










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.32 no.4 Bogotá Oct./Dec. 2012
https://doi.org/10.7705/biomedica.v32i4.568
PRESENTACIÓN DE CASO
doi: http://dx.doi.org/10.7705/biomedica.v32i4.568
1Servicio de Medicina Interna, Universidad de La Sabana, Bogotá, D.C., Colombia
2Servicio de Medicina Interna, Hospital Universitario de La Samaritana, Bogotá, D.C., Colombia
3Servicio de Medicina Interna, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia
Contribución de los autores:
Martha Alejandra Casallas fue la encargada de la recolección de la información de la historia clínica, imágenes, revisión de la literatura, redacción y escritura del manuscrito.
Carlos Mauricio Calderón se encargó de la asesoría temática y la búsqueda de la descripción de las láminas de histología.
Recibido: 25/10/11; aceptado:05/08/12
Se presenta el caso de una mujer de 47 años con cuadro clínico de siete meses de evolución caracterizado por aumento progresivo de peso, hipertensión arterial y diabetes mellitus de reciente aparición, hirsutismo facial y en tórax, alopecia frontal, alteraciones en la menstruación e hipopotasiemia. Se consideró el diagnóstico de síndrome de Cushing, por lo cual se iniciaron estudios e extensión para establecer su etiología. Durante su hospitalización presentó una evolución tórpida y falleció. En la autopsia clínica se encontró un carcinoma de la glándula suprarrenal izquierda, de 400 g, aproximadamente, con metástasis a hígado y trombosis masiva de la vena cava, lo que finalmente produjo su muerte.
Palabras clave: hirsutismo, alopecia, síndrome de Cushing, corteza suprarrenal, carcinoma, metástasis de la neoplasia.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.568
Cushing´s syndrome due to a left adrenal cortical carcinoma with metastasis to the liver and a massive vena cava tumor thrombus
A 47-year-old woman with a seven-month history of increasing weight, hypertension and recently diagnosed diabetes presented features of hirsutism, frontal baldness, amenorrhea and hypokalemia. These characteristics were considered diagnostic of Cushing´s syndrome, and studies were initialized to identify its etiology. During hospitalization, the patient presented a torpid evolution resulting in death. Clinical autopsy revealed a 400 g carcinoma in the left adrenal gland, liver metastasis and a massive vena cava tumor thrombus which was the final cause of death.
Key words: Hirsutism, alopecia, Cushing syndrome, adrenal cortex, carcinoma, neoplasm metastasis.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.568
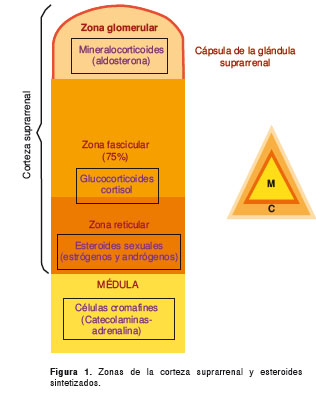
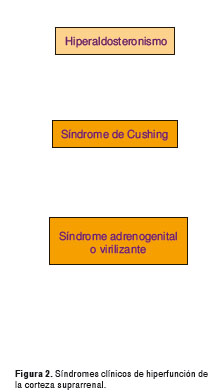
Las enfermedades de la corteza suprarrenal pueden diferenciarse según se asocien a hiperfunción o hipofunción cortical (1,2). La corteza suprarrenal está formada por tres zonas en las cuales se sintetizan tres tipos de esteroides (figura 11) y por cada uno de estos se puede encontrar un síndrome clínico de hiperfunción de la corteza suprarrenal (figura 2); para el objeto de esta revisión, nos enfocaremos en el hipercortisolismo (1,2).
El hipercortisolismo es un trastorno ocasionado por las siguientes causas:
1. Enfermedades hipotalámico-hipofisiarias primarias: hipersecreción de corticotropina (Adrenocorticotropic Hormone, ACTH) o enfermedad de Cushing (más de 50 % de los casos).
