










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157versão On-line ISSN 2590-7379
Biomédica v.32 supl.1 Bogotá mar. 2012
REVISIÓN DE TEMA
Grupo Malaria, Universidad de Antioquia, Medellín, Colombia
Contribución de los autores:
Ambos autores participaron en la revisión bibliográfica y redacción del manuscrito.
Recibido: 31/05/11; aceptado:01/09/11
Se presentan los mecanismos patogénicos más conocidos en la infección por Plasmodium falciparum durante la fase eritrocitaria y extraeritrocitaria. La obstrucción vascular, explicada por los fenómenos de secuestro de glóbulos rojos parasitados y la formación de rosetas, mediados por diversos ligandos y receptores endoteliales, además de los procesos inflamatorios instaurados ante la presencia del parásito, son aspectos centrales en la patogenia de la malaria que permiten explicar.
A partir de eventos como la lesión y la destrucción de eritrocitos, hepatocitos y células endoteliales, la pérdida de integridad del endotelio y la activación de promotores de daño celular y de apoptosis, se explican alteraciones como el aumento de la permeabilidad vascular, la hipoxia y el metabolismo anaerobio, que conducen tanto a lesiones localizadas en órganos como cerebro y pulmón, como a un estado de acidosis generalizada y falla multisistémica.
Palabras clave: malaria/etiología, Plasmodium falciparum, inflamación.
Pathogenic mechanisms in Plasmodium falciparum malaria
The most recognized pathogenic mechanisms of the infection with Plasmodium falciparum, during both the erythrocytic and exo-erithrocytic stages are presented. Vascular obstruction explained by the sequestration of parasitized red blood cells and erythrocyte rosetting, mediated by different endothelial ligands and receptors, in addition to the inflammatory processes induced by the presence of the parasite, are central aspects in the pathogenesis of malaria that explain the processes of damage, dysfunction and cell death in various organs. Alterations such as increased vascular permeability, hypoxia and anaerobic metabolism leading to localized lesions in organs such as brain and lung, as well as to a generalized acidotic state with multisystem failure can be explained by events such as the injury and destruction of erythrocytes, hepatocytes and endothelial cells, the loss of endothelial integrity, and the activation of cell damage and apoptosis promoters.
Key words: Malaria/ethiology, Plasmodium falciparum, inflamation.
Los signos y síntomas clínicos en la malaria, o paludismo, expresión del daño ocasionado en diversos órganos y sistemas, se exacerban durante el período de multiplicación del parásito en el eritrocito, mientras que el estadio hepático y la presencia de gametocitos tradicionalmente no se han asociado con la sintomatología. La malaria causa lesiones estructurales y alteraciones funcionales y metabólicas, cuya presentación clínica está en función de la edad, el estado inmunitario y las características genéticas del huésped, y por la especie, el genotipo y la virulencia del parásito. Puede conducir a un cuadro clínico grave el cual casi siempre se observa en las infecciones causadas por Plasmodium falciparum. Las complicaciones informadas con mayor frecuencia incluyen anemia grave, malaria cerebral, síndrome de dificultad respiratoria aguda y falla renal (1); en los últimos años se vienen informando estas y otras complicaciones en infecciones por P. vivax, al cual se le atribuía menor virulencia (2).
En la patogenia del paludismo grave por P. falciparum se han estudiado ampliamente los mecanismos implicados en la malaria cerebral y la anemia grave, síndromes que pueden explicarse a partir de dos mecanismos fundamentales: la obstrucción vascular causada por glóbulos rojos parasitados y la destrucción de eritrocitos; la interacción entre los eritrocitos parasitados y el endotelio conlleva a un proceso de activación y estrés endotelial que amplifica la respuesta inflamatoria. Estos y otros mecanismos que intervienen de manera específica en diversos sitios, contribuyen al desarrollo de las complicaciones clínicas al lesionar intensamente órganos como el cerebro, el pulmón, el riñón y el hígado, y conducen a un compromiso multisistémico cuya alteración fundamental es la acidosis metabólica.
En esta revisión se pretende describir los mecanismos más conocidos de la patogenia que explican la enfermedad y las complicaciones en la malaria por P. falciparum.
Patogenia de la infección por Plasmodium falciparum
El ciclo de vida de los parásitos del género Plasmodium es complejo e involucra diferentes estadios en el mosquito y el humano. En este último comienza con la inoculación de esporozoítos móviles durante la picadura del mosquito, formas invasivas que viajan por el torrente sanguíneo al hígado, donde comienza el desarrollo asexual tisular; luego de 10 a 12 días ocurre la ruptura de hepatocitos infectados, liberándose miles de merozoítos que invaden los glóbulos rojos para dar continuidad al ciclo asexual sanguíneo. En el ciclo eritrocitario, el parásito crece, se replica y libera merozoítos a la sangre para invadir nuevas células cada 48 horas. La mayor virulencia de P. falciparum con respecto a las otras especies que infectan el humano, puede estar relacionada con su capacidad de adherirse y ser secuestrado en el sistema capilar de los órganos, su rápida multiplicación y la capacidad de invadir glóbulos rojos de todas las edades (3).
