










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.32 no.4 Bogotá Oct./Dec. 2012
https://doi.org/10.7705/biomedica.v32i4.574
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v32i4.574
1Instituto de Genética Humana, Facultad de Medicina, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia
2Instituto de Nutrición, Genética y Metabolismo, Facultad de Medicina, Universidad El Bosque, Bogotá, D.C., Colombia
Contribución de los autores:
Ana Milena Gómez y Reggie García-Robles: análisis de resultados y escritura del artículo.
Ana Milena Gómez: búsqueda de registros.
Fernando Suárez-Obando: análisis de resultados y edición del artículo.
Recibido: 07/10/11; aceptado:31/07/12
Introducción. Las mucopolisacaridosis son enfermedades poco frecuentes de depósito lisosómico de glucosaminoglucanos, con datos variables sobre su incidencia en diferentes países a nivel mundial. En Latinoamérica hay reportes de frecuencias en Brasil, pero en Colombia la información es escasa.
Objetivos. Estimar las frecuencias de las mucopolisacaridosis mediante un estudio retrospectivo en los departamentos de Cundinamarca y Boyacá, y estimar la agregación espacial de los casos en estos mismos departamentos.
Materiales y métodos. Se revisaron los registros de pacientes de diferentes instituciones de referencia para enfermedades genéticas, así como los registros de nacimientos vivos entre 1998 y 2007. Con base en ellos, se estimaron las frecuencias para cada tipo de mucopolisacaridosis. Se analizó la agregación espacial de los casos utilizando el programa SaTScan™.
Resultados. La frecuencia combinada para todas las mucopolisacaridosis fue de 1,98 casos por 100.000 nacidos vivos. La mayor frecuencia fue para la de tipo IV, con 0,68 casos por 100.000 nacidos vivos, mientras que la III fue la menor, con 0,17 casos. Se encontraron tres posibles áreas de agregación espacial para las mucopolisacaridosis I, III y IV.
Conclusión. La frecuencia combinada para todas las mucopolisacaridosis se encuentra dentro de los rangos reportados en la literatura científica, siendo la de tipo IV la más frecuente y la de tipo VII la menos frecuente. Aunque los datos aquí reportados podrían corresponder a un subregistro, dadas las dificultades inherentes a la recolección de la información en nuestro país, consideramos que son un estimativo válido de las frecuencias de estas enfermedades.
Palabras clave: mucopolisacaridosis/epidemiología, errores innatos del metabolismo, enfermedades por almacenamiento lisosómico, análisis por conglomerados.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.574
Estimation of the mucopolysaccharidoses frequencies and cluster analysis in the Colombian provinces of Cundinamarca and Boyacá
Introduction. Mucopolysaccharidoses are a group of infrequent disorders caused by the lysosomal deposit of glycosaminoglycans. Its incidence is quite variable among thecountries where it has been documented. In Brazil, disorder frequencies have been reported, but in Colombia information on them is scarce.
Objectives. The frequency and spatial aggregations of the mucopolysaccharidoses were estimated by a retrospective study in two central Colombian provinces.
Materials and methods. The records of patients and live newborns between 1998-2007 were reviewed from several reference institutions for genetic diseases. From these records, the frequencies for each mucopolysaccharidosis were estimated. The spatial aggregation of the cases was analyzed using the SaTScan software.
Results. The combined frequency for all the mucopolysaccharidoses was 1.98 cases per 100,000 live newborns. MPS IV had the highest frequency with 0.68 cases per 100,000 live newborns and MPS III showed a lower frequency of 0.17/100,000. Three spatial aggregation areas were indicated for MPS I, MPS III and MPS IV.
Conclusion. The combined frequency for all the mucopolysaccharidoses has been reported, with type IV the most frequent and the type VII in second place. The data herein constitute a record subset and, in spite of the difficulties inherent to the data retrieval in Colombia, they are a valid estimate of the frequencies of these diseases in central Colombia.
Keywords: mucopolysaccharidoses/epidemiology; metabolism, inborn errors; lysosomal storage diseases, cluster analysis.
doi: http://dx.doi.org/10.7705/biomedica.v32i4.574
Las mucopolisacaridosis son un grupo heterogéneo de enfermedades de depósito lisosómico, causadas por el déficit de las enzimas requeridas para el catabolismo de los glucosaminoglucanos, componentes importantes de la matriz del tejido conjuntivo. El acúmulo lisosómico de los glucosaminoglucanos no degradados resulta en disfunción celular, tisular y orgánica (1).