2. Secreción ectópica de ACTH por una neoplasia no endocrina, por ejemplo, carcinoma de células pequeñas de pulmón, tumores carcinoides, carcinomas medulares de tiroides o tumor de los islotes pancreáticos.
3. Hipersecreción de cortisol: se produce por un adenoma, un carcinoma o una hiperplasia nodular suprarrenal o síndrome de Cushing suprarrenal (15 a 30 % de los casos).
4. Exógena: uso de glucocorticoides.
Al inicio, se puede presentar ganancia de peso; además el hipercortisolismo produce una atrofia selectiva de las miofibrillas de contracción rápida (de tipo II), lo cual se refleja en una disminución de la masa muscular con debilidad proximal de las extremidades. Los glucocorticoides inducen gluconeogénesis e inhiben la captación de glucosa por las células, con la consiguiente hiperglucemia, glucosuria y polidipsia; además, ocasionan una inmunosupresión que predispone a diferentes tipos de infecciones. Existe pérdida de colágeno, por lo cual se puede encontrar una piel frágil con tendencia a la formación de hematomas, inadecuada cicatrización y formación de estrías. Hay alteraciones en el estado de ánimo o psicosis.
Es frecuente la hipertensión, existe hipopotasiemia, hipocloremia y alcalosis metabólica, en especial, cuando hay producción ectópica de ACTH (1,2).
Teniendo en cuenta lo anterior, se debe considerar practicar pruebas funcionales, empezando por una cuantificación de corticotropina para establecer si el síndrome de Cushing depende o no de la ACTH; en este último caso, es menor de 5 pg/ml. Si no es dependiente, se debe considerar la posibilidad de lesiones en la glándula supprarrenal. Además, se debe comprobar el síndrome de Cushing midiendo la concentración de cortisol urinario libre en orina de 24 horas, el cual es tres a cuatro veces mayor que el valor superior de referencia (10-50 μg al día), en este síndrome. También, se deben practicar otras pruebas, como la de supresión con dosis bajas de dexametasona (0,5 a 2 mg por vía oral) entre las 11 p.m. y las 12 m. Al día siguiente, entre 8:00 y 9:00 a.m., se mide el cortisol sérico; en condiciones normales habrá supresión en la síntesis de ACTH y cortisol, que no se presenta en el síndrome de Cushing. Si el resultado es un valor mayor de 1,8 μg/dl en suero o de 10 μg en orina de 24 horas, es sugestivo de síndrome de Cushing. Otra prueba es el cortisol sérico a las 11:00 p.m., con el paciente despierto; si resulta una concentración de cortisol sérico mayor de 8,3 μg/dl, se considera el diagnóstico de síndrome de Cushing, con una sensibilidad de 92 % y una especificidad de 96,4 %.
En las neoplasias suprarrenales primarias se encuentra un funcionamiento autónomo de la glándula; son más frecuentes en mujeres entre los 30 y 60 años de edad (1,2).
Los adenomas suprarrenales son pequeños (con un tamaño aproximado de dos cm y un peso de 10 g), bien circunscritos, con una superficie lisa y sin áreas de necrosis o hemorragia (3,4).
Los carcinomas son infrecuentes, con una incidencia de 0,6 a 2 casos por un millón de habitantes, y tienden a producir un hipercortisolismo más acentuado. Son tumores no encapsulados, grandes (con un tamaño aproximado de 14 cm y un peso de 200 a 500 g), de bordes indefinidos, nodulares con una superficie gruesa, calcificaciones, actividad mitótica, áreas de necrosis, hemorragia y con compromiso vascular; que muestran las características anaplásicas de los carcinomas (5). Son neoplasias heterogéneas con pobre pronóstico y cuya patogénesis incierta puede ser funcional o no, considerando la síntesis hormonal y los signos clínicos (6,7).
Las mujeres se afectans con mayor frecuencia que los hombres, en una proporción de 1:5 (8); esto se debe, tal vez, al mayor número de procedimientos diagnósticos abdominales practicados en ellas, lo que permite el hallazgo incidental de esta neoplasia. Según la edad, se presenta en adolescentes y en personas en la cuarta y quinta décadas de la vida (9).