Patogenia durante el ciclo eritrocitario
Secuestro y citoadherencia. La exportación y anclaje de proteínas parasitarias en la membrana del glóbulo rojo parasitado induce la formación de protuberancias electrodensas llamadas knobs. Estas estructuras participan en la patogenia de la infección por concentrar la proteína 1 de membrana del eritrocito de P. falciparum (Plasmodium falciparum Erythrocyte Membrane Protein 1, PfEMP1), que participa en la variación antigénica y en el secuestro de los glóbulos rojos parasitados (3). El secuestro es la unión de glóbulos rojos parasitados al endotelio capilar de órganos profundos y tiene lugar durante el desarrollo de trofozoítos y esquizontes, mientras las formas inmaduras del parásito (anillos) están libres en la circulación periférica. En los estudios de patología se ha demostrado la asociación entre la obstrucción vascular por glóbulos rojos parasitados adheridos al endotelio y la malaria cerebral (4). El secuestro de los glóbulos rojos parasitados también se ha descrito en el sistema capilar de pulmón, corazón, intestino, medula ósea y riñón, lo cual sugiere que las alteraciones en diferentes órganos participan en la gravedad de la infección (5).
El parásito ha desarrollado los mecanismos de adherencia como una estrategia de supervivencia que le permite evadir el paso por el bazo y, con ello, evitar su destrucción y multiplicarse; además, le permite protegerse del reconocimiento inmunitario cubriéndose de eritrocitos no infectados, disminuir el acceso de células migratorias del sistema inmunitario y citocinas a los sitios de secuestro mediante bloqueo de la microcirculación, y ejercer un efecto inmunomodulador para inhibir el procesamiento de antígenos en células dendríticas (6, 7).
Receptores implicados en la adherencia. Se han reconocido diversas moléculas in vitro como receptores que median la adherencia de los glóbulos rojos parasitados (cuadro 1). Los receptores más importantes son la glucoproteína CD36 y la molécula 1 de adhesión intercelular (Intercellular Adhesion Molecule 1, ICAM-1) en el endotelio y el sulfato A de condroitina (Chondroitin Sulfate A, CSA) en placenta. Otros receptores incluyen la molécula 1 de adhesión vascular (Vascular Cell Adhesion Molecule 1, VCAM-1) y la E-selectina en endotelio, las proteínas PCAM-1/CD31 en plaquetas y endotelio, el sistema de antígenos ABO, el sulfato de heparán y el receptor 1 del complemento (CR1) en eritrocitos no parasitados (8). Bajo condiciones fisiológicas, el secuestro puede obedecer a una interacción con múltiples receptores y en parásitos aislados se han observado fenotipos multiadhesivos que se asocian con enfermedad grave (3,8,9).
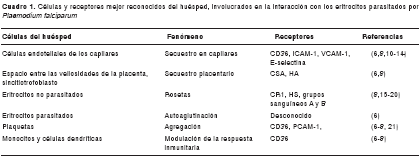
CD36 (Cluster of Differentiation 36). Es el receptor identificado con mayor frecuencia; participa en el secuestro en diferentes órganos como pulmón, riñón, hígado, intestino y músculo; se expresa en endotelio, monocitos/macrófagos, plaquetas y células dendríticas (cuadro 1). La unión a este receptor es común en parásitos aislados en diferentes países de África (9,11,12), Thailandia (13,14) y Brasil (10). La relación entre el CD36 y la gravedad clínica es controversial; en general, se asocia este receptor con enfermedad no complicada (8,9,11,13).
ICAM-1 (Intercellular Adhesion Molecule 1). Participa en la adherencia de glóbulo rojo parasitado al endotelio de cerebro, hígado, riñón y pulmón. La unión a ICAM-1 se ha observado en el 80 % de los parásitos aislados en África y es común en Tailandia y Brasil (8-11,13,14). La interacción con la ICAM-1 se asocia con enfermedad grave, en particular con malaria cerebral (11,13). Los estímulos inflamatorios como el factor de necrosis tumoral alfa (FNTα), cuyos niveles están incrementados durante el paludismo, inducen la expresión de este receptor (22). La adherencia de glóbulos rojos parasitados al endotelio cerebral induce directamente la expresión de ICAM-1 por activación del factor de transcripción nuclear kappa B (NFkB) (23). La ICAM-1 y el CD36 cooperan sinérgicamente, aumentando la adhesión de los glóbulos rojos parasitados al endotelio que expresa ambos receptores.
Otros receptores endoteliales. Representan feno-tipos adherentes menos frecuentes en aislamientos de P. falciparum(8,14). La unión a PCAM-1 se encontró en 50 % de los parásitos aislados en Kenia. Esta molécula se expresa en células endoteliales, leucocitos y plaquetas, lo que sugiere su participación en diferentes fenómenos adhesivos de P. falciparum(9). La unión a VCAM-1, E-selectina y P-selectina fue descrita originalmente en cepas del parásito adaptadas in vitro y es poco frecuente en aislamientos de pacientes (8).
