










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.32 no.4 Bogotá out./dez. 2012
https://doi.org/10.7705/biomedica.v32i4.593
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v32i4.593
1Genética Molecular, Universidad de Antioquia, Medellín, Colombia
2Universidad Pontificia Bolivariana, Medellín, Colombia
3Fundación Integrar, Medellín, Colombia
4Grupo Pediaciencias, Departamento de Pediatría, Escuela de Medicina, Universidad de Antioquia, Medellín, Colombia
5Genética de Poblaciones, Mutacarcinogénesis y Epidemiología Genética, Universidad de Antioquia, Medellín, Colombia
6University College of London, Londres, Inglaterra
Contribución de los autores:
Ana Victoria Valencia y Ana Lucía Páez realizaron la genotipificación, análisis de los resultados y elaboración del manuscrito.
María Elena Sampedro, Clara Ávila, Julio César Cardona, Lina Galvis, Jaime Carrizosa y William Cornejo, contribuyeron en la consecución y el diagnóstico clínico de los pacientes.
Mauricio Camargo realizó los cariotipos y la evaluación de la fragilidad cromosómica.
William Cornejo, Ana Victoria Valencia, Gabriel Bedoya y Andrés Ruiz, contribuyeron con la consecución de financiación, el análisis de los resultados y la elaboración del manuscrito.
El trabajo se llevó a cabo en el Laboratorio de Genética Molecular, Universidad de Antioquia.
Recibido: 03/11/11; aceptado:31/07/12
Introducción. El espectro autista constituye un grupo de trastornos graves del neurodesarrollo, con un fuerte componente genético. Se ha sugerido un papel importante del sistema serotoninérgico en el desarrollo de este grupo de trastornos, con base en los estudios de respuesta a medicamentos y la hiperserotoninemia, característica común en el autismo. Se han implicado múltiples moléculas en el metabolismo y la neurotransmisión de la serotonina; sin embargo, los resultados de los estudios han tenido poca congruencia entre diferentes poblaciones.
Objetivos. Evaluar la relación entre el autismo y el polimorfismo de nucleótido simple (Single Nucleotide Polymorphism, SNP) en los genes SLC6A4, HTR2A e ITGB3, en una muestra de la población antioqueña.
Materiales y métodos. Se genotipificaron 42 núcleos familiares con autismo para 10 variantes en los genes SLC6A4, ITGB3 y HTR2A. Se evaluó la asociación utilizando la prueba de desequilibrio en la transmisión. Se exploró el impacto de la interacción entre estos genes y el autismo, utilizando la reducción multidimensional.
Resultados. Se encontró asociación de las variantes rs4583306 (OR=2,6, p=0,004) y rs2066713 (OR=2,2 p=0,03), en el gen SLC6A4, y asociación de combinaciones genotípicas entre los genes SLC6A4 y HTR2A y el riesgo de autismo (p=0,0001).
Conclusiones. Se encontró asociación significativa con variantes en el gen transportador de serotonina con el autismo, al igual que interacción entre variantes en los genes HTR2A con SLC6A4. Estos resultados concuerdan con los de estudios previos en otras poblaciones y son pruebas a favor del papel del sistema serotoninérgico en la etiología del espectro autista.
Palabras clave: trastorno autístico/genética, estudios de asociación genética, serotonina, polimorfismo de nucleótido simple, polimorfismo genético, epístasis genética.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.593
Evidence for association and epistasis between the genetic markers SLC6A4 and HTR2A in autism etiology
Introduction. Autism spectrum disorders are severe neurodevelopmental disorders with a strong genetic component. The potential role of the serotoninergic system in the development of autistic disorder has been based on the observation of hyperserotoninemia in autistic subjects and the results of drug treatment studies. Multiple molecules involved in serotonin metabolism and neurotransmission have been studied; however, replication studies have been inconsistent. This may be partially related to the marked genetic heterogeneity of autism in different populations.
Objectives. The relationship between autism and single nucleotide polymorphisms of SLC6A4, HTR2A and ITGB3 genes was evaluated in an urban population of northwestern Colombia.
Materials and methods. In Antioquia, Colombia, 42 families with history of autism were screened for 10 SNPs in SLC6A4, HTR2A and ITGB3 genes and evaluated for associations with the transmission disequilibrium test. The interactions among these genes and autism was assessed with multidimensional reduction methods.
Results. A significant main effect was seen among the SLC6A4 gene variants rs4583306 (OR=2.6, p=0.004) and rs2066713 (OR=2.2, p=0.03). No main effect of the ITGB3 or HTR2A variants was found, however, in the interaction effects, the SLC6A4 and HTR2A genes demonstrated significant evidence of association with autism (p<0.001).
Conclusion. Significant association of markers were discovered within the SLC6A4 gene and the combination of SLC6A4 and HTR2A (S-A) genes to autism. These results were consistent with previous studies conducted in other populations and provide further evidence for the implication of the serotoninergic system in the etiology of autistic disorders.
Keywords: Autistic disorder/genetics, genetic association studies, serotonin; polymorphism, single nucleotide; polymorphism, genetic; epístasis, genetic.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.593
El autismo es un trastorno del neurodesarrollo que se hace evidente hacia los tres años de edad y se caracteriza por limitación o ausencia de comunicación verbal, dificultades en la reciprocidad social y comportamientos e intereses restringidos, repetitivos y estereotipados (1,2). Este trastorno hace parte de los trastornos del espectro autista. Los trastornos del espectro autista constituyen un fenotipo más amplio que incluye, además del autismo, el síndrome de Asperger y el trastorno generalizado del desarrollo, no especificado (Pervasive Developmental Disorder, PDD-nos), los cuales se diferencian por la gravedad de los síntomas, el desarrollo temprano del lenguaje y el comportamiento cognitivo y social (1,2).
Los estudios epidemiológicos han demostrado un importante componente genético en la predisposición al autismo. Se estimó una concordancia en gemelos monocigóticos entre 26 y 96 % y, entre 0 y 30 %, para gemelos dicigóticos del mismo sexo (3). Además, se estimó un riesgo de recurrencia para autismo y los trastornos generalizados del desarrollo entre familiares en primer grado de un afectado, 75 veces mayor que el riesgo en la población general. Este riesgo de recurrencia sugiere un patrón de herencia complejo, donde varios genes interactúan sobre el riesgo para desarrollar el trastorno (4).
Uno de los hallazgos más comúnmente replicados en pacientes con autismo es el incremento en los niveles de serotonina (5HT, 5-hidroxitriptamina) en las plaquetas, presente en, aproximadamente, un tercio de los casos y sus familiares en primer grado (5-9).
La serotonina (5-hidroxitriptamina) es un neurotransmisor monoamina, involucrado en la regulación de funciones biológicas como el comportamiento emocional, el sueño, la sensibilidad al dolor y la liberación de hormonas, y juega un papel crucial en la sinaptogénesis y el neurodesarrollo (10,11).
Se ha demostrado que la serotonina promueve el crecimiento de neuritas (axones y dendritas), la sinaptogénesis, y la organización y diferenciación de estructuras límbicas, que incluyen hipotálamo, tálamo, amígdala y núcleo,estriado (12-15). Los cambios en los niveles de serotonina y otros neurotransmisores pueden llevar al desarrollo cortical aberrante, ya que las fibras aferentes provenientes del núcleo del rafe se proyectan a la corteza cerebral durante un periodo crítico en la morfogénesis (16-19).
Varias líneas de investigación apoyan el papel de una disfunción en la neurotransmisión serotoninérgica en la etiología del autismo. Se ha demostrado que algunos niños con autismo carecen del pico de producción de serotonina visto en niños con desarrollo normal; esta reducción se observó específicamente en las vías dentadotálamo- corticales (20).
A pesar del incremento observado en los niveles de serotonina en las plaquetas, se ha demostrado la reducción significativa en la unión del neurotransmisor a sus receptores 5HT2 en la corteza cerebral de individuos con trastornos del espectro autista y sus familiares en primer grado (21,22). Más aún, en los estudios se encontró que esta disminución en la unión a los receptores era inversamente proporcional a los niveles de serotonina en plaquetas (21).
Estas alteraciones en la función serotoninérgica pueden estar determinadas por factores genéticos que, a la vez, pueden ser factores de riesgo para el autismo. Tres de los genes involucrados en la regulación de la neurotransmisión de la serotonina y que, además, son fuertes candidatos posicionales en la etiología genética del autismo, son SLC6A4, ITGB3 y HTR2A.
En los estudios se ha demostrado un importante carácter hereditario de los niveles de serotonina en plaquetas tanto en individuos sanos como en autistas (23-25), y los genes SLC6A4 y ITGB3 fueron identificados como loci que tienen efecto sobre los niveles de serotonina en plaquetas (26-28).