Los pacientes presentan exceso hormonal (virilización en mujeres, y ginecomastia y atrofia testicular en hombres) (10), síndrome de Cushing o efecto local de la masa en un carcinoma hormonalmente inactivo (náuseas, vómito, dolor abdominal o lumbalgia) (11).
Se debe hacer una evaluación bioquímica de la hipersecreción del tumor y una de imaginología, que permitan la más correcta aproximación al diagnóstico y su estadificación, teniendo en cuenta su división en los siguientes estadios: T1N0M0; II: T2N0M0; III T1N1M0 - T2N1M0 - T3N0M0, y IV: T0N0M1 - T3N1- T4, donde T1: tumor de 5 cm o menos, sin invasión; T2: tumor de más de 5 cm, sin invasión; T3: tumor localmente invasivo sin compromiso de órganos adyacentes; T4: tumor con invasión de órganos adyacentes; N0: negativo para ganglios regionales; N1: positivo para ganglios regionales; M0: sin metástasis a distancia; y M1: con metástasis a distancia.
Según se ha reportado en la literatura científica, el carcinoma suprarrenal invade órganos retroperitoneales y adyacentes en cerca del 25 % de los casos. Las metástasis aparecen sobre todo en el hígado y el pulmón, pero pueden ser intravasculares u óseas.
Puede afectar la vena cava inferior (el primer caso descrito fue en 1972) (12) por compresión, invasión directa o por extensión en la luz con la formación de trombos, usualmente, sin lesión de la pared venosa. El trombo tumoral puede subsecuentemente atravesar la unión cavoauricular y progresar a la aurícula derecha. La obstrucción progresiva de la vena cava promueve la formación de venas colaterales, lo que limita las manifestaciones clínicas (13). Para valorar dicha extensión tumoral, se recomienda el uso de cavografía, ecocardiograma transesofágico, tomografía computadorizada (TC) o resonancia magnética, la cual diferencia el trombo tumoral del trombo de fibrinógeno.
La incidencia de esta entidad con extensión a la vena cava inferior ha sido subestimada, ya que muchos de estos casos no se reportan. Por esta razón, no es posible establecer que dicha extensión se pueda considerar como un signo de pobre pronóstico ni que sea un factor predictor negativo para el manejo quirúrgico de esta enfermedad.
Para el manejo de dicha extensión, se han propuesto técnicas que facilitan la resección del tumor, como la trombectomía, la resección parietal de la vena cava inferior lateral o total, sin reconstrucción o con colocación de injerto, la exclusión vascular hepática, el bypass veno-venoso y el bypass cardiopulmonar; en algunos casos, este se lleva a cabo mediante paro circulatorio hipotérmico, en especial, cuando hay compromiso de las cavidades cardiacas derechas (14). Sin embargo, la extensión masiva en la vena cava se considera un factor limitante para la resección tumoral retroperitoneal, debido al pobre pronóstico del tumor y a las dificultades técnicas relacionadas con el control venoso y la reconstrucción vascular (15,16).
Según las características del tumor, se han designado diferentes grados de factibilidad de resección (R0: resección completa; R1: tumor residual microscópico, o R2: tumor residual macroscópico), lo que contribuye a establecer el pronóstico y la conducta que se debe seguir.
La cirugía óptima depende de los límites de la extensión venosa, del tamaño del tumor primario y de la necesidad de procedimientos asociados, como nefrectomía, resección de ganglios linfáticos y hepatectomía por metástasis (17). Con la resección completa del tumor primario y de la extensión venosa, se ha reportado una mejoría en la supervivencia y en la calidad de vida, lo que justifica el manejo quirúrgico agresivo y la práctica de nuevas intervenciones cuando aparecen metástasis o recurrencias de la enfermedad (18).
Además, se considera que el manejo quirúrgico puede tener un carácter paliativo de síntomas tumorales, al controlar la secreción hormonal por el carcinoma y prevenir embolias pulmonares.
Se requieren expertos para este procedimiento, para garantizar una completa resección del tumor y un control adecuado de la extensión tumoral. Sin embargo, la completa resección del tumor es difícil debido a la nueva circulación tumoral y al desarrollo de circulación colateral extensa, especialmente en pacientes con un carcinoma secretor y una exposición prolongada a esteroides.