Ligandos. La PfEMP1 se adhiere a todos los receptores identificados; es una proteína polimorfa de alto peso molecular (200-350 kDa), con tasas de variación antigénica clonal in vitro de 2 % por ciclo de vida (24,25). Esta proteína es codificada por la familia multigénica var,localizada preferencialmente en los subtelómeros de los cromosomas, donde se favorece la recombinación para generar diversidad. Cada genoma de P. falciparum posee entre 40 y 60 genes var, con secuencias muy divergentes en una misma cepa y entre diferentes cepas de P. falciparum. Los genes var se expresan de forma mutuamente excluyente, generando una sola variante de PfEMP1 por ciclo celular; todo el repertorio de genes se transcribe en el estadio de anillo, pero sólo uno se traduce a la proteína que determina el fenotipo adherente y antigénico de glóbulo rojo parasitado (24).
La familia var se clasifica en cuatro grupos según la secuencia de su región promotora 5´ (Ups), ubicación en cromosoma y dirección hacia la cual son transcritos (cuadro 2). Los genes B y C son los más abundantes en el genoma de P. falciparum y codifican para el 80 % de las PfEMP1, las cuales se unen en su mayoría a CD36 y sólo unas pocas a ICAM-1.

Genes VAR, PfEMP1 y enfermedad grave.Se ha observado un incremento significativo en la expresión de transcriptos del grupo B en niños con malaria sintomática (moderada y grave) en Papúa, Nueva Guinea (26,29), Tanzania (28) y Mali (27), mientras los transcriptos de genes C se expresan de forma predominante en los casos de malaria asintomática e hiperparasitemias no asociadas con el desarrollo de manifestaciones graves (26,27,29). Se sugiere la participación de proteínas codificadas por genes de tipo A en el desarrollo de paludismo complicado; dominios DBL-1 del grupo A se han asociado con enfermedad grave y falla multiorgánica en pacientes de Brasil (30). Se ha observado aumento de transcriptos var A en casos sintomáticos y enfermedad grave con respecto a casos asintomáticos y enfermedad moderada, en Tanzania y Mali (26-28); además, la transcripción de genes var de tipo A se relaciona con mayor frecuencia con formación de rosetas (27,29). Sin embargo, en Papúa no se encontró relación entre enfermedad grave y expresión de genes var A, ni con la formación de rosetas (26,29).
La expresión de genes var específicos en diferentes presentaciones clínicas de la enfermedad, sugiere que un subgrupo de genes están asociados con malaria sintomática y complicaciones. Los estudios serológicos respaldan esta hipótesis; los parásitos que causan enfermedad grave en niños no inmunes tienden a expresar subtipos específicos de antígenos de superficie que se expresan con menor frecuencia en niños con cierto grado de inmunidad (31).
Activación endotelial
En pacientes con paludismo hay incremento en la expresión de moléculas de adhesión ICAM-1, VCAM-1 y E-selectina en la superficie endotelial, en tejidos y solubles en plasma (32, 33). En pacientes con malaria complicada, se ha observado que en las células endoteliales con activación inflamatoria o en apoptosis, se forman micropartículas que tienen propiedades proinflamatorias y procoagulantes que pueden ampliar la cascada inflamatoria (34).
En los pacientes con malaria por P. falciparum se incrementan los niveles séricos de marcadores de activación endotelial, como factor de von Willebrand, quimiocina CXCL8/IL-8, endotelina 1 (ET-1) y angiopoyetina 2, moléculas almacenadas y liberadas de los cuerpos de Webel-Palade de las células endoteliales (35-38). El aumento de del factor de von Willebrand se asocia con enfermedad grave y sugiere que la exocitosis de los cuerpos de Webel-Palade participa en la patogenia de la malaria, aunque se desconoce el estímulo que activa esta liberación (37). La angiopoyetina 2 (AGN-2) se encuentra elevada en pacientes con malaria grave en comparación con casos no complicados y controles sanos. Esta molécula participa en la activación endotelial y aumenta la permeabilidad vascular (38); también se ha observado aumento de ET-1 y la disminución del péptido natriurético de tipo C (36,37). La ET-1 es una molécula vasoconstrictora y amplificadora de la inflamación, mientras el péptido natriurético de tipo C produce relajación vascular y reduce la interacción de leucocitos y plaquetas con el endotelio activado. Los hallazgos sugieren que existe alteración en los niveles de sustancias derivadas del endotelio tras la interacción con los glóbulos rojos parasitados, a favor de una actividad vasoconstrictora y proinflamatoria que contribuyen al desarrollo de enfermedad grave.
La citoadherencia entre glóbulos rojos parasi-tados y células endoteliales in vitro promueve un incremento en la expresión de ICAM-1, en la producción de IL-6 y quimiocinas como CCL20/MIP3α (proteína inflamatoria 3α del macrófago), CXCL8 y CCL2/MIP1 (proteína 1 quimioatrayente de monocitos), importantes moléculas que atraen leucocitos a los tejidos inflamados (39). La interacción de los glóbulos rojos parasitados con el endotelio cerebral y pulmonar culmina en la activación del factor de transcripción NF-kB, el cual aumenta la expresión de ICAM-1 y moléculas proinflamatorias (23,40).
Daño y muerte celular. La interacción entre los parásitos aislados de los pacientes con malaria y el endotelio reduce la expresión de las moléculas de adhesión intercelular, como ocludina, vinculina y ZO1, lo que compromete la integridad de la barrera endotelial en el sistema capilar (41). Las células del endotelio pulmonar incrementan la expresión de los genes proinflamatorios y proapoptóticos, y experimentan muerte celular mediada por caspasas en respuesta al contacto con glóbulos rojos parasitados in vitro (42). Los parásitos obtenidos de pacientes con P. falciparum inducen apoptosis de células endoteliales y este fenómeno se relaciona de forma significativa con el desarrollo de manifestaciones neurológicas (43).