El gen SLC6A4 (Solute Carrier Family 6 Member 4), que codifica para la proteína transportadora de serotonina (Serotonin Transporter, SERT), ha sido particularmente estudiado en la etiología genética del autismo, debido a la mejoría de los síntomas conductuales con el uso de inhibidores de la recaptura del neurotransmisor (19,29-32). Además, es un fuerte candidato posicional debido a su localización en la región 17q11.1-q12 (27,33,34), asociada constantemente con el autismo.
La SERT se expresa tanto en cerebro como en plaquetas, y un cambio en su función puede alterar el desarrollo del cerebro y contribuir a la alteración en los niveles de serotonina en plaquetas (35). La SERT controla la duración, disponibilidad y capacidad de la señalización en la sinapsis serotoninérgica; además, juega un papel crítico en la homeostasis, la distribución espacial y la intensidad de la señalización de la serotonina con sus receptores (21).
Se reportó un polimorfismo, 5-HTTLPR (Serotonin- Transporter-Linked Polymorphic Region), en la región promotora del gen SLC6A4, que consiste en una inserción o deleción de 44pb, debida a la presencia de 14 o 16 copias de una secuencia repetida de 20 a 23 pb de longitud (36). El promotor que contiene el alelo de la inserción incrementa su eficiencia en la transcripción, lo que conlleva al incremento en la expresión de la proteína transportadora de serotonina y, por lo tanto, a la mayor eficiencia en la recaptura del neurotransmisor del espacio sináptico. La inserción presenta una actividad de aproximadamente el doble de la del alelo con la deleción, y la captación de serotonina en líneas de linfoblastos homocigotas para la inserción fue 1,9 y 2,2 veces la captación de las líneas con una o dos copias con la deleción en el promotor (37).
Se han llevado a cabo múltiples estudios de asociación con diversos polimorfismos en el gen SLC6A4; particularmente, se ha centrado la atención en el polimorfismo 5-HTTLPR, dada su importancia en la regulación de la expresión, en diferentes poblaciones. Sin embargo, los resultados han sido conflictivos, en tanto que en algunos estudios se reporta la deleción como alelo de riesgo del trastorno (38-42) y en otros estudios se ha encontrado la inserción como la variante de riesgo (10,43).
En el gen SLC6A4 se identificaron variantes raras que "cosegregan" con los rasgos del espectro autista en familias con, al menos, dos casos, y estas variantes se correlacionan con la gravedad del comportamiento compulsivo (44). Además, se observó una correlación del genotipo en 5-HTTLPR con la variabilidad en el fenotipo de autismo, de tal manera que los individuos con una o dos copias de la deleción presentaron una mayor gravedad en el dominio de comunicación no verbal y los individuos homocigotos para la inserción presentaron mayor gravedad en los comportamientos estereotipados y en la agresividad (45).
Se ha identificado asociación entre el genotipo en 5-HTTLPR y la respuesta al tratamiento con inhibidores de la recaptura de serotonina (Selective Serotonin Reuptake Inhibitors, SSRI); los individuos homocigotos para el alelo de la inserción presentan una mejoría de los síntomas de agresión y de los relacionados con el sueño, pero los individuos con una o dos copias de la deleción no la presentan (46,47). Finalmente, no se encontró asociación ni ligamiento genético entre el polimorfismo 5-HTTLPR y el autismo en diferentes estudios (6,15,48,49).
El gen ITGB3 codifica para la proteína integrina beta 3, que es una subunidad del receptor de fibrinógeno específico de plaquetas y megacariocitos. El gen ITGB3 está localizado en la región 17q21.3, para la cual se ha encontrado asociación significativa con los trastornos del espectro autista en diferentes estudios (33,50-52). Se identificaron alelos o haplotipos comunes en el gen ITGB3, que confieren riesgo para el autismo (53,54); sin embargo, esta asociación no se replicó en familias de origen irlandés (55). Además, se identificaron efectos epistáticos significativos entre los genes ITGB3 y SLC6A4 sobre el riesgo de autismo (5,56).
El gen HTR2A es también un candidato posicional; se encuentra en el brazo largo del cromosoma 13q14-q21, una región que ha sido asociada consistentemente con autismo (57-61). Este gen codifica para el receptor 2A de serotonina (5-HT2A), que ha sido implicado en la fisiopatología del autismo mediante diferentes estudios funcionales. Existe una reducción de la densidad de receptores, así como de la respuesta neuroendocrina mediada por receptores 5-HT2, en pacientes con autismo (62). Estas alteraciones en la unión de los receptores 5-HT2 se han observado en familiares en primer grado de los pacientes (22), y en ambos estudios estos hallazgos se correlacionaron negativamente con los niveles de serotonina en plaquetas.
Recientemente, se observó una reducción en el potencial de unión de los receptores 5-HT2 en la corteza cerebral de padres de enfermos con autismo (63).
En dos estudios no se encontró asociación entre variantes en el gen HTR2A y autismo en población caucásica (8), ni en núcleos familiares de origen indio (64). Sin embargo, en una muestra de Corea, se encontró un haplotipo conformado por dos variantes en este gen asociado con la predisposición al autismo (43). Recientemente, un estudio hecho en población coreana, mostró una aumento de la transmisión del alelo G y una asociación del genotipo GG de la variante rs6311 ubicada en el promotor del gen, al comparar 103 pacientes con autismo con un grupo de 214 controles (65).
Las discrepancias entre los estudios que buscan genes de predisposición al autismo, se explicarían por la amplia heterogeneidad genética que subyace en el trastorno en las diferentes poblaciones e, incluso, dentro de una misma población, y la complejidad genética propia del autismo en la que intervienen múltiples genes, cada uno de los cuales aporta un riesgo modesto. Finalmente, los estudios no cubren la amplia variabilidad en los genes candidatos que contribuyen al trastorno; por lo tanto, el encontrar asociación dependerá de los niveles de desequilibrio de ligamiento en el gen, los cuales dependen de la historia de cada población.
La población de Antioquia, Caldas, Risaralda, norte del Valle y norte del Tolima, está conformada por un núcleo fundador común y es producto de una mezcla triétnica, de las poblaciones continentales amerindia, europea y, en menor proporción, africana (66). Esta población alcanzó su mayor expansión demográfica en condiciones de aislamiento, hecho que favorece su homogeneidad genética, como se ha demostrado mediante los niveles de diversidad genética, muy similares a los de otras poblaciones con historia de aislamiento muy bien documentado, como la finlandesa y los judíos ashkenazi (67-70). La mezcla genera desequilibrio de ligamiento entre todos los loci del genoma y, dado que este evento ocurrió recientemente, las vías de desequilibrio de ligamiento en esta población son amplias, lo que permite abarcar una mayor variabilidad con menor número de marcadores genéticos (71). Estas dos características que presenta esta población conocida como "paisa", favorecen la identificación de variantes genéticas de mayor exposición involucradas en algunas enfermedades.
Dados estos antecedentes, en este estudio se evalúa la asociación alélica y de combinaciones alélicas entre variantes en los genes involucrados en la función serotoninérgica y la predisposición al autismo mediante la prueba de desequilibrio en la transmisión en una muestra proveniente de esta población. Además, se evaluó la asociación entre los subfenotipos de autismo con las variantes en los genes SLC6A4, ITGB3 y HTR2A, ajustadas por edad, coeficiente intelectual y sexo.
Materiales y métodos
Población de estudio y evaluación clínica
Los pacientes se seleccionaron mediante la Fundación Integrar de Medellín, remitidos por pediatras, neurólogos, educadores de la ciudad, y mediante la consulta particular de los médicos vinculados a la investigación, por lo cual, el tipo de muestreo es por conveniencia. Se incluyeron los pacientes que cumplieron los criterios definidos y fueron remitidos entre los años 2006 y 2010.
Los pacientes fueron evaluados por un grupo clínico que incluyó neuropediatra, sicólogo infantil y educador especial; se verificaron los criterios del "Manual diagnóstico y estadístico de los trastornos mentales - IV" (DSM-IV) para el autismo o el síndrome de Asperger.
Los criterios de exclusión fueron la presencia de malformaciones típicas de formas del síndrome de autismo, manchas sugestivas de esclerosis tuberosa y neurofobromatosis, retardo mental grave, cariotipo anormal, fragilidad del cromosoma X, prematuridad e hipoxia perinatal.
