










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica v.32 n.2 Bogotá abr./jun. 2012
COMUNICACIÓN BREVE
1Laboratorio de Genética Humana, Facultad de Ciencias, Departamento de Ciencias Biológicas, Universidad de los Andes, Bogotá, D.C., Colombia
2Centro de Investigación y Desarrollo, Facultad de Optometría, Fundación Universitaria del Área Andina, Bogotá, D.C., Colombia
Contribución de los autores:
Diana Sanabria: realizó la parte de genética molecular y redacción del manuscrito.
Helena Groot: colaboración en todas las etapas del proyecto.
Julio Guzmán: médico oftalmólogo que realizó la parte de evaluación de los lentes cosmoprótesicos.
María Claudia Lattig: diseño de la parte experimental y redacción del manuscrito.
Recibido: 03/06/11; aceptado:30/12/11
Introducción. El albinismo óculo-cutáneo es un trastorno hereditario autosómico recesivo relacionado con la pigmentación. Sus manifestaciones clínicas incluyen hipopigmentación en piel, cabello y ojos, hipoplasia en la fóvea y disminución de la agudeza visual. A nivel mundial, hasta el momento, se han reportado, aproximadamente, 230 mutaciones en el gen TYR que causan albinismo óculo-cutáneo de tipo 1. Este gen codifica para la tirosinasa, enzima principal de la biosíntesis de melanina.
Objetivos. Identificar las mutaciones en el gen TYR responsables del albinismo óculo-cutáneo de tipo 1 en cinco individuos colombianos; cuatro de ellos pertenecen a una misma familia y el otro individuo es un caso aislado no relacionado con la familia. Asimismo, se pretende evaluar un sistema oftálmico que permite corregir problemas de refracción, y disminuir el nistagmo y la fotofobia en uno de los casos.
Materiales y métodos. Se hizo la secuenciación de los cinco exones del gen TYR en los cinco individuos de estudio y se buscaron portadores en la familia. Se llevó a cabo la evaluación clínica oftalmológica y la implementación del sistema correctivo en uno de los pacientes.
Resultados. Se encontraron las mutaciones G47D y 1379delTT en la familia, y en el caso aislado, las mutaciones G47D y D42N. Con la implementación del sistema óculo-visual se logró incrementar la agudeza visual y disminuir el nistagmo.
Conclusiones. Por ser el primer estudio en albinismo de este carácter en el país, estos resultados marcan una pauta para estudios posteriores de tamización molecular de albinismo en población colombiana.
Palabras clave: albinismo oculo-cutáneo, monofenol monooxigenasa, mutación, lentes de contacto, Colombia
An overview of oculocutaneous albinism: TYR gene mutations in five Colombian individuals
Introduction. Oculocutaneus albinism is a pigment-related inherited disorder characterized by hypopigmentation of the skin, hair and eyes, foveal hypoplasia and low vision. To date, 230 mutations in the TYR gene have been reported as responsible for oculocutaneus albinism type 1 worldwide. TYR gene encodes the enzyme tyrosinase involved in the metabolic pathway of melanin synthesis.
Objectives. Mutations were identified in the TYR gene as responsible for oculocutaneous albinism type 1 in five Colombian individuals, and a new ophthalmic system was tested that corrected visual defects and symptoms in a patient with oculocutaneous albinism.
Materials and methods. Samples were taken from 5 individuals, four of whom belong to a single family, along with a fifth individual not related to the family. Five exons in the TYR gene were sequenced to search for the gene carriers in the family and in the non-related individual. In addition, clinical ophthalmological evaluation and implementation of an new oculo-visual system was undertaken.
Results. A G47D and 1379delTT mutation was identified in the family. The unrelated individual carried a compound heterozygote for the G47D and D42N mutations. The oculo-visual corrective system was able to increase visual acuity and to diminish the nystagmus and photophobia.
Conclusions. This is the first study in Colombia where albinism mutations are reported. The methods developed will enable future molecular screening studies in Colombian populations.