La apoptosis inducida por el parásito está relacionada con el desarrollo de malaria complicada. Los pacientes con paludismo por P. falciparum tienen aumento de marcadores inflamatorios y apoptóticos de vías extrínsecas, como FNTα, IP10, Fas ligando y receptores solubles para FNTα I y II asociados con enfermedad grave (44, 45). En los casos mortales por malaria cerebral se muestra aumento en estos factores, lo que sugiere que el daño apoptótico inducido por el parásito y una respuesta inflamatoria no regulada participan en su patogenia.
La adherencia de los glóbulos rojos parasitados a las células endoteliales estimula la producción de especies reactivas de oxígeno (Reactive Oxygen Species), como O2 y H2O2, que conllevan a muerte celular; su producción aumenta la expresión de ICAM-1 y VCAM-1 mediante la activación de factores de transcripción sensibles al estado de oxidorreducción de la célula, como NFkB, lo cual induce aumento en los niveles de unión de glóbulos rojos parasitados (46).
Formación de rosetas y otros agregados
Las rosetas son eritrocitos no infectados unidos a uno o más glóbulos rojos parasitados, fenotipo de adherencia que se ha asociado con paludismo complicado. En África se ha informado gran frecuencia de parásitos formadores de rosetas en niños con malaria grave en Gabón (17), Gambia (20), Kenia (9,19), Uganda (18) y Malí (16), mientras en trabajos en Papúa y Malawi se no ha encontrado asociación (12,15). Se postula que este mecanismo facilita la invasión de los eritrocitos adyacentes durante la ruptura de esquizontes, restringe la fagocitosis de los glóbulos rojos parasitados y protege los merozoítos del reconocimiento inmunitario. In vitro se ha observado que la formación de rosetas no influencia las tasas de invasión (47) ni protege los merozoítos del reconocimiento de anticuerpos durante la invasión a eritrocitos no infectados (48). La función biológica de las rosetas permanece desconocida y su papel en la patogenia puede ser la obstrucción vascular.
El ligando del parásito que participa en la formación de rosetas es PfEMP1 a través de su dominio DBL1, y los receptores son el CR1, los sacáridos de los grupos sanguíneos A y B, el glucosaminoglucano sulfato de heparán, las inmunoglobulinas G y M y las inmunoglobulinas no inmunes (8). Los eritrocitos con polimorfismos en CR1, frecuentes en la población africana, presentan reducción en la formación de rosetas y los pacientes con estos polimorfismos se protegen contra la enfermedad grave, lo que sugiere la importancia de este receptor en la patogenia (49).
También se ha descrito que los glóbulos rojos parasitados pueden formar agregados entre ellos y las plaquetas; a través de la unión a CD36 expresado en éstas, este fenotipo de adherencia se ha relacionado fuertemente con malaria grave (21). Otro receptor involucrado en este fenómeno es la molécula gC1qR/HABP1/p32, que también se expresa en el endotelio y es mediadora de adhesión en estas células (50).
Mediadores solubles y respuesta inflamatoria
Las sustancias involucradas en la destrucción y muerte de los parásitos, como citocinas pro-inflamatorias, especies reactivas de oxígeno y óxido nítrico, juegan un papel dual durante la infección por su participación en la patogenia.
Estrés oxidativo. Los pacientes con malaria tienen aumento en la producción de agente s oxidantes y reducción en las defensas antioxidantes, lo que favorece el estrés oxidativo (51). Tres fuentes pueden contribuir a la generación de especies oxidantes en malaria: la degradación de la hemoglobina por los parásitos intraeritrocitarios, su producción por células fagocíticas como mecanismo parasiticida y por células del huésped alteradas en la infección como las células endoteliales en el secuestro y los hepatocitos durante el ciclo hepático del parásito. Los antígenos de P. falciparum pueden activar la producción de especies reactivas de oxígeno por polimorfonucleares sanguíneos, como primera línea de defensa para destruir parásitos; en los niños de Gambia con malaria por P. falciparum, se encontró que los granulocitos tienen gran capacidad de producir especies reactivas de oxígeno y la generación de estas especies reactivas se relacionó con la rápida eliminación de la parasitemia (52).
Estrés oxidativo y hemozoína.En el eritrocito el parásito degrada cerca de 75 % de la hemoglobina, liberando el hemo que es biocristalizado a hemozoína para proteger las membranas de su efecto tóxico; en este proceso los electrones libreados en la oxidación del hierro del grupo hemo forman especies reactivas de oxígeno en presencia de oxígeno. El grupo hemo férrico puede catalizar reacciones de peroxidación lipídica no enzimática, dando lugar a la formación de lipoperóxidos; en los eritrocitos parasitados estos se encuentran en grandes cantidades, en especial, los derivados del ácido araquidónico (20-hydroxyeicosatraenoic acid, HETE). Estos productos parecen tener algún grado de toxicidad sobre los monocitos humanos y eritrocitos infectados (53).