En todos los pacientes se evaluó la presencia de síntomas en los tres dominios para autismo: incapacidad en la interacción social recíproca, en la comunicación y la presencia de patrones de comportamiento restringidos y estereotipados, mediante la Entrevista Diagnóstica para el Autismo - Revisada (ADI-R, Autism Diagnostic Interview- Revised) (72) y el instrumento diagnóstico de Observación Diagnóstica del Autismo (ADOS, Autism Diagnostic Observation Schedule) (73). Estos instrumentos diagnósticos generan una calificación de gravedad en las tres dimensiones, durante el curso de la vida (ADI-R) y en el momento de la evaluación (ADOS).
La ADI-R es una entrevista basada en los criterios del Diagnostic and Statistical Manual of Mental Disorders (DSM-IV, 1994) y la International Classification of Diseases (ICD-10; 1992), la cual debe hacerse a los padres o personas a cargo y su propósito es hacer un diagnóstico de autismo o de alteraciones del espectro autista en pacientes de más de dos años de edad. Está compuesta por 93 ítems y su aplicación lleva un tiempo aproximado de dos horas y media (74). El ADOS es una escala semiestructurada (75) de observación directa del individuo que se basa en la conducta para determinar el posible diagnóstico. En ella se evalúan varias características dependientes del nivel de desarrollo del lenguaje alcanzado por el paciente, ya que se encuentra diseñada tanto para individuos verbales como no verbales, con el fin de abarcar individuos con diferentes niveles de desarrollo (76).
A los casos se les hizo la medición del funcionamiento intelectual apropiada para su edad y se excluyeron los individuos con un puntaje por debajo de 30 para evitar confusiones en el diagnóstico.
Con el fin de minimizar la posible heterogeneidad genética en la muestra de estudio, se incluyeron los individuos cuyos bisabuelos, al menos seis de ocho, pertenecieran a la población "paisa".
Muestras biológicas y genotipificación
Se tomaron 10 ml de sangre periférica de cada uno de los pacientes, con el propósito de hacer un cariotipo y evaluar la fragilidad cromosómica, y para extraer el ADN.
Se estableció el cariotipo con bandeo cromosómico R de tipo replicativo de alta resolución y se evaluó la fragilidad del cromosoma X mediante el cultivo en medio pobre en ácido fólico. Se evaluaron 100 mitosis para determinar el número, la morfología y la presencia de rupturas cromosómicas.
El ADN se extrajo por métodos estándar de fenolcloroformo y se genotipificaron las variantes genéticas seleccionadas. Se obtuvo una muestra de sangre de los dos padres del caso índice para su genotipificación.
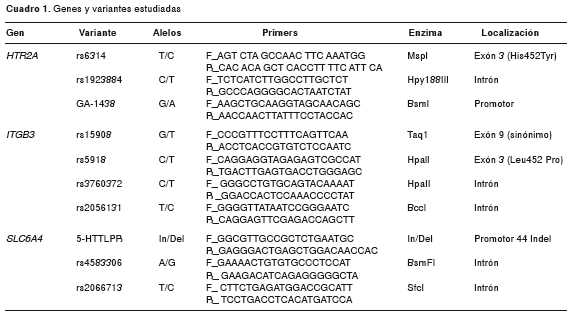
Se amplificaron las regiones cromosómicas que albergan las variantes seleccionadas mediante la reacción en cadena de la polimerasa (PCR), en un volumen final de 25 μl, que contenían 50 ng de ADN genómico, 0,25 μM de cada iniciador (primer), 200 μM dNTP, 0,5 unidades de Taq DNA polimerasa Invitrogen®, 1X de solución tampón (MgCl2: 1,5 Mm, KCl: 50 mM, Tris: 10 mM, pH: 8,3) proveído con la Taq polimerasa. El perfil de temperaturas utilizadas en esta amplificación fue: 94 °C durante 5 segundos, seguido de 35 ciclos de amplificación a 94 °C durante 30 segundos, 50 a 60 °C durante 30 segundos, 72 °C por 30 segundos y, finalmente, un ciclo de extensión final a 72 °C durante 10 minutos. Se comprobó esta amplificación en geles de agarosa al 2 % y el producto amplificado se sometió a digestión con la enzima específica que reconoce uno de los dos alelos de cada variante. Las variantes evaluadas, la secuencia de los cebadores para la amplificación y la enzima de restricción específica, se presentan en el cuadro 1.
Las secuencias de los SNP se tomaron de la base de datos http://ncbi.nlm.nih.gov y las secuencias de los iniciadores y condiciones de PCR se obtuvieron en el software PRIMER3plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi.) y de los reportes en otros estudios.
La genotipificación de los SNP (cuadro 1) se realizó por medio de digestión con enzimas de restricción específicas para cada polimorfismo, bajo las condiciones recomendadas. Las enzimas fueron escogidas empleando el software NEBcutter V2.0 (http://tools.neb.com/NEBcutter2) y los genotipos se resolvieron en geles de agarosa entre 2 y 3 %, según el tamaño del fragmento por resolver (figura 1). En cada gel se utilizaron controles homocigotos para la digestión, para verificar la digestión total del amplicón. Se hizo doble lectura independiente de los genotipos y, en caso de que no hubiera consenso, se repitió la genotipificación.

Las muestras fueron codificadas de tal manera que el proceso de genotipificación fuera ciego a la condición de afección y a la estructura de las familias nucleares. Se utilizaron duplicados para verificar la calidad de los genotipos y, en el caso de genotipos dudosos, se repitieron para confirmarlos.
Se verificaron inconsistencias mendelianas y se excluyeron aquellas familias que las presentaron.
Análisis estadístico
Se calcularon los datos estadísticos descriptivos de los subfenotipos de autismo para los pacientes y por grupos definidos según el sexo, y se evaluó la diferencia entre estos grupos, así como la correlación entre los subfenotipos clínicos, mediante pruebas paramétricas o no paramétricas, según se cumplió el supuesto de distribución normal de los datos, con el software SPSS™, versión 18.
El cálculo de las frecuencias alélicas y genotípicas, la evaluación de la transmisión mendeliana, así como el equilibrio de Hardy-Weinberg para los casos índice y los padres, se llevaron a cabo con el software PLINK, disponible en http://pngu.mgh.harvard.edu/~purcell/plink/index.shtml (77). Se evaluó el desequilibrio de ligamiento entre pares de variantes con el software HAPLOVIEW (versión 4.0) disponible en http://www.broadinstitute.org/scientific-community/science/programs/medicaland-population-genetics/haploview/downloads (78).
La transmisión diferencial de alelos de padres a hijos afectados se evaluó con la prueba de desequilibrio en la transmisión (79). El diseño de tríos presenta ventajas sobre los estudios de casos y controles y sobre los métodos de ligamiento genético paramétricos, ya que es sólido a la estratificación de la población y tiene más poder para detectar genes con efecto genético moderado que los análisis de ligamiento, a la vez que no requiere de la obtención de familias extensas con múltiples individuos afectados. La prueba de desequilibrio en la transmisión se practicó mediante el programa UNPHASED, disponible en http://www.mrcbsu.cam.ac.uk/personal/frank/software/unphased/ (80), el cual permite hacer una regresión logística condicional de los alelos y haplotipos transmitidos frente a los no transmitidos a los casos. Para cada análisis de asociación se hicieron 10.000 permutaciones, para evitar el error generado debido a múltiples pruebas.
Se hizo una aproximación a la posible interacción de las diferentes variantes estudiadas sobre el riesgo de autismo, utilizando el análisis de reducción dimensional multifactor (81,82). La reducción dimensional multifactor es un método libre de modelo, que no asume un patrón de herencia particular y no estima ningún parámetro, en contraste con la regresión logística, por ejemplo. El método de reducción dimensional multifactor agrupa los genotipos (para uno o múltiples loci) en grupos de alto y bajo riesgo, reduciendo de esta manera las dimensiones del genotipo que se espera prediga el rasgo de interés. Luego, se evalúa la capacidad de este nuevo genotipo, que es unidimensional aunque incluya información de múltiples loci, para clasificar y predecir el rasgo de interés utilizando pruebas de validación cruzada y permutaciones (83).
El método de reducción dimensional multifactor fue diseñado para datos de casos y controles; por lo tanto, se extrajo de cada familia el individuo caso y se construyó el genotipo de un "seudocontrol", a partir de los alelos de los padres que no fueron transmitidos a los hijos afectados (84,85).
Para evaluar el papel de los polimorfismos sobre la gravedad de los síntomas en las tres dimensiones del autismo, se consideró la calificación obtenida por la ADI-R como la variable dependiente en un modelo de regresión lineal, en el cual el genotipo, la edad, el sexo y el coeficiente intelectual fueron las variables independientes, utilizando el software estadístico SPSS™, versión 18.