El riesgo quirúrgico es alto, con una mortalidad de 10 a 15 %. La cirugía debe evitarse en pacientes con múltiples enfermedades concomitantes o metástasis extensas.
Es de anotar que el pronóstico de estos pacientes es pobre, incluso después de la cirugía, con una supervivencia a cinco años entre 16 y 38 % en diferentes series (19).
El principal fármaco para su tratamiento es el mitotano (op´-DDD), un isómero del insecticida DDT, el cual inhibe la elaboración de cortisol y disminuye las concentraciones de esteroides en el plasma y la orina. Aunque su acción citotóxica es bastante selectiva sobre la zona secretora de los glucocorticoides de la corteza suprarrenal, también puede inhibir la zona glomerular y la esteroideogénesis pero no producir regresión de las metástasis. Además, altera el metabolismo extrasuprarrenal del cortisol, por lo que es necesario evaluar los niveles de cortisol en plasma y orina para ajustar los efectos del fármaco. Los pacientes tratados con mitotano deben someterse a un tratamiento de mantenimiento con glucocorticoides a largo plazo y en algunos conviene administrar mineralocorticoides sustitutivos (1,2). Aproximadamente, en un tercio de los pacientes se consigue un retroceso del tumor primario y las metástasis, pero la supervivencia a largo plazo no se modifica.
Las metástasis óseas no suelen responder al fármaco y es necesario el uso de radioterapia. Se han implementado enfoques alternativos de quimioterapia basada en el platino. Sin embargo, en el momento aún no existen datos que indiquen que la supervivencia se prolongue (1,2).
A largo plazo, el pronóstico es pobre a causa de la falta de diagnóstico y de manejo adyuvante efectivo (20). Sin embargo, se ha considerado la implementación de esquemas como la resección quirúrgica más el uso de mitotano o, en pacientes de alto riesgo quirúrgico, el uso de mitotano más quimioterapia.
Reporte de caso
Se presenta el caso de una paciente de sexo femenino de 47 años de edad, que ingresó por un cuadro clínico iniciado en junio de 2009 y caracterizado por astenia, adinamia y dificultad para la deambulación secundaria a disnea y edema de los miembros inferiores. Posteriormente, presentó edema progresivo que se extendió a la cara y se asoció con palidez cutánea y aumento de 32 kg de peso, aproximadamente, en los últimos seis meses, sin cambios en la dieta, por lo cual asistió a un hospital local donde le diagnosticaron hipertensión y diabetes de novo, y le iniciaron manejo farmacológico. En octubre de 2009, presentó hirsutismo en los pómulos, el mentón, los hombros y el tercio superior del tórax, alopecia y caída del tercio externo de las cejas. Fue valorada por el Servicio de Endocrinología, donde consideraron el síndrome de Cushing y solicitaron exámenes de laboratorio.
Persistió con edema progresivo e incapacidad para la deambulación secundaria a este, por lo que asistió a un hospital local donde le diagnosticaron infección de vías urinarias por Enterococcus spp. y Escherichia coli, iniciaron antibioticoterapia y documentaron hipopotasiemia persistente a pesar de la reposición intravenosa. Además, presentaba lesiones equimóticas y petequiales en las extremidades, con tiempos de coagulación y recuento de plaquetas normales, Se practicaron exámenes de laboratorio que reportaron: tirotropina (TSH), 1,12 UI/ml; hemograma con descenso de hemoglobina (12/01/2010, 12,5 g/dl y 14/12/2010, 8,5 g/dl) con volúmenes normales; deshidrogenasa láctica (LDH), 2.220 UI/L; hiperazoemia prerrenal, y lesiones hepáticas por ecografía abdominal total, por lo cual se decidió remitir la paciente a un hospital de mayor nivel de complejidad, para su estudio y manejo.