Hasta el momento se han descrito seis tipos de mucopolisacaridosis, algunas de ellas con subtipos clínicos, determinados por diferentes defectos enzimáticos, pero con características fenotípicas semejantes entre ellas. Estas enfermedades tienen un mecanismo de herencia autosómico recesivo, excepto la mucopolisacaridosis II (síndrome de Hunter) que es recesiva ligada al X.
La alteración en el catabolismo de los glucosaminoglucanos produce diferentes tipos de compromiso clínico, según el órgano de depósito principal: el sulfato de heparán produce síntomas predominantemente neurológicos, como en las enfermedades de Hurler, la de Hunter y el síndrome de Sanfilippo; el sulfato de quetarán produce opacidades de córnea y alteraciones esqueléticas sin compromiso neurológico, como en el síndrome de Morquio, y el sulfato de dermatán produce miocardiopatía y valvulopatía, como en las mucopolisacaridosis I, II y VI (2).
Los pacientes presentan un amplio rango de características clínicas, que comprenden deformidades esqueléticas, cardiopatías, alteraciones oculares y auditivas, infecciones respiratorias a repetición, organomegalias y compromiso mental, entre otras, las cuales afectan de manera significativa su calidad de vida (2). Sin embargo, el curso de las mucopolisacaridosis ha cambiado durante las últimas décadas debido al tratamiento de reemplazo enzimático para, al menos, tres de estas enfermedades: las de tipo I, II y VI (3). Otra aproximación terapéutica es el trasplante de células madre hematopoyéticas, que ha demostrado mejoría clínica en la de tipo I (1) y el tratamiento de reemplazo enzimático para la de tipo IV se encuentra en las últimas fases de estudio (3).
Los estudios previos sobre la incidencia de los diferentes tipos de mucopolisacaridosis han demostrado variaciones considerables en diversas poblaciones. La menor incidencia para todas las mucopolisacaridosis es la reportada por Malm, et al., en Suecia, con 1,75 casos por 100.000 nacimientos (4), y la mayor, en los Países Bajos por Poorthuis, et al., con 4,5 casos por 100.000 nacimientos (5). Sin embargo, se considera que la prevalencia general está entre 3,4 y 4,5 por 100.000 nacimientos (6).
En Latinoamérica, Coelho, et al., encontraron la mucopolisacaridosis I como la más frecuente en un análisis de pacientes con sospecha de errores innatos del metabolismo, la cual correspondió al 25 % de todas las mucopolisacaridosis y al 8,3 % de todos los casos de sospecha de errores innatos del metabolismo. En segundo lugar de frecuencia, encontraron a la mucopolisacaridosis VI, correspondiente al 18 % del total de las mucopolisacaridosis y al 6 % de todos los casos de sospecha de errores innatos del metabolismo (7).
En Colombia, la información concerniente a la frecuencia de estas enfermedades es escasa. Barrera reportó que la mucopolisacaridosis de tipo IV, o síndrome de Morquio, es la más prevalente en Colombia, mientras que la de tipo III es menos frecuente, en contraste con lo reportado a nivel mundial (8); no obstante, en este reporte no se precisan valores de frecuencias. Bernal y Briceño hallaron figuras con características físicas que sugieren mucopolisacaridosis de tipo I y de tipo IV en un estudio sobre cerámica de la cultura Tumaco-La Tolita, cuyos integrantes habitaron en los actuales límites entre Colombia y Ecuador desde mediados del primer milenio antes de Cristo hasta el tercer siglo de nuestra era (9). En 1990, Briceño presentó un estudio sobre genética de poblaciones en Boyacá, donde encontró dos familias de primos con siete casos de síndrome de Morquio en el municipio de Saboyá y otra familia con tres hermanos con la misma enfermedad en el municipio de Villapinzón (Cundinamarca) (10). Por otro lado, dentro del grupo de médicos genetistas existe la impresión de que hay una cantidad importante de pacientes con estas enfermedades, originarios de los departamentos de Cundinamarca y Boyacá.