Consideraciones éticas
El protocolo de este estudio fue aprobado por el Comité de Ética de la Sede de Investigación Universitaria de la Universidad de Antioquia. Todos los participantes o su representante legal, leyeron y comprendieron el documento informativo del estudio y posteriormente firmaron un consentimiento informado.
Resultados
Se identificaron 42 casos de origen antioqueño, de los cuales, cuatro tuvieron diagnóstico de síndrome de Asperger y 38 de autismo clásico. De los casos, 32 eran hombres y 10 mujeres, con una razón de 3,2 hombres por una mujer afectada. Las características fenotípicas, con relación a la deficiencia en las tres dimensiones de síntomas autistas y desempeño intelectual, se presentan en el cuadro 2. No hubo diferencias significativas para la edad entre hombres y mujeres, pero sí se encontró una mayor deficiencia en la interacción social recíproca medida con la ADI-R en las mujeres en comparación con el sexo masculino, con promedios de 26,4 versus 22,8, para este subfenotipo. Los hombres tuvieron un mejor desempeño intelectual, aunque no fue significativamente diferente (70,5 Vs. 57,6; p=0,18).

Para las dimensiones de síntomas de autismo, se encontró alta correlación entre las deficiencias en la interacción social recíproca y las deficiencias en la comunicación y coeficiente intelectual, con coeficientes de correlación de 0,66 (p=0,001) y -0,62 (p=0,001), respectivamente. Por el contrario, la dimensión de comportamientos e intereses restringidos y estereotipados, no se correlacionó con la interacción social (r=0,23; p=0,15) ni con el desempeño intelectual (r=-0,01; p=0,97) y tuvo una leve correlación con las deficiencias en la comunicación (r=0,35; p=0,04).
El marcador rs6314 presentó una frecuencia del alelo menor T, de 0,07; hubo ausencia del genotipo homocigoto para el alelo T (cuadro 3); y, dado que los padres que se tienen en cuenta para la prueba de asociación basada en desequilibrio en la transmisión son solo los heterocigotos, cuya frecuencia es del 13 % para este marcador, este fue descartado para el análisis.
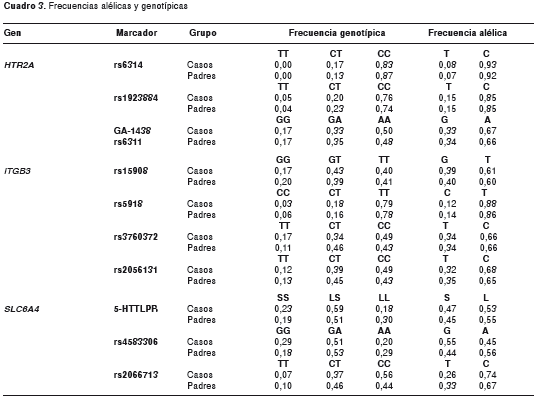
Las frecuencias alélicas y genotípicas para los casos y sus padres, fueron similares y se presentan en el cuadro 3. Los genotipos se distribuyeron de acuerdo con lo esperado bajo el modelo del equilibrio de Hardy-Weinberg.
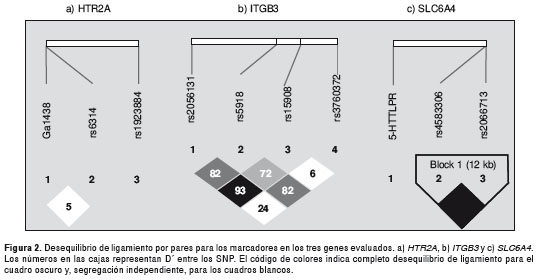
Se evidenció un exceso significativo en la transmisión de alelos para dos polimorfismos en el gen SLC6A4, del transportador de serotonina. El alelo G para el marcador rs4583306 (OR=2,64, p=0,004) y el alelo C de la variante rs2066713 (OR=2,18, p=0,026), se transmitieron con mayor frecuencia a los descendientes afectados que el alelo complementario (cuadro 4). Esta asociación permaneció aún después del ajuste por las permutaciones. Los dos marcadores rs4583306 y rs2066713 se encontraron en el mismo bloque de ligamiento (figura 2c). Además, se observó una tendencia a la asociación para el polimorfismo de la inserción/deleción en la región promotora de este mismo gen 5HTTLPR, que segrega independientemente del bloque haplotípico de los marcadores rs4583306 y rs2066713. Para el marcador 5HTTLPR se observó una mayor transmisión del alelo de la deleción S, con el número de veces que este alelo se dejó de transmitir al descendiente afectado.
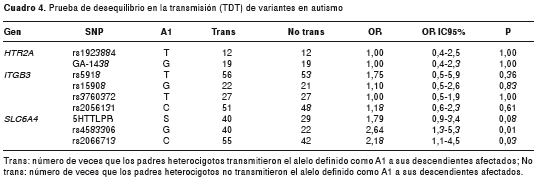
No se encontraron pruebas de asociación para ninguno de los polimorfismos independiente en los genes ITGB3 ni HTR2A, en la muestra evaluada.
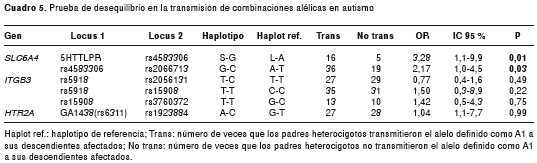
Debido a las limitaciones en el tamaño de muestra, solo se evaluaron haplotipos conformados por pares de marcadores; estos resultados se presentan en el cuadro 5. El haplotipo S-G, conformado por los polimorfismos 5HTTLPR y rs4583306 del gen del transportador de serotonina SLC6A4, se asoció con un incremento de tres veces el riesgo de autismo (OR=3,4; IC95%1,1-9,9; p=0,01) y el haplotipo G-C de los SNP rs4583306-rs2066713 presentaron un incremento de dos veces para el riesgo de padecer autismo; nuevamente, ambos polimorfismos estaban en el gen SLC6A4. No se identificó asociación significativa con otros haplotipos
Para evaluar el impacto de combinaciones de dos y tres SNP en los tres genes evaluados en la etiología del autismo, se utilizó el método de reducción dimensional multifactor. Los tres mejores modelos que explican el autismo se presentan en el cuadro 6.

Los dos primeros modelos están constituidos básicamente por los polimorfismos en el gen SLC6A4, 5HTTLPR. El modelo que incluye estos dos polimorfismos tiene una prueba de precisión balanceada máxima de 0,78 (p<0,0001), lo que indica que este modelo tiene la capacidad de clasificar adecuadamente el 78 % de los individuos incluidos en el análisis como casos o controles y tiene una validación cruzada de 10/10. El mejor modelo de tres SNP incluye, además de los polimorfismos anteriores, al SNP GA-1436 en el receptor de serotonina, HTR2A. Este último modelo incrementó la capacidad de clasificación en el 7 %. Este modelo clasifica adecuadamente el 85 % de los individuos usados en el análisis (prueba de precisión balanceada de 0,85, p<0,0001) y tiene una validación cruzada de 6/10. Esto indica que un modelo de interacción entre los genes SLC6A4 y HTR2A está asociado con el incremento en el riesgo de autismo.
Estas variantes podrían participar en la modificación del efecto de otras variantes y estar relacionadas con la gravedad del trastorno, más que con el trastorno en sí. Para probar esta hipótesis, se modeló el efecto de los polimorfismos sobre la gravedad de los subfenotipos de autismo, ajustando por sexo, edad y coeficiente intelectual. Los resultados más significativos se presentan en el cuadro 7.
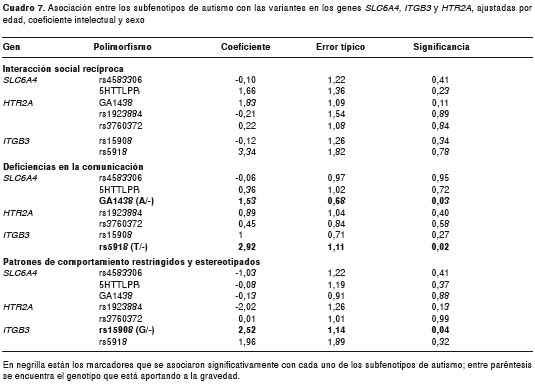
El alelo A de la variante GA-1438 en el gen HTR2A y el alelo T de rs5918 del gen ITGB3, se relacionaron con un incremento en la deficiencia en la comunicación; sin embargo, no se asociaron con los síntomas en la interacción social recíproca, a pesar de la alta correlación que presentaron los síntomas de estos dos subfenotipos del autismo. El alelo T del SNP rs15908 de ITGB3 se asoció significativamente con los patrones de comportamiento restringidos y estereotipados.