Al ingresar a nuestra institución, se encontró una paciente de 117 kg de peso con un índice de masa corporal (IMC) de 37,7 kg/m2, taquicárdica, con oxígeno por cánula nasal a 2 L/minuto, con adecuada oxigenación y equilibrio ácido-basico. Presentaba facies cushingoide y signos de virilización, edema generalizado de predominio en los miembros inferiores, y lesiones equimóticas en los sitios de venopunción, la cara anterior de las glándulas mamarias, el hipocondrio derecho, los muslos y la cara anterior medial de los brazos. Presentaba ideas delirantes de persecución (“Una mujer me hizo brujería”) y fuerza 2/5 en miembros superiores y 1/5 en miembros inferiores.
Por el cuadro clínico y los hallazgos en el examen físico, se consideró que cursaba con un síndrome de Cushing suprarrenal, por lo que se solicitó concentración urinaria y sérica de cortisol, con un valor de cortisol sérico de 38,9 μg/dl (valores de referencia: 5-25 μg/dl), TC de tórax y abdomen (protocolo para suprarrenal) y valoración por endocrinología. Se continuó con tratamiento antihipertensivo, insulina subcutánea y anticoagulación plena por cursar con dolor torácico asociado a disnea; se descartó un síndrome coronario agudo y se sospechó muy probable tromboembolia pulmonar, para lo cual se solicitó angiotomografía de tórax y monitorización continua.
El 19 de enero de 2010, durante su traslado para una TC abdominal, la paciente presentó paro cardiorrespiratorio sin respuesta a las maniobras de reanimación y falleció.
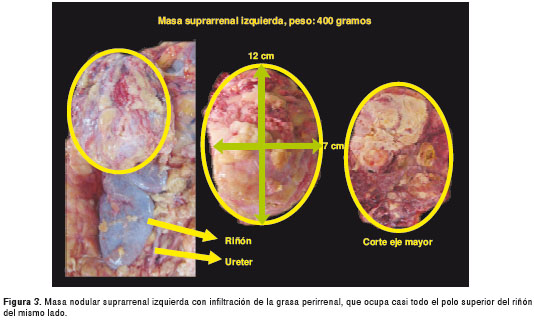
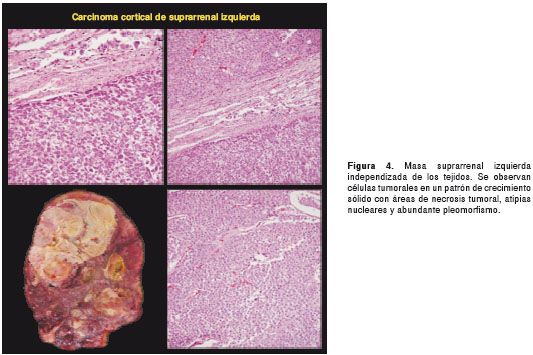
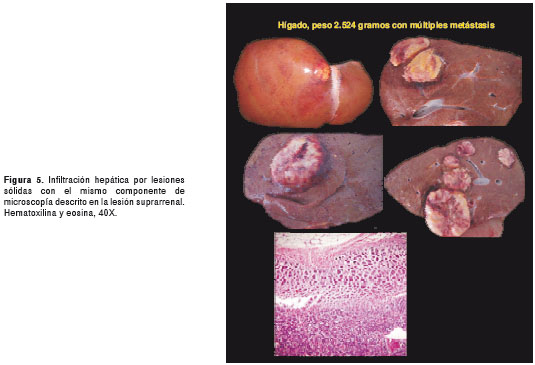
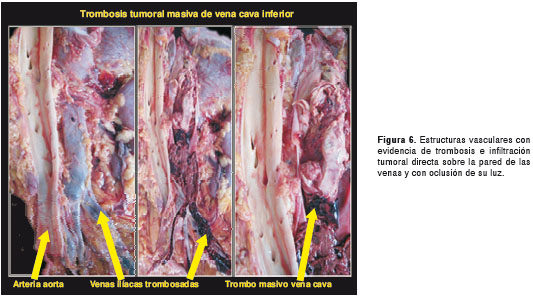
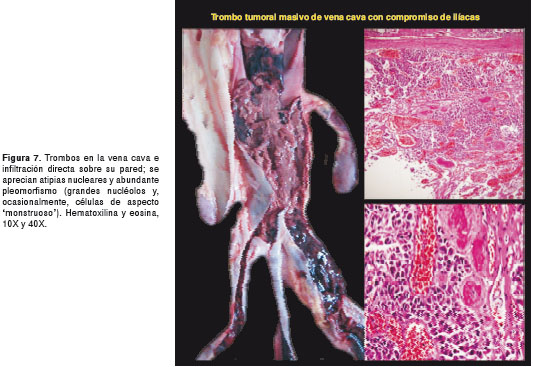
Por tratarse de una paciente sin diagnóstico etiológico, se solicitó practicar la autopsia Clínica. El Servicio de Patología reportó un carcinoma de la glándula suprarrenal izquierda, de 400 g de peso (figura 3 y figura 4), con metástasis al hígado (figura 5) y extensión a la vena cava (figura 6 y figura 7); esta última desencadenó una trombosis masiva de dicha vena, lo cual ocasionó un estado de bajo gasto e hipoperfusión, con el posterior deceso de la paciente.