Key words: Albinism, oculocutaneus; monofenol monooxigenasa, mutation, contact lenses, Colombia
El albinismo óculo-cutáneo es una condición clínica y genéticamente heterogénea. Se caracteriza por ser un trastorno de herencia autosómica recesiva, definida clínicamente por la ausencia parcial o total de melanina, pigmento producido en los melanocitos que se encuentra en pelo, piel y ojos (1-3). La ausencia de melanina en la zona de la úvea del ojo (iris, coroides y cuerpo ciliar) y en el epitelio pigmentario de la retina, ocasiona hipoplasia en la fóvea y, a su vez, una dirección errónea de las fibras del nervio óptico en el quiasma durante el desarrollo embrionario (4,5). Lo anterior se traduce en una reducción considerable de la agudeza visual, además de errores de refracción de la luz (miopía, astigmatismo e hipermetropía) (6), y un movimiento de carácter oscilatorio horizontal involuntario, denominado nistagmo, que reduce la capacidad para enfocar un punto fijo, lo cual contribuye a la disminución de la visión característica de individuos con albinismo (7).
Hasta el momento, el albinismo óculo-cutáneo se ha clasificado clínicamente en cuatro tipos: 1 A/B, 2, 3 y 4. El de tipo 1A (MIM 203100) es el más grave; se caracteriza por una carencia total en la producción de melanina y da como resultado el color blanco en cabello, pestañas, cejas y piel. El iris es de color azul claro casi rosa y totalmente translúcido. Los otros tipos son más difíciles de determinar a nivel clínico, siendo el diagnóstico molecular una herramienta indispensable para la correcta caracterización de este trastorno (8).
El albinismo óculo-cutáneo ha sido reportado en casi todas las poblaciones humanas, con una frecuencia promedio de 1 en 17.000 personas, es decir que, aproximadamente, uno de cada setenta pacientes es portador de mutaciones en los genes implicados. Por otro lado, el de tipo 1 presenta una prevalencia de 1 en 40.000 pacientes, aproximadamente, en la mayoría de poblaciones humanas (9). En Colombia, se han reportado casos de albinismo óculo-cutáneo en poblaciones indígenas, como los kuna del Chocó (10), y en municipios de Antioquia, como Marinilla (11) y Santuario (12,13), que se caracterizan por tener poblaciones con frecuente endogamia. En los departamentos de Cundinamarca y Boyacá no se han encontrado reportes en la literatura científica hasta el momento.
El gen tirosinasa (TYR, MIM 606933), localizado en el cromosoma 11q14.3 (14), es el que está comprometido en el albinismo óculo-cutáneo de tipo 1 A y B. Este gen está compuesto por cinco exones que codifican para una proteína de 529 aminoácidos, denominada enzima tirosinasa (15). Esta enzima es una proteína que utiliza el cobre como cofactor y está encargada de catalizar la conversión de tirosina a L-dihidroxifenilalanina (DOPA) y de DOPA a dopaquinona en la ruta metabólica de la biosíntesis de la melanina (16).
Hasta el momento, se han reportado unas 230 mutaciones del gen TYR como responsables del albinismo óculo-cutáneo de tipo 1. Entre estas mutaciones existen aquellas que ocasionan una disrupción total en la estructura o en la función de esta enzima y son responsables del albinismo óculo-cutáneo de tipo 1A, mientras que las mutaciones en que la enzima es parcialmente funcional son responsables del albinismo de tipo 1B, lo cual permite la acumulación de pigmento en el transcurso de la vida (9). En ambos tipos se ha reportado que la mayoría de los pacientes son heterocigotos compuestos, es decir que contienen dos mutaciones diferentes en el gen TYR y, en menor frecuencia, se encuentran casos en estado homocigoto. En algunos pacientes, únicamente se ha encontrado una mutación en un alelo, lo que sugiere que mutaciones en otros genes podrían estar implicadas en esta condición y que de alguna forma están interactuando con el gen TYR. Por otra parte, es usual hallar mutaciones fundadoras o propias de ciertas poblaciones (17,18).
