings. Stat Med. 2010 Dec 1. [ Links ]
29. Nijs J, Meeus M, De Meirleir K. Chronic musculoskeletal pain in chronic fatigue syndrome: recent developments and therapeutic implications. Man Ther. 2006;11:187-91. [ Links ]
30. Jammes Y, Steinberg JG, Mambrini O, Bregeon F, Delliaux S. Chronic fatigue syndrome: assessment of increased oxidative stress and altered muscle excitability in response to incremental exercise. J Intern Med. 2005;257:299-310. [ Links ]
31. Naranch K, Park YJ, Repka-Ramirez MS, Velarde A, Clauw D, Baraniuk JN. A tender sinus does not always mean rhinosinusitis. Otolaryngol Head Neck Surg. 2002;127:387-97. [ Links ]
32. Baraniuk JN, Clauw DJ, Gaumond E. Rhinitis symptoms in chronic fatigue syndrome. Ann Allergy Asthma Immunol. 1998;81:359-65. [ Links ]
33. Bhattacharjee M, Botting CH, Sillanpaa MJ. Bayesian biomarker identification based on marker-expression proteomics data. Genomics. 2008;92:384-92. [ Links ]
34. Melville P, Mooney R. Constructing diverse classifier ensembles using artificial training examples. Proceedings of the IJCA. 2003:505-10. [ Links ]
35. Camillo F, Liberati C. The kernel approach in the future of data mining: many subjective choices in a complex landscape. Bologna: Universita di Bologna; 2008. [ Links ]
36. Cawley G, Talbot N. Efficient Model Selection for Kernel Logistic Regression. Norwich, United Kingdom: School of Computing Sciences, University of East Anglia; 2008. [ Links ]
37. Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. 1994;121:953-9. [ Links ]
38. Reeves WC, Wagner D, Nisenbaum R, Jones JF, Gurbaxani B, Solomon L, et al. Chronic fatigue syndrome- -a clinically empirical approach to its definition and study. BMC Med. 2005;3:19. [ Links ]
39. Presson AP, Sobel EM, Papp JC, Suarez CJ, Whistler T, Rajeevan MS, et al. Integrated weighted gene coexpression network analysis with an application to chronic fatigue syndrome. BMC Syst Biol. 2008;2:95. [ Links ]