Discusión
Aproximadamente, 20 a 25 % de los pacientes con síndrome de Cushing tienen una neoplasia suprarrenal. Sin embargo, su presentación oligosintomática es pasada por alto o se hace manifiesta como el hallazgo esporádico de masas en estudios imaginológicos ("incidentaloma", 5 %) (21).
En este caso, la paciente presentó la mayoría de los signos y síntomas descritos en la literatura científica para el síndrome de Cushing, con virilización, una presentación clínica característica de un tumor funcional con alta carga hormonal. En la autopsia, se observó un carcinoma de la glándula suprarrenal izquierda, de rápida evolución, con múltiples metástasis hepáticas y extensión a la vena cava con trombosis.
La prevalencia real es desconocida y mucha de la información citada proviene de autopsias en las que se describen masas suprarrenales no diagnosticadas en 1,4 a 6 % de los casos (22).
La determinación de la conducta terapéutica según lo descrito en la literatura científica, se debe basar en las enfermedades concomitantes asociadas, el tamaño tumoral, la extensión venosa del carcinoma y la necesidad de procedimientos asociados.
En este caso, se encontró un carcinoma de 400 g, de 12 x 7 cm de longitud, con múltiples metástasis hepáticas. Para estas, hubiese requerido una hepatectomía y, además, el extenso compromiso de la vena cava hubiera representado una gran dificultad para la adecuada resección quirúrgica de la lesión principal.
Debido a la tórpida evolución asociada con rápida progresión del tumor, la paciente falleció. Queremos destacar que se trató de una entidad que se manifestó durante siete meses, aproximadamente, a lo largo de los cuales mostró sutiles o grandes indicios (según sean vistos por el médico), que son ayudas fundamentales para el diagnóstico del síndrome de la enfermedad ante la cual nos encontrábamos; esto debe hacer parte fundamental del esquema mental del médico para el diagnóstico de las enfermedades.
Declaramos no tener conflictos de interés.
Esta publicación fue financiada por los autores.