Por este motivo, se planteó un estudio retrospectivo para establecer la frecuencia de las mucopolisacaridosis en los departamentos de Cundinamarca y Boyacá, y en Bogotá, estimando la frecuencia de los eventos y evaluando la agregación espacial de los casos. La agregación espacial, que hace parte de la epidemiología espacial, la cual consiste en la descripción y análisis de las variaciones geográficas de las enfermedades con respecto a diversos factores, permite identificar conglomerados (clusters), término que implica un exceso de casos en una situación en particular limitada por tiempo y espacio, y ayuda a determinar factores subyacentes que influyen en la aparición de estas agregaciones (11).
Materiales y métodos
Se hizo un estudio retrospectivo mediante la búsqueda de registros de pacientes con diagnóstico de mucopolisacaridosis, nacidos en el período comprendido entre enero de 1998 y diciembre de 2007. Se obtuvieron los datos de registros de historias clínicas y del Laboratorio de Genética del Hospital de La Victoria, de Bogotá (Cundinamarca), centro de referencia de pacientes con enfermedades genéticas de la Secretaría Distrital de Salud con atención a pacientes del régimen subsidiado; de historias clínicas del Hospital El Salvador, de Ubaté (Cundinamarca), hospital con servicio de consulta externa en Genética Clínica, y con atención a pacientes del régimen subsidiado y área de influencia sobre los municipios de Fúquene, Simijaca, Sutatausa, Susa, Carmen de Carupa, Cucunubá, Lenguazaque, Tausa y Guachetá, así como pacientes provenientes del departamento de Boyacá; de historias clínicas del Instituto de Genética Humana de la Pontificia Universidad Javeriana, de Bogotá, institución de referencia a nivel nacional, que atiende a pacientes de los regímenes contributivo y subsidiado, y de las bases de datos de registros de pacientes afiliados a ACOPEL (Asociación Colombiana de Pacientes con Enfermedades de Depósito Lisosomal), entidad de referencia en el país que los asocia, sin importar su afiliación al régimen de salud, con el objetivo de ayudar en la consecución de su tratamiento integral.
Los criterios de inclusión fueron los siguientes:
Pacientes originarios de los departamentos de Cundinamarca y Boyacá. Se definieron como tal aquellos individuos nacidos en estos departamentos y con, al menos, alguno de los padres naturales de los mismos. Las poblaciones de Cundinamarca y Boyacá fueron analizadas como un grupo debido a que comparten una gran área geográfica (altiplano cundiboyacense), aspectos socioculturales, fenómenos históricos y de poblamiento, así como bagaje genético (12-14).
Pacientes con diagnóstico de mucopolisacaridosis. En todos los registros clínicos se corroboró la valoración por un médico especialista en Genética Clínica. El diagnóstico fue confirmado por electroforesis de glucosaminoglucanos en orina, ensayo de determinación de niveles enzimáticos en leucocitos realizado en laboratorios especializados, o por ambas técnicas, usadas ampliamente en la práctica clínica para el diagnóstico de mucopolisacaridosis (15,16).
La frecuencia de cada una y de todas las mucopolisacaridosis se calculó dividiendo el total del número de casos por el total del número de nacimientos vivos durante el período de estudio. Esta misma metodología ha sido utilizada por los diferentes autores que han publicado datos sobre las frecuencias de estas enfermedades en diferentes países (4,5,15-21), a excepción de Hutchesson, et al., quienes reportaron frecuencias de mucopolisacaridosis I y III basándose en los datos de un programa de tamización neonatal (22).
Los datos de los nacimientos vivos en los departamentos de Cundinamarca y Boyacá, durante el período comprendido entre enero de 1998 y diciembre de 2007, se obtuvieron del Departamento Administrativo Nacional de Estadística (DANE). Se consideró este período debido a la disponibilidad de los datos suministrados por el DANE (23).
Utilizando los datos de los pacientes con mucopolisacaridosis y los datos estadísticos y geográficos de nacimientos durante el período en estudio, se hizo el análisis de agregación espacial mediante el programa SaTScan™, versión 8.1.1.1 (24), metodología que ha sido usada en la evaluación de agregaciones espaciales de otras enfermedades (25). Esta aplicación informática evalúa la agregación espacial asumiendo una distribución de Poisson para la presentación de casos en el área geográfica evaluada y de acuerdo con las frecuencias de los eventos encontrados en el total de la población, con significancia estadística (ρ<0,05).