Este trabajo se desarrolló con el propósito de identificar mutaciones en el gen TYR que sean responsables del albinismo óculo-cutáneo de tipo 1 en cinco individuos. Igualmente, se pretende evaluar un sistema correctivo óculo-visual en uno de ellos. Según el conocimiento de los autores, hasta el momento no existen reportes de mutaciones implicadas en albinismo óculo-cutáneo en casos colombianos, siendo este el inicio de estudios genéticos para albinismo óculo-cutáneo en el país.
Materiales y métodos
Casos de estudio
En este trabajo se estudiaron cinco individuos con albinismo óculo-cutáneo, de los cuales, cuatro pertenecen a una misma familia. El individuo propósito era una mujer de 24 años de edad, habitante de Bogotá, con diagnóstico primario de albinismo óculo-cutáneo de tipo 1A, con la piel y el cabello blanco, ojos violeta y nistagmo. A esta paciente, además del estudio molecular, se le evaluó un sistema oftálmico para mejorar la calidad visual.
El segundo individuo era un niño de 12 años de edad, primo en tercer grado de la individuo propósito, habitante de Bogotá, con diagnóstico primario de albinismo óculo-cutáneo de tipo 1B, con cabello rubio, piel blanca, ojos azules y nistagmo.
Por otro lado, en el municipio de Ciénega (Boyacá), de donde es oriundo el padre de la individuo propósito, residen dos mujeres hermanas de 52 y 53 años, que tienen cabello blanco, piel hipopigmentada, ojos claros y nistagmo.
El quinto individuo, era un hombre de 22 años, habitante de Bogotá, sin parentesco familiar con los cuatro pacientes anteriores y con diagnóstico primario de albinismo óculo-cutáneo de tipo IA, con piel y cabello blanco, ojos violeta y nistagmo.
Análisis molecular
Para la búsqueda de mutaciones en el gen TYR, se tomó una muestra de 10 ml, aproximadamente, de sangre periférica a los cinco pacientes y al mayor número de familiares que dieron su consentimiento o asentimiento informado previo para participar en este estudio.
El ADN genómico se extrajo mediante la técnica de salting out, utilizando el kit Corpogen DNA 2000TM (Corpogen, Colombia) y siguiendo las especificaciones de manufactura. Se amplificaron los tres primeros exones del gen TYR mediante reacción en cadena de la polimerasa (PCR), empleando los cebadores que se muestran en el cuadro 1 y utilizando las siguientes condiciones de PCR: un ciclo inicial de desnaturalización a 95 °C por cinco minutos, seguido de 30 ciclos que constan de una desnaturalización a 95 °C por 45 s, anillamiento a 60 °C por 45 s y extensión a 72 °C por un minuto. Al terminar los 30 ciclos se hizo una extensión final a 72 °C por cuatro minutos.
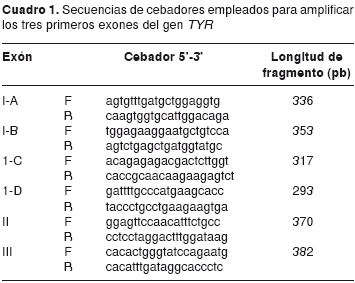
La reacción se llevó a un volumen final de 25 μl (12,5 μl de GoTaq® Green Master Mix) (Promega, USA), 50-100 ng ADN genómico y 0,2 μM de cada cebador.
Dada la alta homología que los exones IV y V presentan con el seudogen TYRL, se emplearon los cebadores propuestos por Chaki, et al., (19).
Los productos amplificados fueron enviados a secuenciar a Macrogen Sequencing Service (Macrogen, Korea). Una vez se obtuvieron las secuencias, se compararon con la secuencia referencia del gen TYR (NG_008748.1) disponible en GenBank.
Después de identificar las mutaciones en el paciente propósito y en los otros cuatro pacientes, se procedió a identificar los portadores en el grupo familiar que participó en el estudio mediante secuenciación o análisis de perfil de restricción (RFLP) con la enzima HaeIII.