Discusión
En el presente estudio se evalúa la asociación de variantes en genes involucrados en la vía de la serotonina con el autismo: SLC6A4, ITGB3 y HTR2A. Las variantes independientes que mostraron asociación alélica significativa se encuentran ubicadas en el gen transportador de serotonina SLC6A4 (rs4583306 y rs2066713). Estas dos variantes se encuentran en completo desequilibrio de ligamiento en la muestra evaluada, lo cual hace muy probable que la variante que confiere predisposición al autismo se encuentre en este bloque de ligamiento, ya que ambas variantes se encuentran en intrones y no hay pruebas de que tengan un efecto en la regulación del gen.
En un estudio de 2005, se encontró asociación del alelo T de la variante rs2066713 con los niveles de serotonina en sangre (p=0,0191) y este efecto fue mayor entre los casos de sexo masculino (27). Hasta el momento no hay estudios reportados en que se evalúe el efecto que tiene la variante rs4583306 en la propensión al autismo.
La variante 5-HTTLPR, ubicada en este mismo gen, mostró una tendencia a la asociación (p=0,07), donde el alelo de la deleción (S) se transmitió 40 veces en relación con las 29 veces que se transmitió el alelo alternativo de la inserción (cuadro 3). Este polimorfismo se transmite en forma independiente del bloque de ligamiento donde se encuentran los polimorfismos rs4583306 y rs2066713, como se observa en la figura 2. Por lo tanto, la posible asociación obtenida con el polimorfismo 5HTTLPR puede deberse a un riesgo aportado por esta variante u otra en desequilibrio de ligamiento con ella pero independiente del bloque de las variantes rs4583306 y rs2066713.
Se ha reportado un efecto funcional del polimorfismo 5-HTTLPR, para el cual el alelo S en el promotor del gen SLC6A4 presenta, aproximadamente, la mitad de la actividad basal del promotor con el alelo L de la inserción (37). El alelo de la deleción ya ha sido previamente asociado con autismo en estudios realizados en diferentes poblaciones (39-42). Sin embargo, los hallazgos no han sido constantes, pues otros estudios han reportado el alelo L como de riesgo para autismo en poblaciones de Alemania, Israel y Portugal (10,24,86).
Estas inconsistencias pueden deberse a la heterogeneidad genética para el autismo en las diferentes poblaciones estudiadas o a que las muestras incluidas en diferentes estudios no tengan los mismos criterios diagnósticos o se consideren casos con un fenotipo amplio de autismo, lo cual hace la muestra muy heterogénea y, por lo tanto, las causales de predisposición no serán las mismas o tendrán efectos diferentes. Finalmente, las inconsistencias en relación con el alelo de riesgo para autismo pueden deberse al contexto genético en el cual se presenta la variante. En rasgos complejos se esperan múltiples variantes aportando al riesgo total del fenotipo; por lo tanto, el riesgo del polimorfismo 5HTTLPR puede estar modificado por otras variantes que presentan diferencias en frecuencia alélica en las diferentes poblaciones.
Es posible que en este estudio no se encontrara una asociación significativa de la variante S debido al pequeño tamaño de la muestra. Pero el efecto que esta variante tiene sobre la predisposición al autismo se puede evidenciar al evaluar los haplotipos, donde se encontró un riesgo de 3,28 (p=0,01) cuando el alelo S está presente junto a la variante G de rs4583306.
Por otra lado, el haplotipo G-C de las variantes rs4583306 y rs2066713 presenta un riesgo de 2,17 (p=0,026) para la predisposición al autismo (cuadro 5), muy similar al riesgo identificado para las variantes independientemente. Ambas variantes se encuentran en regiones no codificantes y no hay pruebas de que presenten un efecto funcional, por lo que es muy probable que la variante que está contribuyendo a la predisposición al autismo sea otra que sí tenga un efecto funcional y esté en desequilibrio de ligamiento con este bloque. La variante rs2066713 se encuentra en desequilibrio de ligamiento con rs6355, una sustitución no sinónima, Gly56Ala ubicada en el exón 3 del gen que incrementa la actividad de la proteína SERT (44). En un estudio de 2005, se encontró asociación significativa entre el alelo Ala56 en forma homocigótica o heterocigótica y autismo, así como una mayor gravedad de los comportamientos restringidos y estereotipados (p=0,0085). En este mismo estudio se evaluó el efecto del polimorfismo 5HTTLPR en una muestra de 341 familias con, al menos, dos afectados y 43 tríos, y no se encontró una asociación significativa, pero, al restringir el análisis a las familias cuyo caso era hombre, sí se encontró asociación significativa del alelo corto (p=0,03) (44). En un reciente reporte de un paciente con autismo atípico, se encontró que era heterocigoto para Gly56Ala y homocigoto L/L en el polimorfismo 5HTTLPR. La madre de este sujeto, sin síntomas, tenía genotipos Gly56Ala, pero era heterocigótica S/L para 5HTTLPR. Se postuló que el genotipo Gly56Ala-L/L predispone a un aumento en la recaptura de serotonina de la hendidura sináptica, lo cual puede ser un factor decisivo para el fenotipo autista (87).
Además, las variantes intrónicas o regiones no codificantes pueden constituir un factor de riesgo con relevancia biológica, al afectar la transcripción, la estabilidad del transcripto o el procesamiento del ARN (88).
El análisis de reducción dimensional de datos encontró efectos epistáticos entre las variantes 5HTTLPR y rs4583306 en un modelo de dos vías; ambas variantes se encuentran en el gen SLC6A4 (p<0,0001). Ya se había demostrado la presencia de múltiples alelos de predisposición al autismo en el gen SLC6A4 en una muestra de 340 familias de Estados Unidos. Por lo tanto, no es raro que en la muestra analizada, dos variantes diferentes, la 5HTTLPR en el promotor del gen y otra en desequilibrio de ligamiento con rs4583306, estén confiriendo riesgo para el autismo. Esta hipótesis de múltiples alelos de riesgo en el gen SLC6A4 puede explicar las inconsistencias entre los diferentes estudios en relación con si existe o no asociación a este gen o el alelo que confiere predisposición. Es necesario abarcar una mayor variabilidad del gen, evaluando polimorfismos en diferentes sus regiones, y posiblemente este sea un mejor factor predictor del riesgo a autismo. En el cuadro 6, se muestra cómo el modelo de un solo locus rs4583306, tiene la capacidad de clasificar adecuadamente el 70 % de los casos, según su estado de afección, mientras que incluyendo el polimorfismo 5HTTLPR se incrementa la precisión en la clasificación de 78 % de los sujetos.
En un modelo de tres vías se identificó la interacción significativa: además de las dos variantes 5HTTLPR y rs4583306, se incluyó la variante GA- 1438 en el gen HTR2A (p < 0,0001). Este modelo aporta un 7 % a la precisión en la clasificación de los individuos del estudio. Este hallazgo sugiere la interacción de múltiples genes involucrados en la función serotoninérgica sobre el riesgo de autismo en la población antioqueña
Se observó una modificación en la gravedad de las dimensiones de síntomas de autismo por parte de variantes ITGB3 y HTR2A.
La gravedad de las deficiencias en la comunicación se encontró modificada por el genotipo en las variantes rs5918 de ITGB3 y GA-1438 de HTR2A, después de ajustar por sexo, edad y coeficiente intelectual. Para estas variantes no se detectaron efectos principales sobre el fenotipo de autismo; solo el alelo A del marcador GA-1438 se encontró confiriendo riesgo cuando está acompañado de las variantes S de 5HTTLPR y el alelo G de rs4583306 en el gen SLC6A4.
En el 2008, Carneiro, et al., encontraron una evidencia funcional de la interacción entre las proteínas codificadas por los genes ITGB3 (αIIbβ3) y SLC6A4 (SERT) (89).
La disrupción homocigota de ITGB3 en ratones evita la captura de serotonina por parte de las plaquetas, lo cual sugiere que la magnitud del gen ITGB3 impacta la actividad de la proteína SERT. Estos resultados señalan que se requiere una adecuada expresión o señalización de αIIbβ3 para la función normal de la SERT (89). Las plaquetas que portan el polimorfismo Pro33 en ITGB3, cercano al polimorfismo evaluado en este trabajo, Leu59Pro, mostraron un incremento en la activación αIIbβ3, por la unión a fibrinógeno. Se observó un incremento del 260 % en la actividad de captura de la proteína SERT por la presencia del alelo Pro33 en ITGB3 en relación con el homocigoto para Leu33 (89).
No existen estudios que demuestren la interacción a nivel funcional de las proteínas ITGB3 con HTR2A, que apoyen los efectos conjuntos de variantes en estos dos genes sobre la gravedad de las deficiencias en comunicación identificadas en este estudio.