Daño celular por peroxidación lipídica. La descom-posición de ácidos grasos insaturados de las membranas por acción de especies reactivas de oxígeno, genera productos finales como el aldehído malónico y 4-hidroxialqueno; el aldehído malónico es un compuesto muy reactivo capaz de formar aductos en proteínas y ácidos nucleicos, induciendo daño celular. Este marcador de estrés oxidativo se ha encontrado aumentado en el suero de pacientes con malaria no complicada de Colombia (54) y en el de pacientes con malaria grave (55).
Estrés oxidativo y anemia. Los productos de la peroxidación lipídica están incrementados en los glóbulos rojos parasitados, participan en el daño oxidativo de membranas y disminuyen la capacidad de deformación de los eritrocitos infectados. En los pacientes con malaria por P. falciparum se aumentan los niveles de especies reactivas derivadas del ácido tiobarbitúrico eritrocitario, indicador de daño en la membrana de eritrocitos (51). El plasma de pacientes con malaria contienen prooxidantes que, en conjunto con especies reactivas de oxígeno producidas en las células fagocíticas, alteran la membrana de los eritrocitos y contribuyen a la anemia (56,57).
Estrés nitrosidativo. El óxido nítrico se produce en forma constitutiva en algunos tejidos, donde cumple funciones como neurotransmisor y regulador del tono vascular y, en forma inducible, con estímulos proinflamatorios como el FNTα, en los leucocitos para la defensa contra microorganismos intracelulares, y en el endotelio para el control de la permeabilidad y la activación celular. Su papel durante la malaria no es claro; algunos autores sugieren su importancia como defensa contra el parásito, mientras otros argumentan que su producción exagerada contri-buye a la patogenia (22).
Defensa contra Plasmodium falciparum. Las grandes concentraciones de óxido nítrico en los pacientes con malaria se relacionan con la rápida eliminación de la parasitemia. Se ha encontrado que las especies derivadas del óxido nítrico de monocitos y hepatocitos, inhiben el desarrollo de los parásitos de la malaria (58, 59); sin embargo, en estudios recientes se cuestiona el papel antiparasitario de los monocitos humanos mediante el óxido nítrico, ya que se ha observado que estas células son incapaces de producirlo en presencia de diferentes estímulos, incluyendo hemozoína, lipopolisacárido bacteriano o interferón gamma (IFN-γ) (60).
Participación en la patogenia. La producción local de óxido nítrico puede contribuir a la patogenia de la malaria cerebral por causar neurotoxicidad, vasodilatación y aumento de la presión cerebral. Se han detectado grandes concentraciones de intermediarios de óxido nítrico en el líquido cefalorraquídeo y el suero de sujetos con casos graves y mortales de malaria cerebral (61), y los análisis inmunohisotológicos revelan la presencia de sintasa inducible de óxido nítrico (iNOS) en cerebro, en casos fatales de malaria, asociándose su cantidad con la gravedad (62); esto sugiere que el óxido nítrico activa mecanismos neuropatológicos. En pacientes con malaria y anemia grave existe incremento en los niveles de óxido nítrico y en la actividad de la iNOS (63, 64); se cree que el óxido nítrico participa en la supresión de la hematopoyesis e induce la destrucción de los eritrocitos. Además, se ha implicado en la acidosis metabólica, ya que inhibe la respiración mitocondrial mediante la interacción con enzimas como la aconitasa y los complejos I y II, generando un estado de hipoxia celular que desvía el metabolismo hacia la degradación anaerobia de la glucosa con la producción de lactato y acumulación de hidrogeniones (22). En otros estudios no se ha encontrado asociación entre la producción de óxido nítrico y la enfermedad grave (65-67), y se ha encontrado una relación inversa entre la gravedad y los marcadores de la producción de óxido nítrico y los niveles circulantes de iNOS en los monocitos de sangre periférica (68-70).
Hipótesis de la biodisponibilidad del óxido nítrico. La arginina es el sustrato para la síntesis de óxido nítrico; los pacientes con malaria presentan bajos niveles de este aminoácido en sangre que se asocian con malaria grave y muerte (70,71). Durante la hemólisis de los glóbulos rojos parasitados se libera arginasa eritrocitaria y hemoglobina, que reducen la disponibilidad del óxido nítrico, la primera, por degradar la arginina y la segunda, por secuestrar el óxido nítrico disponible. El óxido nítrico regula la permeabilidad vascular, reduce el tránsito de leucocitos y la activación endotelial y plaquetaria; una escasa disponibilidad de óxido nítrico mantiene la activación del endotelio, leucocitos y plaquetas, lo que contribuye en la patogenia al aumentar el secuestro de glóbulos rojos parasitados y leucocitos, activar cascadas de coagulación y alterar el flujo sanguíneo. La disfunción endotelial observada en los casos complicados es reversible con el suministro de L-arginina, lo cual sugiere la importancia de mejorar la disponibilidad del óxido nítrico en el tratamiento de la malaria grave (71).
Activación de la respuesta inflamatoria
Los productos derivados del parásito durante la liberación de merozoítos en el ciclo eritocítico, inducen la producción de mediadores pro-inflamatorios, como FNTα, IL1 e IL6. Estos mediadores inducen la producción de otras citocinas y enzimas que amplían la cascada inflamatoria y activan la defensa celular en el huésped; cuando la producción de mediadores inflamatorios se excede, altera la fisiología del huésped y es causante de enfermedad.