Los eventos fueron evaluados como número de casos encontrados en los registros por el período de observación, dentro de un área geográfica específica. Se ubicaron las áreas de agregación espacial en un mapa de los departamentos de Cundinamarca y Boyacá, usando el sistema de información geográfica ArcGIS Explorer™, versión 1200, que permite visualizar la distribución espacial de las enfermedades usando las coordenadas y el área de influencia que indica el programa SaTScan™ para las posibles agregaciones espaciales encontradas (26).
Resultados
En el período de enero de 1998 a diciembre de 2007, se encontraron en los registros evaluados 35 niños originarios de Cundinamarca, Boyacá y Bogotá, a quienes se les diagnosticó mucopolisacaridosis. En el cuadro 1 se observa el lugar de nacimiento de los pacientes. Durante este período hubo un total de 1’767.059 nacidos vivos en la misma región (12). En el cuadro 2 se presentan los datos de las frecuencias correspondientes a cada tipo de mucopolisacaridosis. La frecuencia combinada de todos los casos de mucopolisacaridosis fue de 1,98 casos por 100.000 nacidos vivos. La de tipo IV obtuvo la mayor frecuencia, con 0,68 casos por 100.000 nacidos vivos, mientras que la de tipo III fue la menor, con 0,17 casos por 100.000. Las mucopolisacaridosis I, II y VI arrojaron frecuencias de 0,45, 0,45 y 0,23 casos por 100.000 nacimientos, respectivamente. No se registraron casos de la de tipo VII.
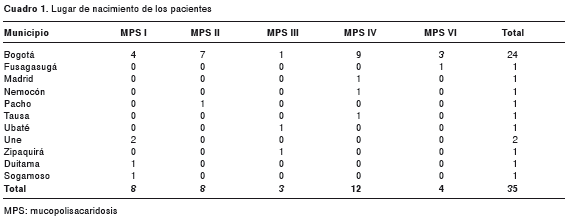

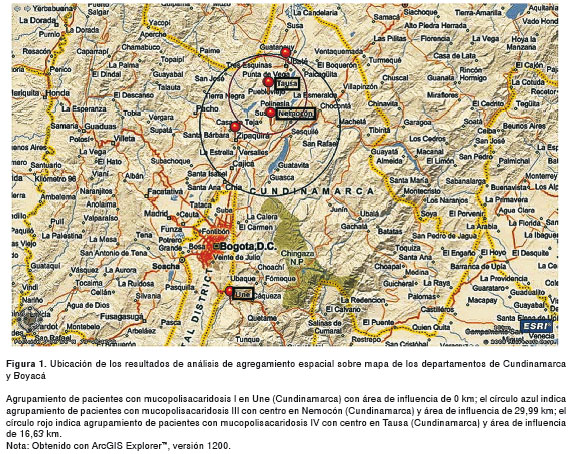
En el análisis de la agregación espacial, se encontraron tres probables agrupamientos (todos en el departamento de Cundinamarca): en Une (04°24′11″N, 74°01′31″W), con dos casos de mucopolisacaridosis I para 0,0015 casos esperados (ρ=0,001); en el área comprendida por Zipaquirá (05°01′42″N, 74°00′21″W), Nemocón (05°03′00″N, 73°53′00″W), Tausa (05°11′58″N, 73°53′26″W) y Ubaté (05°18′44″N, 73°49′09″W), con dos casos de mucopolisacaridosis III para 0,098 casos esperados (ρ=0,003); y en el área de Tausa, Ubaté y Nemocón, con dos casos de la de tipo IV para 0,14 casos esperados (ρ=0,019) (cuadro 3 y figura 1).

Discusión
Los estudios para la estimación de la frecuencia de enfermedades raras, como las mucopolisacaridosis, tienen dificultades inherentes (16), debido a que no existe un reporte obligatorio, un sistema de registro de este tipo de enfermedades, ni un programa de tamización neonatal dirigido a estas enfermedades. Además, es posible que haya un subdiagnóstico por falta de personal médico entrenado para su diagnóstico y las dificultades propias del sistema de salud. Debido a la gran variabilidad de las manifestaciones clínicas en los pacientes afectados, el diagnóstico usualmente es difícil y puede pasarse por alto, lo que suele conducir a la subestimación de la frecuencia de algunos tipos más sutiles clínicamente (15). Otro problema asociado es el hecho de que no existe un centro de referencia para el diagnóstico y manejo de estas enfermedades, por lo que hay un gran número de instituciones diferentes responsables de estos pacientes y se hace extremadamente difícil acceder a todos los registros. A pesar de las dificultades asociadas a este tipo de estudios, consideramos que los resultados obtenidos son una adecuada estimación de la frecuencia de mucopolisacaridosis para la población de estudio, ya que los registros de los pacientes se obtuvieron de los principales centros de referencia de la región y las frecuencias encontradas están dentro de las reportadas en la literatura científica.