Evaluación oftalmológica e implementación del sistema de corrección óculo-visual
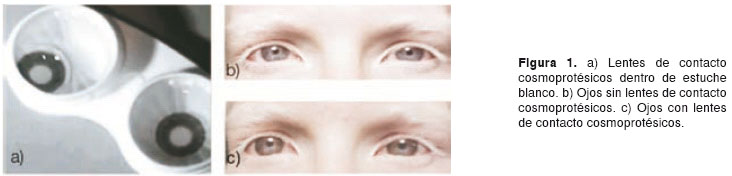
A la individuo propósito se le hizo un diagnóstico ocular, el cual evalúa fotofobia, defectos de refracción, transiluminación del iris, movimientos anormales del globo ocular (nistagmo, estrabismo), disminución en la agudeza visual e hipoplasia de la fóvea. Para elaborar el sistema óculo-visual de la paciente, se implementó la fórmula de corrección de la miopía en lentes de contacto blandos cosmoprotésicos de la casa comercial Keratos para ambos ojos, simulando la presencia de un iris opaco con pupila transparente, en la cara posterior (figura 1). Para la corrección del astigmatismo, se implementaron anteojos con filtro para luz ultravioleta y la respectiva fórmula.
Resultados
Análisis molecular
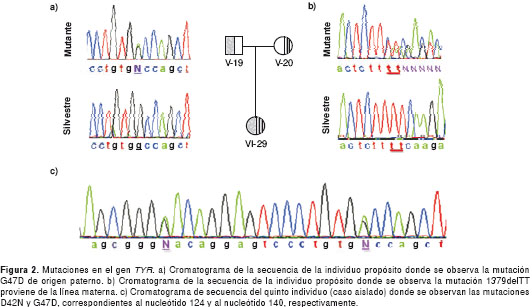
La individuo propósito es heterocigota compuesta para las mutaciones G47D y la 1379delTT en el gen TYR (figura 2a y figura 2b). Ambas mutaciones han sido reportadas en la literatura científica. Por el lado paterno, se identificó la sustitución de guanina por adenina en el nucleótido 140 localizado en el primer exón, la cual resulta en un cambio en el aminoácido 47 de glicina por ácido aspártico (G47D) (20). Por el lado materno, se identificó una deleción de dos timinas en el nucleótido 1379 ubicado en el exón V (1379delTT). Esta mutación cambia el marco de lectura produciendo una proteína no funcional (21).
La figura 3 corresponde al árbol genealógico de la familia en cuestión, en la que se muestran los individuos que portan las mutaciones G47D y 1379delTT. Por el lado paterno de dicha paciente (VI-29), se encontró que el padre (V-19) y la abuela (IV-11) eran portadores de la mutación G47D. Al individuo con albinismo óculo-cutáneo 1B (VI-1) solo se le encontró una copia de la mutación G47D, sin presentar más mutaciones en ninguno de los cinco exones del gen TYR.
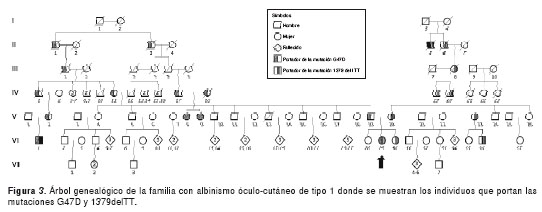
Dado que la línea paterna de la individuo propósito es oriunda del municipio de Ciénega (Boyacá), se decidió hacer una visita en la cual se tuvo conocimiento de dos hermanas albinas. Luego de indagar con familiares de la individuo propósito y de las dos hermanas con albinismo, se sospechó un posible parentesco familiar que fue confirmado al revisar las partidas de bautizo. Se trata de una raíz parental (II-3 y II-4) que data del año 1850. En el árbol genealógico, las hermanas se ubicaron en las posiciones V-8 y V-9. Al amplificar y secuenciar el gen TYR, se encontró que las dos presentan la mutación G47D en estado homocigoto.