Factor de necrosis tumoral alfa (FNTα). Los eritrocitos parasitados y los productos del parásito, como glicosil-fosfatidil-inositol y hemozoína, estimulan la producción de FNTα en monocitos (72). Los elevados niveles de FNTα interfieren con la multiplicación del parásito y su producción temprana durante la infección es protectora por su capacidad de estimular los mecanismos microbicidas de los fagocitos, mientras altos y prolongados niveles pueden ser deletéreos en el curso de la enfermedad, por inducir fiebre, hipoglucemia, supresión de la médula ósea, coagulopatía, hipergammaglobulinemia, hipotensión y aumento en los niveles séricos de reactantes de fase aguda (22,73,74).
En los pacientes africanos con malaria se encuentra incremento en los niveles de FNTα que se relacionan con la presentación de fiebre y la gravedad de la infección (75-77). En los modelos de ratón, el exceso de FNTα contribuye al desarrollo de malaria cerebral; sin embargo, en los ensayos clínicos con agentes como anticuerpos anti-FNTα y pentoxilina (interfiere en la producción), no se ha demostrado reducción de la mortalidad y presentación de malaria cerebral en humanos (74). El incremento en los niveles de FNTα ocasiona una respuesta sistémica relacionada con las complicaciones palúdicas (cuadro 3); se han encontrado altos niveles de esta citocina en pacientes con anemia palúdica grave, falla renal, hipoglucemia y falla multiorgánica. Se sugiere que la producción local de citocinas, en especial de FNTα, juega un papel muy importante en el desarrollo de las complicaciones, que no puede ser evidenciado en sus niveles plasmáticos (74).
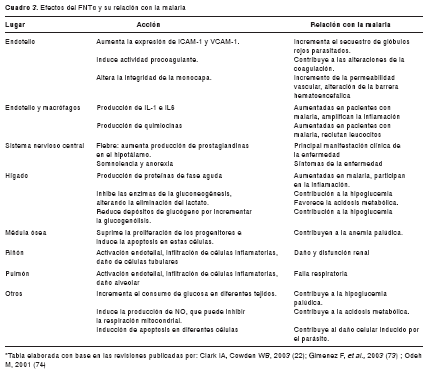
Otras citocinas. Los niveles séricos de las citocinas proinflamatorias IL-1 e IL-6 se encuentran incrementados en los pacientes con malaria y se han relacionado con enfermedad grave y malaria cerebral (75,78). Estas citocinas complementan muchas de las acciones mediadas por el FNTα, como la inducción de fiebre, la activación endotelial por IL-1 y el incremento en la producción de proteínas de fase aguda por IL-6, amplificando la respuesta inflamatoria. El IFN-γ se considera una citocina proinflamatoria; una de sus principales funciones es activar la respuesta celular, potenciando los mecanismos microbicidas de las células fagocíticas. Es producida por linfocitos T y células asesinas naturales; sus niveles se incrementan durante la malaria y protege de altas parasitemias y enfermedad clínica (79), aunque también puede tener un papel en la patogenia de la infección.
Citocinas antiinflamatorias. La IL-10 regula la respuesta proinflamatoria inducida por el parásito, ya que inhibe la producción de FNTα protegiendo al huésped de una respuesta exacerbada (80); una disminución en los niveles de esta citocina podría participar en el desarrollo de complicaciones. Se han evidenciado una respuesta insuficiente de IL-10 a altas concentraciones de FNTα en malaria grave, y altos niveles de esta citocina en niños con enfermedad moderada y no complicada (78,80,81).
Factor beta de crecimiento transformador (TGFb).Esta citocina regula el equilibrio entre las citocinas proinflamatorias y antiinflamatorias; está disminuida en el plasma de niños y adultos con malaria grave, mientras que el FNTα y la IL-10 están aumentados, lo que apoya la hipótesis de que un desequilibrio entre ambos perfiles de citocinas contribuye al desarrollo de enfermedad grave (82).
Quimiocinas. Son citocinas quimioatrayentes que reclutan y movilizan leucocitos a los sitios de inflamación. Los pacientes con enfermedad moderada y grave tienen incremento en la producción de quimiocinas, como CCL2/MCP-1, CCL3/MIP-1α y CCL4/MIP-1b, CXCL8/IL-8 e CXC10/IP-10, y disminución en los niveles de RANTES (78,83). MCP-1, MIP1α y MIP1b son potentes atrayentes de monocitos, mientras la IL-8 atrae polimorfonucleares. La quimiocina IP-10 se encuentra elevada en el líquido cefalorraquídeo de pacientes con malaria cerebral y se propone que juega un papel al atraer linfocitos T activados al cerebro.
Obstrucción vascular e inflamación
Los lugares de secuestro constituyen sitios ricos en material del parásito, capaz de activar una respuesta inflamatoria. Como parte de un ciclo, el incremento en la producción de mediadores inflamatorios favorece el secuestro, por incrementar la expresión de las moléculas de adhesión que son usadas por el parásito para adherirse al endotelio. La interacción entre glóbulos rojos parasitados y el endotelio conlleva a un proceso de activación y estrés endotelial caracterizado por la activación de vías de señalización, producción de mediadores inflamatorios, estrés oxidativo y apoptosis celular, daño que contribuye al desarrollo de las complicaciones palúdicas y amplía la respuesta inflamatoria.