En el período de estos diez años (1998-2007), la frecuencia estimada de mucopolisacaridosis en los departamentos de Cundinamarca y Boyacá fue de 1,98 por 100.000 nacidos vivos, que, a pesar de encontrarse dentro del rango reportado probablemente sea mayor, debido al probable subdiagnóstico y subregistro de los pacientes. Sin embargo, se encuentra una incidencia para mucopolisacaridosis IV de 0,68 por 100.000 nacimientos; este dato es superado solamente por las frecuencias encontradas en Noruega, con 0,76 (4), e Irlanda del Norte, con 1,3 (17), y para mucopolisacaridosis VI, 0,23 por 100.000 nacimientos, que iguala a la de Alemania (16) y es solamente sobrepasada por la de Australia, con 0,4 casos por 100.000 nacidos vivos (18). Analizando estos datos se observa que aun teniendo una incidencia global baja, se encuentran frecuencias relativamente altas para la de tipo IV y la de tipo VI en la región de estudio, al compararlas con lo reportado en la literatura científica.
En cuanto a las frecuencias para mucopolisacaridosis I y II, se encontraron 0,45 casos por 100.000 nacimientos para cada una de ellas. Al comparar estos resultados con lo reportado, se observa que la del tipo I se encuentra por debajo de lo hallado en los diferentes estudios, encontrándose solo en Taiwan un dato de incidencia menor que el del presente reporte (15), y la frecuencia encontrada para la de tipo II es comparable con lo reportado.
La frecuencia hallada para la mucopolisacaridosis III en la región de estudio, 0,17 casos por 100.000 nacidos vivos, es la menor al compararla con los estudios de diferentes países, y es este tipo la reportada como más frecuente en la mayoría de las publicaciones. Este hallazgo puede deberse a una menor frecuencia de este tipo de mucopolisacaridosis en la población estudiada. Sin embargo, hay que tener en cuenta que esta enfermedad es la que menos características físicas presenta y su mayor compromiso es neurológico y psiquiátrico, por lo cual se puede estar frente a un alto subdiagnóstico de este tipo de mucopolisacaridosis.
Finalmente, no se encontró ningún paciente con la de tipo VII, lo cual es comparable con la mayoría de los datos publicados. La forma de presentación más común de la mucopolisacaridosis VII es la eritroblastosis fetal (hydrops fetalis) (27). Sin embargo hay algunos reportes de pacientes con esta enfermedad que muestran características extremadamente leves (28). Esta gran heterogeneidad clínica podría causar un subdiagnóstico de este tipo de mucopolisacaridosis, pero, a pesar de esto, los datos parecen concordar en cuanto a que esta es la forma de mucopolisacaridosis menos frecuente en la mayoría de las poblaciones (cuadro 4).
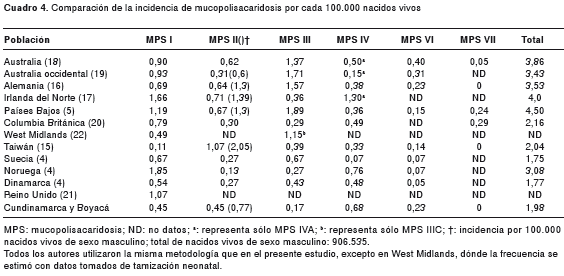
Considerando que es posible que haya una frecuencia variable de las mucopolisacaridosis entre poblaciones diferentes, es razonable que no se encuentre un patrón muy similar en su frecuencia al compararlas con los estudios de poblaciones europeas, asiáticas y australianas. Al comparar los datos con lo que se reporta en Latinoamérica, se encuentra una alta frecuencia de mucopolisacaridosis VI, lo que concuerda con lo descrito en Brasil por Coelho, et al., pero contrasta con una mayor proporción de la de tipo I encontrada en este país (7). Además, se observa que los resultados concuerdan con lo reportado por Barrera, et al., para Colombia, observándose la mayor frecuencia para la de tipo IV y, la menor, para la de tipo III (8).