La búsqueda de la mutación 1379delTT en la línea materna de la individuo propósito arrojó como resultado que los familiares IV-19, V-20, V-23 y VI- 30 y VI-36 son portadores. En el árbol genealógico se puede observar que el tío abuelo (IV-19) es portador y descendiente de un ancestro (II-5), quien por tradición oral de la familia se dice que tuvo albinismo. De este hecho se pueden inferir dos cosas interesantes: la primera es que posiblemente el ancestro sí tuvo albinismo, y la segunda es que muy probablemente el abuelo fallecido (IV-20) fue también portador de esta mutación; por lo tanto, la pudo heredar a la madre (V-20) y ella a su hija que es la individuo propósito (VI-29).
El quinto individuo, aunque no está relacionado con la familia anterior, también presentó la mutación G47D en estado heterocigoto y, además, tiene otra mutación reportada en la literatura científica como D42N (22) (figura 2c). La mutación D42N es una sustitución de guanina por adenina en el nucleótido 124 ubicado en el primer exón y traduce un cambio en el aminoácido 42 de ácido aspártico por asparagina. Este paciente también presenta un polimorfismo reportado como Y/S192, que consiste en una sustitución de citocina por adenina en el nucleótido 575 del primer exón del gen TYR. Este polimorfismo genera un cambio sinónimo de serina por tirosina en el aminoácido 192, sin alterar la viabilidad de la proteína (23).
Análisis del haplotipo asociado con la mutación G47D
Diferentes estudios basados en análisis de haplotipos en población judía sefardita (24) y en población puertorriqueña (25,26), han demostrado que la mutación G47D en ambas poblaciones parece tener el mismo origen ancestral. Con el fin de explorar si la mutación G47D en nuestra población también está asociada al haplotipo judía sefardita, se analizaron dos de los cuatro sitios polimórficos del gen tirosinasa utilizados en los estudios anteriores. El primer sitio polimórfico denominado Y/S192, localizado en el primer exón del gen, fue observado por secuenciación. El segundo polimorfismo, ubicado en el nucleótido -199 de la región promotora, contiene una variación de tipo SNP (Single Nucleotide Polymorphism) de C/A qué se puede diferenciar mediante la enzima de restricción TaqI (25).
Individuos albinos con la mutación G47D e individuos no albinos portadores de la mutación, que se han estudiados en poblaciones judía sefardita y puertorriqueña, presentan la variante S (serina) en el codón 192 y la variante C en el polimorfismo -199 (25). En el estudio presente, la propósito y sus familiares que tienen la mutación G47D, y el quinto individuo, también presentan la variante S en el codón 192 y la variante C en la posición -199 en estado homocigoto. Respecto al individuo no relacionado con la familia, se encontró que es heterocigoto para la variante Y/S192 y homocigoto para la variante C en la posición -199. De lo anterior se sugiere que la mutación G47D reportada en casos colombianos, puede tener el mismo origen ancestral de la encontrada en los judíos sefarditas y en los puertorriqueños. Sin embargo, es necesario hacer un estudio más profundo de haplotipos del gen TYR para poder concluir esta posibilidad de forma contundente.
Evaluación clínica-oftalmológica e implementación del sistema de corrección óculo-visual
A la paciente propósito se le hizo un examen oftalmológico completo en el cual se concluyó que presentaba un nistagmo congénito y no refería sensación subjetiva de movimiento del campo visual u oscilopsia. La nistagmoscopia evidenció la presencia de un nistagmo horizontal pendular en la mirada horizontal, inconstante, que se hacía constante en las miradas arriba y abajo, y, más aún, en las laterales. En cuanto a la condición de refracción, presentó un grado importante de astigmatismo miópico compuesto: ojo derecho, -3,75 dioptrías de miopía, -6,00 de astigmatismo en 0°, y agudeza visual de 20/400; ojo izquierdo, -3,00 de miopía, -6,10 de astigmatismo en 120°, y agudeza visual de 20/400.