Es notorio que, a pesar de la estrecha correlación entre la gravedad de los síntomas en las dimensiones de interacción social recíproca y comunicación (r=0,63, p=0,0001), la asociación vista con las variantes rs5918 y GA-1438 para la gravedad de los síntomas en la comunicación no se replicara para la dimensión de interacción social. Esto sugiere que, aunque seguramente habrá variantes de predisposición o modificadoras comunes a las dos dimensiones, no es el caso de las variantes estudiadas en los genes ITGB3 y HTR2A.
Además, se observó una significativa asociación entre el polimorfismo rs15908; específicamente, el alelo G contribuye a la gravedad de los comportamientos restringidos y estereotipados. Esta variante es una sustitución sinónima presente en el exón 9 del gen ITGB3, y es poco probable que sea esta la variante que esté aportando a la gravedad de estos síntomas; sin embargo, es posible que otra variante, posiblemente en el mismo exón o en una región en desequilibrio de ligamiento con ésta, sea la variante directamente implicada en este subfenotipo.
Existen varias limitaciones en este estudio. La primera es el reducido tamaño de la muestra. La potencia estadística que tiene este estudio para detectar un factor de riesgo con un OR de 2, es del 60 %, para un marcador completamente polimórfico (MAF=0,5). Sin embargo, en un rasgo complejo los riesgos aportados por cada variante independiente pueden ser mucho menores que este; por lo tanto, es muy probable que en el presente estudio no se estén detectando asociaciones de factores de riesgo moderados. Este bajo poder de la muestra limita la posibilidad de evaluar interacciones entre variantes y entre diferentes genes, efectos que pueden ser de gran importancia en estos rasgos complejos y determinar en gran medida su capacidad de heredarse. No obstante, los casos incluidos en este estudio fueron meticulosamente diagnosticados, con el fin de reducir la posible heterogeneidad genética para el rasgo, y la población a la cual pertenecían las familias tenía una historia de aislamiento genético durante la mayor expansión demográfica. Ambos hechos favorecen la identificación de factores de riesgo que son compartidos por una buena proporción de los casos.
Finalmente, los SNP elegidos no abarcan la variabilidad genética total de cada uno de los genes estudiados; por esto, es probable que haya variantes en estos genes que aporten a la predisposición a la enfermedad, que no están en desequilibrio de ligamiento con aquellas que se estudiaron y, por lo tanto, no se está detectando la asociación.
Nuestros hallazgos soportan el papel que tiene la regulación de la serotonina sobre la etiología del autismo. Se identificaron diferentes variantes en el gen SLC6A4 que presentan un efecto principal o en forma epistática con otros genes del sistema sobre el riesgo de autismo en población antioqueña. Se encontró interacción significativa sobre el autismo entre variantes en los genes SLC6A4 y el del receptor de serotonina HTR2A. Además, se evidenció que variantes en los genes ITGB3 y HTR2A modifican la gravedad de los síntomas en las dimensiones de comunicación y la presencia de comportamientos restringidos y estereotipados, en pacientes de origen antioqueño.
Agradecemos a las familias participantes, quienes hicieron posible este estudio. A la Fundación Integrar (Medellín), la Fundación Prisma (Cali), ASOPAHINES (Bello) y la Fundación CEREVIDI (Sincelejo); a Nicolás Lassa, de Barranquilla, y a los profesionales de Cúcuta, Valledupar y Cartagena que nos remitieron sus pacientes a la investigación. Nuestro agradecimiento a Olga Zea, por su colaboración en el procesamiento de los cariotipos.
Los autores declaran que no existe conflicto de intereses y que no hubo injerencia externa en el desarrollo de esta investigación.
El estudio fue financiado por Colciencias (proyecto No. 111534319139). A. Valencia fue financiada por la beca para el apoyo a doctorados nacionales de Colciencias, convocatoria 2007. Las evaluaciones clínicas fueron realizadas en los consultorios de W.
Cornejo, J. Cardona y la Fundación Integrar, sin ningún costo.
Correspondencia: Ana Victoria Valencia, Laboratorio de Genética Molecular, Universidad de Antioquia, Sede de Investigación Universitaria, Calle 62 N° 52-59, torre 2, laboratorio 430, Medellín, Colombia Teléfono: (574) 219 6467; fax: (574) 219 6469 valenciaduarte@gmail.com
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Washington, D.C.: American Psychiatric Publishing; 1994. [ Links ]
2. World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders. Geneve: WHO; 1992. [ Links ]
3. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol Med. 1995;25:63-77. http://dx.doi.org/10.1017/S0033291700028099 [ Links ]
4. Bolton P, Macdonald H, Pickles A, Rios P, Goode S, Crowson M, et al. A case-control family history study of autism. J Child Psychol Psychiatry. 1994;35:877-900. http://dx.doi.org/10.1111/j.1469-7610.1994.tb02300.x [ Links ]
5. Coutinho AM, Sousa I, Martins M, Correia C, Morgadinho T, Bento C, et al. Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum Genet. 2007;121:243-56. http://dx.doi.org/10.1007/s00439-006-0301-3 [ Links ]
6. Ramoz N, Cai G, Reichert JG, Corwin TE, Kryzak LA, Smith CJ, et al. Family-based association study of TPH1 and TPH2 polymorphisms in autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:861-7. http://dx.doi.org/10.1002/ajmg.b.30356 [ Links ]
7. Janusonis S. Statistical distribution of blood serotonin as a predictor of early autistic brain abnormalities. Theor Biol Med Model. 2005;2:27. http://dx.doi.org/10.1186/1742-4682-2-27 [ Links ]
8. Veenstra-van der Weele J, Kim SJ, Lord C, Courchesne R, Akshoomoff N, Leventhal BL, et al. Transmission disequilibrium studies of the serotonin 5-HT2A receptor gene (HTR2A) in autism. Am J Med Genet. 2002;114:277- 83. http://dx.doi.org/10.1002/ajmg.10192 [ Links ]
9. Burgess NK, Sweeten TL, McMahon WM, Fujinami RS. Hyperserotoninemia and altered immunity in autism. J Autism Dev Disord. 2006;36:697-704. http://dx.doi.org/10.1007/s10803-006-0100-7 [ Links ]
10. Klauck SM, Poustka F, Benner A, Lesch KP, Poustka A. Serotonin transporter (5-HTT) gene variants associated with autism? Hum Mol Genet. 1997;6:2233-8. http://dx.doi.org/10.1093/hmg/6.13.2233 [ Links ]
11. Guhathakurta S, Ghosh S, Sinha S, Chatterjee A, Ahmed S, Chowdhury SR, et al. Serotonin transporter promoter variants: Analysis in Indian autistic and control population. Brain Res. 2006;1092:28-35. http://dx.doi.org/10.1016/j.brainres.2006.03.078 [ Links ]
12. Fricker AD, Rios C, Devi LA, Gomes I. Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Brain Res Mol Brain Res. 2005;138:228-35. http://dx.doi.org/10.1016/j.molbrainres.2005.04.016. [ Links ]
13. Mazer C, Muneyyirci J, Taheny K, Raio N, Borella A, Whitaker-Azmitia P. Serotonin depletion during synaptogenesis leads to decreased synaptic density and learning deficits in the adult rat: A possible model of neurodevelopmental disorders with cognitive deficits. Brain Res. 1997;760:68-73. http://dx.doi.org/10.1016/S0006-8993(97)00297-7. [ Links ]
14. Faber KM, Haring JH. Synaptogenesis in the postnatal rat fascia dentata is influenced by 5-HT1a receptor activation. Brain Res Dev Brain Res. 1999;114:245-52. http://dx.doi.org/10.1016/S0165-3806(99)00036-X [ Links ]
15. Persico AM, Di Pino G, Levitt P. Multiple receptors mediate the trophic effects of serotonin on ventroposterior thalamic neurons in vitro. Brain Res. 2006;1095:17-25. http://dx.doi.org/10.1016/j.brainres.2006.04.006 [ Links ]
16. Blue ME, Molliver ME. 6-Hydroxydopamine induces serotonergic axon sprouting in cerebral cortex of newborn rat. Brain Res. 1987;429:255-69. http://dx.doi.org/10.1016/0165-3806(87)90106-4 [ Links ]