El análisis de agregación espacial muestra agregaciones con más casos de los esperados en el área comprendida por Zipaquirá, Nemocón, Tausa y Ubaté para mucopolisacaridosis III, y el área que comprenden Nemocón, Tausa y Ubaté, para la de tipo IV, y en Une, una mayor cantidad de casos de la de tipo I (figura 1). Esto indica que es probable que existan áreas geográficas donde prevalecen algunos tipos de estas enfermedades, con lo cual se puede dirigir especialmente la atención hacia las poblaciones de estos lugares entrenando mejor al personal de salud que allí trabaja y, si es posible, llevando a cabo brigadas que apunten a encontrar nuevos casos de estas enfermedades. Se debe considerar la consanguinidad como un posible factor determinante de la frecuencia y agrupación de los eventos en estas poblaciones, tal como se ha descrito para otras enfermedades autosómicas recesivas en diferentes estudios (29,30). Por otro lado, el reporte de Bernal y Briceño sobre el hallazgo de cerámicas que parecen representar enfermedades genéticas entre las cuales se encuentran las mucopolisacaridosis, sugiere que estas enfermedades existían antes del advenimiento de los españoles (9). Surge, entonces, la pregunta sobre si nos encontramos ante un efecto fundador en estas enfermedades; esta inquietud debe ser abordada en próximos estudios de análisis molecular.
Los resultados de este estudio apoyan la sospecha surgida desde la práctica de la genética clínica sobre la frecuencia de mucopolisacaridosis en nuestra población, encontrándose que la mucopolisacaridosis más frecuente es la de tipo IV.
Con esto se quiere alertar a la comunidad médica sobre la importancia de reconocer y diagnosticar oportunamente estas enfermedades, esperando también que sirva como soporte para considerar un programa de tamización que las evalúe, ya que en la actualidad se cuenta con tratamiento para algunas de ellas, sumado además a la importancia que reviste poder brindar manejo y asesoría adecuados para cada uno de estos pacientes y sus familias.
Al profesor Fernando Poletta, del Estudio Colaborativo de Malformaciones Congénitas (ECLAMC) y Centro de Educación Medica e Investigaciones Clínicas (CEMIC), por su asesoría para el análisis metodológico.
A la Asociación Colombiana de Pacientes con Enfermedades de Depósito Lisosomal (ACOPEL), por su colaboración en la ubicación de los pacientes.
Los autores declaran no tener ningún conflicto de intereses.
Este fue un trabajo realizado en el Instituto de Genética Humana de la Pontificia Universidad Javeriana.
Correspondencia: Reggie García-Robles, Instituto de Genética Humana, Facultad de Medicina, Pontificia Universidad Javeriana, Carrera 7ª Nº 40- 62, Bogotá, D.C., Colombia.
Teléfono: (571) 320 8320, extensión 2791; fax: (571) 320 8320, extensión 2793 rgarcia@javeriana.edu.co
1. Muenzer J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(Suppl.):S27-34. http://dx.doi.org/10.1016/j.jpeds.2004.01.052 [ Links ]
2. Correa LN. Mucopolisacaridosis. Precop. 2005;3:30-7. [ Links ]
3. Giugliani R, Federhen A, Munoz MV, Vieira TA, Artigalas O, Pinto LL, et al. Enzyme replacement therapy for mucopolysaccharidoses I, II and VI: Recommendations from a group of Brazilian F experts. Rev Assoc Med Bras. 2010;56:271-7. http://dx.doi.org/10.1590/S0104-42302010000300009 [ Links ]
4. Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: Incidence and prevalence. Acta Paediatr. 2008;97:1577-81. http://dx.doi.org/10.1111/j.1651-2227.2008.00965.x [ Links ]
5. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151-6. http://dx.doi.org/10.1007/s004399900075 [ Links ]
6. Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Munoz V, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121:e377-86. http://dx.doi.org/10.1542/peds.2007-1350 [ Links ]
7. Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr. 1997;156:650-4. http://dx.doi.org/10.1007/s004310050685 [ Links ]