El secuestro de glóbulos rojos parasitados, la citoadherencia y la formación de rosetas, producen obstrucción capilar con reducción del flujo sanguíneo, que conduce a hipoxia, disminución del intercambio de metabolitos y liberación de mediadores inflamatorios. Estos mecanismos se han asociado con síndromes clínicos como la malaria cerebral y la acidosis metabólica, y con lesión en diversos órganos, pero, ha sido difícil establecer una relación directa con las complicaciones (84). En la malaria cerebral estos cambios pueden explicar el edema y el aumento de la presión intracraneal; sin embargo, la heterogeneidad de los procesos que llevan al coma no ha permitido establecer una relación de causa-efecto entre esta complicación y el secuestro de glóbulos rojos parasitados. De igual manera, ha sido difícil probar una relación directa entre los fenotipos adherentes y los síndromes clínicos, tanto por la variabilidad genética de los parásitos como de los casos clínicos; incluso, la formación de rosetas no tiene una relación constante con la expresión de la enfermedad (84).
En el pulmón, el secuestro de glóbulos rojos parasitados en el espacio alvéolo-capilar y la presencia de macrófagos y trombos de fibrina, se consideran las causas de la obstrucción vascular, edema, daño al endotelio capilar y a las células alveolares, los cuales permiten explicar alteraciones en el intercambio de gases; este proceso se empeora por la respuesta inflamatoria desencadenada, incluso después de suministrar el tratamiento para la malaria (85). El incremento en la permeabilidad alveolar se considera la alteración más prominente en la lesión pulmonar aguda y el síndrome de dificultad respiratoria aguda en el paludismo; el daño pulmonar se refleja tanto en lesiones estructurales observables en el alvéolo pulmonar, como en la alteración funcional debida a los cambios en la permeabilidad de la membrana endotelial.
Tras la activación de los linfocitos T CD8+ por los antígenos de Plasmodium, se sintetizan IFN-γ, factores quimiotácticos, IL-1, IL-2 y FNTα, que promueven en el tejido pulmonar la migración y activación de macrófagos, células asesinas naturales y neutrófilos. La activación de neutrófilos y la generación de especies reactivas de oxígeno funcionan sinérgicamente con proteasas derivadas de neutrófilos para causar disrupción de las células endoteliales, con aumento de la permeabilidad capilar pulmonar (86,87). Se tiene evidencia de que el óxido nítrico y las especies reactivas de oxígeno se combinan para formar peroxinitrito, que causa disfunción mitocondrial, y ésta puede llevar a una falla en la contracción muscular; los altos niveles de iNOS en la musculatura de la pared torácica observada en la sepsis y la malaria, podrían apoyar la teoría de que la debilidad de los músculos respiratorios restringe la capacidad de compensar la acidosis que presenta el paciente (22).
Además de adquirir un fenotipo proinflamatorio, por acción del FNTα y la IL-1, la célula endotelial se torna protrombótica y la monocapa endotelial pierde su función de barrera, permitiendo el paso de factores plasmáticos y células sanguíneas. La oclusión trombótica vascular pulmonar es responsable del aumento en la presión de la arteria pulmonar, junto con la vasoconstricción y el edema intersticial; se ha observado que las lesiones vasculares más consistentes del síndrome de dificultad respiratorio agudo son los microémbolos (87).
Hemólisis y diseritropoyésis
La patogenia de la anemia en la malaria involucra diversos mecanismos.
Apoptosis eritrocitaria. El estrés oxidativo generado por la invasión del glóbulo rojo por P. falciparum, activa diferentes canales aniónicos y un canal de permeabilidad al Ca2+ no selectivo que permite la entrada de Ca2+ y Na+. La entrada de Ca2+ estimula el paso bidireccional de fosfolípidos a través de la bicapa, proceso que conduce a la pérdida de la asimetría de la fosfatidilserina de la membrana celular y a su exposición en la superficie externa de la membrana; esta modificación podría facilitar la unión a receptores de fosfatidilserina en los macrófagos, con la posterior fagocitosis del eritrocito infectado (88). La activación de canales iónicos permite la entrada de iones necesarios el parásito, pero también es un mecanismo de aceleración de la apoptosis eritrocitaria que sirve como línea de defensa del huésped (88).
Destrucción de eritrocitos parasitados. Tanto los glóbulos rojos parasitados como los no parasitados son destruidos por diversos mecanismos. La destrucción directa se produce cuando los parásitos completan su ciclo intraeritrocitario de maduración, al alcanzar el estado de esquizonte maduro en un período de 42 horas; se considera que ello contribuye poco a la anemia (89) y que aportaría sólo 10 % de la disminución total del hematocrito (90). La destrucción indirecta de glóbulos rojos parasitados se produce por acción de los macrófagos mediante mecanismos de fagocitosis por opsonización con inmunoglobulinas o factores del complemento, citotoxicidad mediada por anticuerpos y células asesinas naturales; otras señales de reconocimiento por células fagocíticas incluyen anormalidades en la rigidez de la membrana y exposición de antígenos parasitados y residuos de fosfatidil serina en la superficie de eritrocitos infectados (90-92). El FNTα también se ha sdo involucrado en la eritrofagocitosis, ya que esta citocina potencia la actividad fagocítica de macrófagos, acelera la eliminación de eritrocitos senescentes o dañados y potencia la eritrofagocitosis (81,93,94).