17. Blue ME, Erzurumlu RS, Jhaveri S. A comparison of pattern formation by thalamocortical and serotonergic afferents in the rat barrel field cortex. Cereb Cortex. 1991;1:380-9. http://dx.doi.org/10.1093/cercor/1.5.380 [ Links ]
18. Fujimiya M, Hosoda S, Kitahama K, Kimura H, Maeda T. Early development of serotonin neuron in the rat brain as studied by immunohistochemistry combined with tryptophan administration. Brain Dev. 1986;8:335-42. http://dx.doi.org/10.1016/S0387-7604(86)80053-5 [ Links ]
19. Rhoades RW, Mooney RD, Chiaia NL, Bennett-Clarke CA. Development and plasticity of the serotoninergic projection to the hamster´s superior colliculus. J Comp Neurol. 1990;299:151-66. [ Links ]
20. Chandana SR, Behen ME, Juhasz C, Muzik O, Rothermel RD, Mangner TJ, et al. Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int J Dev Neurosci. 2005;23:171-82. http://dx.doi.org/10.1016/j.ijdevneu.2004.08.002 [ Links ]
21. Murphy DG, Daly E, Schmitz N, Toal F, Murphy K, Curran S, et al. Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger´s syndrome: An in vivo SPECT study. Am J Psychiatry. 2006;163:934-6. [ Links ]
22. Cook EH Jr, Arora RC, Anderson GM, Berry-Kravis EM, Yan SY, Yeoh HC, et al. Platelet serotonin studies in hyperserotonemic relatives of children with autistic disorder. Life Sci. 1993;52:2005-15. http://dx.doi.org/10.1016/0024-3205(93)90685-V [ Links ]
23. Abney M, McPeek MS, Ober C. Broad and narrow heritabilities of quantitative traits in a founder population. Am J Hum Genet. 2001;68:1302-7. [ Links ]
24. Coutinho AM, Oliveira G, Morgadinho T, Fesel C, Macedo TR, Bento C, et al. Variants of the serotonin transporter gene (SLC6A4) significantly contribute to hyperserotonemia in autism. Mol Psychiatry. 2004;9:264-71. http://dx.doi.org/10.1038/sj.mp.4001409 [ Links ]
25. Ober C, Abney M, McPeek MS. The genetic dissection of complex traits in a founder population. Am J Hum Genet. 2001;69:1068-79. http://dx.doi.org/10.1086/324025 [ Links ]
26. Weiss LA, Abney M, Parry R, Scanu AM, Cook EH Jr, Ober C. Variation in ITGB3 has sex-specific associations with plasma lipoprotein(a) and whole blood serotonin levels in a population-based sample. Hum Genet. 2005;117:81-7. http://dx.doi.org/10.1007/s00439-004-1250-3 [ Links ]
27. Weiss LA, Abney M, Cook EH Jr, Ober C. Sexspecific genetic architecture of whole blood serotonin levels. Am J Hum Genet. 2005;76:33-41. http://dx.doi.org/10.1086/426697 [ Links ]
28. Weiss LA, Veenstra-van der Weele J, Newman DL, Kim SJ, Dytch H, McPeek MS, et al. Genome-wide association study identifies ITGB3 as a QTL for whole blood serotonin. Eur J Hum Genet. 2004;12:949-54. http://dx.doi.org/10.1038/ sj.ejhg.5201239 [ Links ]
29. Baghdadli A, Gonnier V, Aussilloux C. Review of psychopharmacological treatments in adolescents and adults with autistic disorders. Encephale. 2002;28:248-54. http://dx.doi.org/ENC-06-2002-28-3-0013-7006-101019-ART9 [ Links ]
30. Buchsbaum MS, Hollander E, Haznedar MM, Tang C, Spiegel-Cohen J, Wei TC, et al. Effect of fluoxetine on regional cerebral metabolism in autistic spectrum disorders: A pilot study. Int J Neuropsychopharmacol. 2001;4:119-25. http://dx.doi.org/10.1017/S1461145701002280 [ Links ]
31. Fatemi SH, Realmuto GM, Khan L, Thuras P. Fluoxetine in treatment of adolescent patients with autism: A longitudinal open trial. J Autism Dev Disord. 1998;28:303-7. [ Links ]
32. Cook EH Jr, Rowlett R, Jaselskis C, Leventhal BL. Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry. 1992;31:739-45. http://dx.doi.org/10.1097/00004583-199207000-00024 [ Links ]
33. Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, et al. Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet. 2004;75:1117-23. http://dx.doi.org/10.1086/426034 [ Links ]
34. Cantor RM, Kono N, Duvall JA, Álvarez-Retuerto A, Stone JL, Alarcón M, et al. Replication of autism linkage: Finemapping peak at 17q21. Am J Hum Genet. 2005;76:1050-6. http://dx.doi.org/10.1086/430278 [ Links ]
35. Lesch KP, Wolozin BL, Murphy DL, Reiderer P. Primary structure of the human platelet serotonin uptake site: Identity with the brain serotonin transporter. J Neurochem. 1993;60:2319-22. http://dx.doi.org/10.1111/j.1471-4159.1993.tb03522.x [ Links ]
36. Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996;66:2621-4. http://dx.doi.org/10.1046/j.1471-4159.1996.66062621.x [ Links ]
37. Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527-31. http://dx.doi.org/10.1126/science.274.5292.1527 [ Links ]
38. Conroy J, Meally E, Kearney G, Fitzgerald M, Gill M, Gallagher L. Serotonin transporter gene and autism: A haplotype analysis in an Irish autistic population. Mol Psychiatry. 2004;9:587-93. http://dx.doi.org/10.1038/ sj.mp.4001459 [ Links ]
39. Arieff Z, Kaur M, Gameeldien H, Merwe LV, Bajic VB. 5-HTTLPR polymorphism: Analysis in South African autistic individuals. Hum Biol. 2010;82:291-300. http://dx.doi.org/10.1038/sj.mp.4001459 [ Links ]
40. Cook EH Jr, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, et al. Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry. 1997;2:247-50. [ Links ]
41. Kistner-Griffin E, Brune CW, Davis LK, Sutcliffe JS, Cox NJ, Cook EH Jr. Parent-of-origin effects of the serotonin transporter gene associated with autism. Am J Med Genet B Neuropsychiatr Genet. 2010;156:139-44. http://dx.doi.org/10.1002/ajmg.b.31146 [ Links ]
42. McCauley JL, Olson LM, Dowd M, Amin T, Steele A, Blakely RD, et al. Linkage and association analysis at the serotonin transporter (SLC6A4) locus in a rigid-compulsive subset of autism. Am J Med Genet B Neuropsychiatr Genet. 2004;127B:104-12. http://dx.doi.org/10.1002/ajmg.b.20151 [ Links ]
43. Cho IH, Yoo HJ, Park M, Lee YS, Kim SA. Family-based association study of 5-HTTLPR and the 5-HT2A receptor gene polymorphisms with autism spectrum disorder in Korean trios. Brain Res. 2007;1139:34-41. http://dx.doi.org/10.1016/j.brainres.2007.01.002 [ Links ]
44. Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L, et al. Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am J Hum Genet. 2005;77:265-79. http://dx.doi.org/10.1086/432648 [ Links ]
45. Brune CW, Kim SJ, Salt J, Leventhal BL, Lord C, Cook EH Jr. 5-HTTLPR genotype-specific phenotype in children and adolescents with autism. Am J Psychiatry. 2006; 163:2148-56. [ Links ]
46. Pollock BG, Ferrell RE, Mulsant BH, Mazumdar S, Miller M, Sweet RA, et al. Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology. 2000;23:587-90. http://dx.doi.org/10.1016/S0893-133X (00)00132-9 [ Links ]
47. White KJ, Walline CC, Barker EL. Serotonin transporters: Implications for antidepressant drug development. AAPS J. 2005;7:E421-33. http://dx.doi.org/10.1208/aapsj070242 [ Links ]
48. Maestrini E, Lai C, Marlow A, Matthews N, Wallace S, Bailey A, et al. Serotonin transporter (5-HTT) and gammaaminobutyric acid receptor subunit beta3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. The International Molecular Genetic Study of Autism Consortium. Am J Med Genet. 1999;88:492-6. http://dx.doi.org/10.1002/(SICI)1096-8628(19991015)88:5<492::AIDAJMG11> 3.0.CO;2-X [ Links ]
49. Longo D, Schuler-Faccini L, Brandalize AP, dos Santos Riesgo R, Bau CH. Influence of the 5-HTTLPR polymorphism and environmental risk factors in a Brazilian sample of patients with autism spectrum disorders. Brain Res. 2009;1267:9-17. http://dx.doi.org/10.1016/j.brainres.2009.02.072 [ Links ]
50. Wassink TH, Hazlett HC, Epping EA, Arndt S, Dager SR, Schellenberg GD, et al. Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch Gen Psychiatry. 2007;64:709-17. [ Links ]
51. Kim SJ, Cox N, Courchesne R, Lord C, Corsello C, Akshoomoff N, et al. Transmission disequilibrium mapping at the serotonin transporter gene (SLC6A4) region in autistic disorder. Mol Psychiatry. 2002;7:278-88. http://dx.doi.org/ 10.1038/sj/mp/4001033 [ Links ]