Como resultado del uso del sistema óculo-visual, se logró una agudeza visual final de 20/80 en ambos ojos y una disminución considerable del nistagmo en términos de amplitud y frecuencia de los movimientos. La paciente manifestó experimentar mejor agudeza visual, haciendo lectura a mayor distancia que sin el sistema. No obstante, expresó la sensación de campo visual reducido lateralmente. Tiempo después, la paciente fue valorada para contemplar la posibilidad de corregir los errores de refracción mediante una intervención quirúrgica con láser en ambos ojos, LASEK (Laser Assisted Subepithelial Keratomileusis). Actualmente, la paciente se encuentra en el periodo de cicatrización, que tarda dos años. Una vez transcurra este tiempo será valorada nuevamente para determinar los resultados definitivos de la cirugía e implementar de nuevo el lente de contacto cosmoprotésico sin correcciones de refracción.
Discusión
Según el conocimiento de los autores, este es el primer trabajo en el que se reportan mutaciones relacionadas con albinismo óculo-cutáneo en Colombia. Las mutaciones G47D y 1379delTT han sido reportadas en diferentes lugares del mundo y ambas son responsables de albinismo óculo-cutáneo 1A, bien sea en forma homocigota o heterocigota compuesta. La mutación G47D se ha reportado en población de judíos marroquíes y sefarditas (26), en las Islas Canarias y Puerto Rico (24), México, Cuba y en caucásicos de origen no hispano residentes en Estados Unidos (8,27,28), mientras que la mutación 1379delTT ha sido únicamente reportada en población bengalí occidental en la India (21,22).
El encontrar mutaciones que ya se han reportado en otras poblaciones humanas genera un indicio de posibles eventos pasados de migración. Este es el caso de la mutación G47D y la población judía marroquí y judía sefardita. Esta población tuvo varias oleadas de emigración hacia el norte de África y al sur de Europa en el siglo VII y en el siglo XIV, y llegaron a América en el siglo XV huyendo de las persecuciones. Algunos de ellos se convirtieron al cristianismo. Las Islas Canarias y Puerto Rico hacían parte de los lugares que visitaron en sus viajes de exploración (29); de esta forma, probablemente, trajeron consigo la mutación G47D. Esta mutación se ha encontrado asociada a un mismo haplotipo genético en las poblaciones de Puerto Rico, Islas Canarias y judía sefardita; por tanto, se concluye que, muy probablemente, la mutación G47D tuvo origen en la población judía sefardita-marroquí y se dispersó en las oleadas de emigración a estas poblaciones.
En este estudio, los resultados de los análisis parciales para este haplotipo sugieren que la mutación G47D colombiana hace parte de este mismo ancestro común. No obstante, aún quedan interrogantes que se espera sean resueltos acerca de si la mutación G47D es una mutación antigua que fue traída a nuestro país cuando los judíos sefarditas-marroquíes fueron expulsados de España y empezaron sus viajes exploratorios o si, por el contrario, esta es una mutación que apareció de novo en nuestro país. Por otro lado, tanto la mutación 1379delTT como la mutación D42N, únicamente han sido reportadas en población bengalí occidental, donde aún no se han realizado estudios de haplotipos, dejando muchos interrogantes sobre cómo llegaron estas mutaciones a nuestro país o si se originaron espontáneamente en nuestra población.
Respecto al problema oftalmológico que presentan los pacientes, hasta el presente no se han difundido tratamientos para el albinismo que permitan generar pigmento o mejorar significativamente la capacidad visual, pero es posible tomar medidas preventivas y correctivas. Al respecto, la National Organization for Albinism and Hypopigmentation (NOAH) (30), sugiere evitar la exposición al sol o aplicar protectores solares de factor alto. En cuanto a los ojos, es muy recomendado usar anteojos con protección ultravioleta y emplear fórmulas oftálmicas cuando se presentan problemas de refracción; incluso, es posible corregirlos mediante cirugía láser, como es el caso de la paciente de este estudio (31).