Correspondencia: Martha Alejandra Casallas, Servicio de Medicina Interna, Universidad de La Sabana, Hospital Universitario de La Samaritana, Bogotá, D.C., Colombia Teléfono: (571) 407 7075, extensión 10554; celular: (300) 292 0337 m_aleja99@hotmail.com
1. Contran R, Kumar V, CollinsT. Robbins: Patología estructural y funcional; 6a edición. Madrid: Mc Graw Hill; 2005. p. 1197-201. [ Links ]
2. Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson L. Harrison: Principios de medicina interna. 17ª edición. Madrid: Mc Graw Hill; 2008. p. 2446-54. [ Links ]
3. Boscaro M, Arnaldi G. Approach to the patient with possible Cushing´s syndrome. J Clin Endocrinol Metab. 2009;94:3121-31. http://dx.doi.org/10.1210/jc.2009-0612 [ Links ]
4. Dluhy R, Maher M, Wu C. Case 7-2005: A 59 year old woman with an incidentally discovered adrenal nodule. N Engl J Med. 2005;352:1025-32. [ Links ]
5. Hedican SP, Marshall FF. Adrenocortical carcinoma with intracaval extension. J Urol. 1997;158:2056-61. http://dx.doi.org/10.1016/S0022-5347(01)68152-7 [ Links ]
6. Allolio B, Fassnacht M. Adrenocortical carcinoma. Clinical review. J Clin Endocrinol Metab. 2006;91:2027-37. http:// dx.doi.org/10.1210/jc.2005-2639 [ Links ]
7. Reincke M, Karl M, Travis W. P53 mutations in human adrenocortical neoplasms: Immunohistochemical and molecular studies. J Clin Endocrinol Metab. 1994;78:790-4. http://dx.doi.org/10.1210/jc.78.3.790 [ Links ]
8. Del Gaudio AD, Del Gaudio GA. Virilizing adrenocortical tumors in an adult woman. Report of 10 patients, 2 of whom each had a tumor secreting only testosterone. Cancer. 1993;72:1997-2003. [ Links ]
9. Wooten M, King D. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72:3145-55. [ Links ]
10. Vassilopoulou-Sellin R, Schultz P. Adrenocortical carcinoma. Clinical outcome at the end of the 20th century. Cancer. 2001;92:1113-21. [ Links ]
11. Crucitti F, Bellantone R, Ferrante A, Bischerini M, Crucitti P. The Italian registry for adrenal cortical carcinoma: Analysis of a multiinstitutional series of 129 patients. The ACC Italian registry study group. Surgery. 1996;119:161-70. [ Links ]
12. Castleman B, Scully R, McNeely B. Case records of the Massachusetts General Hospital. Case 46-1972. N Engl J Med. 1972;287:1033-40. [ Links ]
13. Golub RM, Parsons RE, Sigel B, Barnes AU. A review of venous collaterals in inferior vena cava obstruction. Clinical Anatomy. 1992;5:441-51. http://dx.doi.org/10.1002/ca.980050604 [ Links ]
14. Shahian DM, Nieh PT, Libertino JA. Resection of atriocaval carcinoma using hypothermic circulatory arrest. Ann Thorac Surg. 1989;48:421-2. http://dx.doi.org/10.1016/S0003-4975(10) 62870-6 [ Links ]
15. Bower T, Nagorney D, Toomey B, Gloviczki P, Pairolero P, Hallett J. Vena cava replacement for malignant disease: Is there a role? Ann Vasc Surg. 1993;7:51-62. http://dx.doi.org/10.1007/BF02042660 [ Links ]
16. Eilber F, Gelabert H, Quinones-Baldrich W. Prosthetic replacement of the inferior vena cava for malignancy. J Vasc Surg. 1998;28:75-83. [ Links ]
17. Smith B, Mulherin J, Sawyers J, Turner B, Prager R, Dean R. Suprarenal vena caval occlusion. Principles of operative management. Ann Surg. 1984;199:656-67. [ Links ]
18. Roizen M. Anesthetic implications of concurrent diseases. Adrenocortical malfunction. 4th edition. New York: Churchill Livingstone; 1994. p. 917-8. [ Links ]
19. Icard P, Goudet P, Charpenay C. Adrenocortical carcinoma: Surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons Study Group. World J Surg. 2001;25:891-7. http://dx.doi.org/10.1007/s00268-001-0047-y [ Links ]
20. Chiche L, Dousset B, Kieffer E, Chapuis Y. Adrenocortical carcinoma extending into the inferior vena cava: Presentation of a 15-patient series and review of the literature. Surgery. 2006;139:15-27. http://dx.doi.org/10.1016/j.surg.2005. 05.014 [ Links ]
21. Song J, Chaudry F, Mayo-Smith W. The incidental adrenal mass on CT: Prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol. 2008;190:1163-8. http://dx.doi.org/10.2214/AJR.07.2799 [ Links ]
22. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein S. The clinically inapparent adrenal mass: pdate in the diagnosis and management. Endocr Rev. 2004;25:309-40. http://dx.doi.org/10.1210/er.2002-0031 [ Links ]