8. Barrera LA. Estudios bioquímicos de los errores innatos del metabolismo en Colombia, durante dos décadas. Rev Acad Colomb Cienc Exactas Fis Nat. 2009;33:377-94. [ Links ]
9. Bernal JE, Briceño I. Genetic and other diseases in the pottery of Tumaco-La Tolita culture in Colombia-Ecuador. Clin Genet. 2006;70:188-91. http://dx.doi.org/10.1111/j.1399-0004.2006.00670.x [ Links ]
10. Briceño I. Estudio de genética de poblaciones en Saboyá (Boyacá). Bogotá: Pontificia Universidad Javeriana; 1990. [ Links ]
11. Elliott P, Wartenberg D. Spatial epidemiology: Current approaches and future challenges. Environ Health Perspect. 2004;112:998-1006. http://dx.doi.org/10.1289/ehp.6735 [ Links ]
12. Ramírez R, Tobasura I. Migración boyacense en la cordillera central, 1876-1945 del altiplano cundiboyacense a los espacios de homogeneización antioqueña. Bull Inst Fr Etudes Andin. 2004;33:225-53. [ Links ]
13. Gracia LJ, Jiménez LC. Poblamiento y marginalidad en el altiplano cundiboyacense de Colombia. Anais do X Encontro de Geógrafos da América Latina; Brasil: Universidad Nacional de Colombia; 2005. p. 6599-624. [ Links ]
14. Rojas KM, Roa M, Briceño I, Guaneme C, Gómez A. Polimorfismos de 17 marcadores STR del cromosoma-Y en una muestra poblacional del altiplano cundiboyacense. Colomb Med. 2011;42:88-97. [ Links ]
15. Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A. 2009;149A:960-4. http://dx.doi.org/10.1002/ajmg.a.32781 [ Links ]
16. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011-7. http://dx.doi.org/10.1007/s10545-005-0112-z [ Links ]
17. Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101:355-8. http://dx.doi.org/10.1007/s004390050641 [ Links ]
18. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54. [ Links ]
19. Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123A:310-3. http://dx.doi.org/10.1002/ajmg.a.20314 [ Links ]
20. Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105:e10. [ Links ]
21. Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in mucopolysaccharidosis I: Hurler, Hurler- Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008;3:24. http://dx.doi.org/10.1186/1750-1172-3-24 [ Links ]
22. Hutchesson AC, Bundey S, Preece MA, Hall SK, Green A. A comparison of disease and gene frequencies of inborn errors of metabolism among different ethnic groups in the West Midlands, UK. J Med Genet. 1998;35:366-70. [ Links ]
23. Departamento Administrativo Nacional de Estadística. Estadísticas vitales. Fecha de consulta: 22 de julio de 2010. Disponible en: http://www.dane.gov.co/daneweb_V09/index.php?option=com_content&view=article&id=786&Item id=119. [ Links ]
24. Kulldorf M. SaTScan. Fecha de consulta: 22 de julio de 2010. Disponible en: http://www.satscan.org. [ Links ]
25. Forand SP, Talbot TO, Druschel C, Cross PK. Data quality and the spatial analysis of disease rates: Congenital malformations in New York State. Health Place. 2002;8:191-9. http://dx.doi.org/10.1016/S1353-8292(01)00037-5, [ Links ]
26. esri. ArcGIS Explorer. Fecha de consulta: 22 de julio del 2010. Disponible en: http://www.esri.com/software/arcgis/explorer/index.html. [ Links ]
27. Vervoort R, Islam MR, Sly WS, Zabot MT, Kleijer WJ, Chabas A, et al. Molecular analysis of patients with betaglucuronidase deficiency presenting as hydrops fetalis or as early mucopolysaccharidosis VII. Am J Hum Genet. 1996;58:457-71. [ Links ]
28. Vervoort R, Gitzelmann R, Bosshard N, Maire I, Liebaers I, Lissens W. Low beta-glucuronidase enzyme activity and mutations in the human beta-glucuronidase gene in mild mucopolysaccharidosis type VII, pseudodeficiency and a heterozygote. Hum Genet. 1998;102:69-78. [ Links ]
29. Khlat M, Khoury M. Inbreeding and diseases: Demographic, genetic, and epidemiologic perspectives. Epidemiol Rev. 1991;13:28-41. [ Links ]
30. Stoll C, Alembik Y, Dott B, Feingold J. Parental consanguinity as a cause of increased incidence of birth defects in a study of 131,760 consecutive births. Am J Med Genet. 1994;49:114-7. http://dx.doi.org/10.1002/ajmg.1320490123 [ Links ]