El método óculo-visual propuesto en este trabajo, plantea corregir defectos de refracción y crear una barrera artificial para moderar la entrada de luz al ojo, generando un mejor contraste de las imágenes. Esta función la suele cumplir el pigmento presente en el iris y en las pestañas, y que está ausente en pacientes albinos. La barrera es ensamblada en lentes cosmoprotésicos que son diseñados con una máscara opaca, cuya función es disminuir la intensidad de la luz actuando igual que anteojos de sol, pero con la ventaja de ser menos llamativos y permitir la combinación con fórmula correctiva de errores de refracción (32). Todo ello contribuye a atenuar el nistagmo y mejorar así la agudeza visual.
Este es el primer reporte del uso de lentes cosmoprotésicos en un paciente con albinismo óculocutáneo que demostró una mejoría de la capacidad visual. Aunque cabe aclarar, el resultado depende de cada paciente y de factores como su disciplina y su capacidad de adaptación al tratamiento en sí, en especial a la reducción del campo visual.
Agradecimientos
Agradecemos a todas las personas que participaron en este estudio y que hicieron posible la realización del mismo.
Conflicto de interés
Los autores manifiestan no tener conflicto de intereses en esta publicación.
Financiación
El presente proyecto fue financiado por el proyecto fondo de profesores asistentes de la Universidad de los Andes otorgado a María Claudia Lattig y por fondos del grupo OPTOS de la Fundación Universitaria del Área Andina adscrito a Colciencias.
Correspondencia: María Claudia Lattig, Laboratorio de Genética Humana, Facultad de Ciencias, Universidad de los Andes, Carrera 1 N°18A-12, bloque M, Bogotá, D.C., Colombia Teléfono: (571) 339 4949, extensión 3230; fax: (571) 339 4949 extensión 2771 mlattig@uniandes.edu.co
Referencias
1. Zhao H, Boissy RE. Distinguishing between the catalytic potential and apparent expression of tyrosinase activities. Am J Med Sci. 1994;308:322-30. [ Links ]
2. Witkop CJ. Albinism: Hematologic-storage disease, susceptibility to skin cancer, and optic neuronal defects shared in all types of oculocutaneous and ocular albinism. Ala J Med Sci. 1979;16:327-30. [ Links ]
3. Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43. [ Links ]
4. Meyer CH, Lapolice DJ, Freedman SF. Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence tomography. Am J Ophthalmol. 2002;133:409-10. [ Links ]
5. Creel D. Problems of ocular miswiring in albinism, Duane´s syndrome, and Marcus Gunn phenomenon. Int Ophthalmol Clin. 1984;24:165-76. [ Links ]
6. Creel DJ, Summers CG, King RA. Visual anomalies associated with albinism. Ophthalmic Paediatr Genet. 1990;11:193-200. [ Links ]
7. Abadi RV. Mechanisms underlying nystagmus. J R Soc Med. 2002;95:231-4. [ Links ]
8. King RA, Pietsch J, Fryer JP, Savage S, Brott MJ, Russell-Eggitt I, et al. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003;113:502-13. [ Links ]
9. King RA, Hearing VJ, Creel D, Oetting WS. Albinism. In: Scriver CR, Beaudet AL, Sly WS, Valle DY, editors. The metabolic and molecular basis of inherited disease. Séptima edición. New York: McGraw-Hill; 1995. p. 4353-92. [ Links ]
10. Sinclair FG. El albinismo entre los amerindios de Panamá. Scientia (Panama). 1989;4:41-50. [ Links ]
11. Soto-Calderón ID. Evaluación histórica y genética del origen de Marinilla y su zona de influencia. Medellin: Universidad de Antioquia; 2003. [ Links ]
12. Tirado-Muñoz MC. Estudio de la isonimia en el albinismo de la población de El Santuario. Medellin: Universidad de Antioquia; 1987. [ Links ]