52. Devlin B, Cook EH, Jr., Coon H, Dawson G, Grigorenko EL, McMahon W, et al. Autism and the serotonin transporter: The long and short of it. Mol Psychiatry. 2005;10:1110-6. http://dx.doi.org/10.1038/sj.mp.4001724 [ Links ]
53. Napolioni V, Lombardi F, Sacco R, Curatolo P, Manzi B, Alessandrelli R, et al. Family-based association study of ITGB3 in autism spectrum disorder and its endophenotypes. Eur J Hum Genet. 2011;19:353-9. http://dx.doi.org/10.1038/ ejhg.2010.180 [ Links ]
54. Ma DQ, Rabionet R, Konidari I, Jaworski J, Cukier HN, Wright HH, et al. Association and gene-gene interaction of SLC6A4 and ITGB3 in autism. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:477-83. http://dx.doi.org/ 10.1002/ajmg.b.31003 [ Links ]
55. Cochrane LE, Tansey KE, Gill M, Gallagher L, Anney RJ. Lack of association between markers in the ITGA3, ITGAV, ITGA6 and ITGB3 and autism in an Irish sample. Autism Res. 2010;3:342-4. http://dx.doi.org/10.1002/aur.157 [ Links ]
56. Mei H, Cuccaro ML, Martin ER. Multifactor dimensionality reduction-phenomics: a novel method to capture genetic heterogeneity with use of phenotypic variables. Am J Hum Genet. 2007;81:1251-61. [ Links ]
57. Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. Am J Med Genet. 1999;88:609-15. http://dx.doi.org/10.1002/(SICI)1096-8628 (19991215)88:6<609::AID-AJMG7>3.0.CO;2-L [ Links ]
58. Badner JA, Gershon ES. Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Mol Psychiatry. 2002;7:56-66. http://dx.doi.org/10.1038/sj/mp/4000922 [ Links ]
59. IMGSAC IMgSoAC. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Hum Mol Genet. 1998;7:571-8. http://dx.doi.org/10.1093/ hmg/7.3.571 [ Links ]
60. Philippe A, Martínez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, et al. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805-12. http://dx.doi.org/10.1093/hmg/8.5.805 [ Links ]
61. Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, et al. A genomic screen of autism: Evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493- 507. http://dx.doi.org/10.1086/302497 [ Links ]
62. McBride PA, Anderson GM, Hertzig ME, Sweeney JA, Kream J, Cohen DJ, et al. Serotonergic responsivity in male young adults with autistic disorder. Results of a pilot study. Arch Gen Psychiatry. 1989;46:213-21. [ Links ]
63. Goldberg J, Anderson GM, Zwaigenbaum L, Hall GB, Nahmias C, Thompson A, et al. Cortical serotonin type-2 receptor density in parents of children with autism spectrum disorders. J Autism Dev Disord. 2009;39:97-104. http://dx.doi.org/10.1007/s10803-008-0604-4 [ Links ]
64. Guhathakurta S, Singh AS, Sinha S, Chatterjee A, Ahmed S, Ghosh S, et al. Analysis of serotonin receptor 2A gene (HTR2A): Association study with autism spectrum disorder in the Indian population and investigation of the gene expression in peripheral blood leukocytes. Neurochem Int. 2009;55:754-9. http://dx.doi.org/10.1016/j.neuint.2009.07.008 [ Links ]
65. Hranilovic D, Blazevic S, Babic M, Smurinic M, Bujas-Petkovic Z, Jernej B. 5-HT2A receptor gene polymorphisms in Croatian subjects with autistic disorder. Psychiatry Res. 2010;178:556-8. http://dx.doi.org/10.1016/j.psychres.2010.04.007 [ Links ]
66. Carvajal-Carmona LG, Ophoff R, Service S, Hartiala J, Molina J, León P, et al. Genetic demography of Antioquia (Colombia) and the Central Valley of Costa Rica. Hum Genet. 2003;112:534-41. http://dx.doi.org/10.1007/s00439-002-0899-8 [ Links ]
67. Bedoya G, Montoya P, García J, Soto I, Bourgeois S, Carvajal L, et al. Admixture dynamics in Hispanics: A shift in the nuclear genetic ancestry of a South American population isolate. Proc Natl Acad Sci USA. 2006;103:7234-9. http://dx.doi.org/10.1073/pnas.0508716103 [ Links ]
68. Carvajal-Carmona LG, Soto ID, Pineda N, Ortiz-Barrientos D, Duque C, Ospina-Duque J, et al. Strong Amerind/white sex bias and a possible Sephardic contribution among the founders of a population in northwest Colombia. Am J Hum Genet. 2000;67:1287-95. http://dx.doi.org/10.1016/S0002-9297(07)62956-5 [ Links ]
69. Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton, NJ: Princeton Unviersity Press; 1994. p. 302-430. [ Links ]
70. Freimer NB, Reus VI, Escamilla M, Spesny M, Smith L, Service S, et al. An approach to investigating linkage for bipolar disorder using large Costa Rican pedigrees. Am J Med Genet. 1996;67:254-63. [ Links ]
71. Service S, DeYoung J, Karayiorgou M, Roos JL, Pretorious H, Bedoya G, et al. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet. 2006;38:556-60. http://dx.doi.org/10.1038/ng1770 [ Links ]
72. Lord C, Rutter M, Le Couteur A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659-85. http://dx.doi.org/10.1007/BF02172145 [ Links ]
73. Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, et al. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205-23. http://dx.doi.org/10.1023/A:1005592401947 [ Links ]
74. Rutter M, LeCouteur A, Lord C. Autism diagnostic interview revised (ADI-R). Torrance, CA: Western Psychological Services; 2003. p. 1-5. [ Links ]
75. Lord C, Rutter M, DiLavore PC, Risi S. Autism diagnostic observation schedule. ADOS. 3th edition. Torrance, CA: Western Psychological Services; 2003. p. 2-8. [ Links ]
76. Filipek PA, Accardo PJ, Baranek GT, Cook EH Jr, Dawson G, Gordon B, et al. The screening and diagnosis of autistic spectrum disorders. J Autism Dev Disord. 1999;29:439-84. [ Links ]
77. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: A tool set for wholegenome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559-75. http://dx.doi.org/10.1086/519795 [ Links ]
78. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263-5. http://dx.doi.org/10.1093/bioinformatics/bth457 [ Links ]
79. Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983-9. [ Links ]
80. Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66:87-98. http://dx.doi.org/10.1159/000119108 [ Links ]
81. Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69:138-47. http://dx.doi.org/10.1086/321276, [ Links ]
82. Moore JH. Computational analysis of gene-gene interactions using multifactor dimensionality reduction. Expert Rev Mol Diagn. 2004;4:795-803. http://dx.doi.org/10.1586/14737159.4.6.795 [ Links ]
83. Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and geneenvironment interactions. Bioinformatics. 2003;19:376-82. http://dx.doi.org/10.1093/bioinformatics/btf869 [ Links ]
84. Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley- Koch AE, Mei H, et al. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet. 2005;77:377-88. http://dx.doi.org/10.1086/433195 [ Links ]
85. Anderson BM, Schnetz-Boutaud NC, Bartlett J, Wotawa AM, Wright HH, Abramson RK, et al. Examination of association of genes in the serotonin system to autism. Neurogenetics. 2009;10:209-16. http://dx.doi.org/10.1007/ s10048-009-0171-7 [ Links ]
86. Yirmiya N, Pilowsky T, Nemanov L, Arbelle S, Feinsilver T, Fried I, et al. Evidence for an association with the serotonin transporter promoter region polymorphism and autism. Am J Med Genet. 2001;105:381-6. http://dx.doi.org/10.1002/ajmg.1365 [ Links ]
87. Adamsen D, Meili D, Blau N, Thony B, Ramaekers V. Autism associated with low 5-hydroxyindolacetic acid in CSF and the heterozygous SLC6A4 gene Gly56Ala plus 5-HTTLPR L/L promoter variants. Mol Genet Metab. 2010;102:368-73. http://dx.doi.org/10.1016/j.ymgme.2010.11.162 [ Links ]
88. Pagani F, Baralle FE. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat Rev Genet. 2004;5:389-96. http://dx.doi.org/10.1038/nrg1327 [ Links ]
89. Carneiro AM, Cook EH, Murphy DL, Blakely RD. Interactions between integrin alpha-II-beta-3 and the serotonin transporter regulate serotonin transport and platelet aggregation in mice and humans. J Clin Invest. 2008;118:1544-52. http://dx.doi.org/10.1172/JCI33374. [ Links ]