13. Bravo-Aguilar ML. Estudio genético epidemiológico del albinismo como un pro regional de El Santuario 1990 (tesis). Medellín: Universidad de Antioquia; 1990. [ Links ]
14. Barton DE, Kwon BS, Francke U. Human tyrosinase gene, mapped to chromosome 11 (q14----q21), defines second region of homology with mouse chromosome 7. Genomics. 1988;3:17-24. [ Links ]
15. Kwon BS, Haq AK, Kim GS, Pomerantz SH, Halaban R. Cloning and characterization of a human tyrosinase cDNA. Prog Clin Biol Res. 1988;256:273-82. [ Links ]
16. Cooksey CJ, Garratt PJ, Land EJ, Pavel S, Ramsden CA, Riley PA, et al. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J Biol Chem. 1997;272:26226-35. [ Links ]
17. Oetting WS, Fryer JP, Shriram S, King RA. Oculocutaneous albinism type 1: The last 100 years. Pigment cell research/ sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. 2003;16:307-11. [ Links ]
18. Oetting WS, Garrett SS, Brott M, King RA. P gene mutations associated with oculocutaneous albinism type II (OCA2). Hum Mutat. 2005;25:323. [ Links ]
19. Chaki M, Mukhopadhyay A, Ray K. Determination of variants in the 3´-region of the tyrosinase gene requires locus specific amplification. Hum Mutat. 2005;26:53-8. [ Links ]
20. Oetting WS, King RA. Molecular basis of albinism: Mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat. 1999;13:99-115. [ Links ]
21. Sundaresan P, Sil AK, Philp AR, Randolph MA, Natchiar G, Namperumalsamy P. Genetic analysis of oculocutaneous albinism type 1 (OCA1) in Indian families: Two novel frameshift mutations in the TYR Gene. Mol Vis. 2004;10:1005-10. [ Links ]
22. Chaki M, Mukhopadhyay A, Chatterjee S, Das M, Samanta S, Ray K. Higher prevalence of OCA1 in an ethnic group of eastern India is due to a founder mutation in the tyrosinase gene. Mol Vis. 2005;11:531-4. [ Links ]
23. Giebel LB, Spritz RA. RFLP for MboI in the human tyrosinase (TYR) gene detected by PCR. Nucleic Acids Res. 1990;18:3103. [ Links ]
24. Gershoni-Baruch R, Rosenmann A, Droetto S, Holmes S, Tripathi RK, Spritz RA. Mutations of the tyrosinase gene in patients with oculocutaneous albinism from various ethnic groups in Israel. Am J Hum Gen. 1994;54:586-94. [ Links ]
25. Oetting WS, Witkop CJ, Jr., Brown SA, Colomer R, Fryer JP, Bloom KE, et al. A frequent tyrosinase gene mutation associated with type I-A (tyrosinase-negative) oculocutaneous albinism in Puerto Rico. Am J Hum Gen. 1993;52:17-23. [ Links ]
26. Santiago PJ, Rodríguez-Pérez Y, Renta JY, Izquierdo NJ, Del Fierro L, Munoz D, et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky- Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J Invest Dermatol. 2006;126:85-90. [ Links ]
27. Opitz S, Kasmann-Kellner B, Kaufmann M, Schwinger E, Zuhlke C. Detection of 53 novel DNA variations within the tyrosinase gene and accumulation of mutations in 17 patients with albinism. Hum Mutat. 2004;23:630-1. [ Links ]
28. Hutton SM, Spritz RA. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J Invest Dermatol. 2008;128:2442-50. [ Links ]
29. Ladero-Quesada MA, Valdeón BJ, Alcala-Galve J. Judíos sefarditas conversos: la expulsión de 1492 y sus consecuencias. Valladolid: Ámbito Editores S.A.; 1995. [ Links ]
30. Wilson R. The National Organization for Albinism and Hypopigmentation (NOAH). Insight. 1993;18:8. [ Links ]
31. Hertle RW, Anninger W, Yang D, Shatnawi R, Hill VM. Effects of extraocular muscle surgery on 15 patients with oculo-cutaneous albinism (OCA) and infantile nystagmus syndrome (INS). Am J Ophthalmol. 2004;138:978-87. [ Links ]
32. Gauthier CA, Grant T, Holden BA. Clinical performance of two opaque, tinted soft contact lenses. J Am Optom Assoc. 1992;63:344-9. [ Links ]