Destrucción de eritrocitos no parasitados. Las alteraciones en la morfología del eritrocito no parasitado, como el daño de la membrana, la reducción de la capacidad de deformación y el depósito de inmunocomplejos, ocasionan reducción en su vida media por destrucción acelerada, eritrólisis o acción de los macrófagos (93). Entre los mecanismos que explican la destrucción esplénica prematura de eritrocitos no parasitados, están: el daño de la membrana eritrocitaria causado por la respuesta inflamatoria, por el estrés oxidativo generado por la producción de radicales libres de oxígeno con exposición de fosfatidilserinas y por la disminución de la elasticidad de la membrana (93); el depósito de anticuerpos sobre la membrana del eritrocito que pueden estar dirigidos contra elementos modificados de la pared del glóbulo rojo y actuar como autoanticuerpos, o contra proteínas de estadios asexuales del parásito depositadas sobre la célula durante la ruptura periódica de esquizontes; y la producción de alteraciones en las proteínas reguladoras del complemento sobre la membrana del eritrocito (CR1, DAF) que pueden predisponer al depósito de complejos inmunitarios y complemento sobre el glóbulo rojo y facilitar su destrucción en el bazo (92,95,96).
Diseritropoyesis. El estudio histopatológico de la médula ósea durante la infección palúdica revela una médula hipercelular con incremento significativo en el número de precursores eritroides, pero el proceso de maduración es anormal (94), lo que disminuye el número de reticulocitos producidos a pesar de encontrarse niveles adecuados de hierro y ácido fólico. La eritropoyetina es el principal estímulo para la proliferación y maduración de los precursores eritroides; su deficiencia puede ser resultado de la acción de citocinas como FNTα e IL10 y se ha asociado con la depresión de la función medular; además, se considera que el desequilibrio de FNTα, IFN-γ e IL10, con altos niveles de FNTα (más de 1 ng/ml), puede suprimir la eritropoyesis (74,93). Aun con niveles adecuados de eritropoyetina puede haber una respuesta medular insuficiente, lo cual podría explicarse por alteraciones de la eritropoyetina o de sus receptores en las células precursoras eritroides (93,97). En pacientes con malaria y anemia grave se ha visto una maduración inadecuada de los precursores eritroides, con alteraciones en el citoesqueleto que conducen a marcada diseritropoyesis (97).
Destrucción esplénica. El bazo remueve los glóbulos rojos parasitados por su menor capacidad de deformación y los eritrocitos sensibilizados con inmunoglobulinas durante la infección por P. falciparum; por ello, el secuestro de glóbulos rojos parasitados con formas maduras permite evadir la circulación esplénica y la destrucción de estas células alteradas. El secuestro de glóbulos rojos parasitados parece ser un mecanismo relacionado con la presencia del bazo, porque se ha descrito que parásitos obtenidos de pacientes con esplenectomía no producen citoadherencia, ya que se observan estadios maduros circulantes e incapacidad de unirse a receptores endoteliales in vitro (98,99). A pesar de que la destrucción esplénica de glóbulos rojos parasitados contribuye a la anemia, el bazo parece jugar un papel benéfico contra las complicaciones; en los estudios de perfusión ex vivo de bazo humano se demuestran que un 10 % de los anillos quedan retenidos en cada paso por el bazo, lo que indica que este órgano de filtración puede controlar la biomasa de parásitos que puede secuestrarse (100).
Fases del ciclo extraeritrocitario y patogenia
Aunque menos estudiados, los mecanismos de la patogenia relacionados con el ciclo extra-eritrocitario parecen tener importancia en la génesis del daño hepático y pulmonar, como lo sugieren las investigaciones preliminares en modelos de ratón.
Travesía del esporozoíto de la dermis hasta el hígado. Los esporozoítos inoculados en la dermis permanecen en ella entre tres y cuatro horas, tiempo durante el cual migran a través de las células; algunos de ellos son destruidos, otros se movilizan por los ganglios linfáticos donde podrían iniciar una respuesta inmunológica adaptativa y la mayoría alcanzan los vasos sanguíneos (101). La obtención de imágenes in vivo de cultivos en modelos de infección por P. berghei en ratones, ha permitido evidenciar detalles de la invasión del plasmodio aún no conocidos en el humano; los esporozoítos inoculados ingresan al hígado, donde reconocen proteoglucanos de heparán-sulfato, se desplazan entre las células del endotelio de los sinusoides hepáticos evadiendo las células de Kupffer; después de migrar a través de varios hepatocitos, el esporozoíto permanece en uno de ellos, proceso que causa lesiones en la membrana celular e induce liberación de factor de crecimiento de hepatocitos, implicado en la regeneración tisular, y preserva los hepatocitos temporalmente mediante la activación de la cinasa-PI3, una vía antiapoptótica (102). En muchos hepatocitos se produce la muerte, dependiendo de si se sella o no la membrana celular después del paso del esporozoíto; tanto este proceso como la maduración y la ruptura de esquizontes originan cambios patológicos que contribuyen al daño tisular y al desarrollo de disfunción hepática, pero todavía la patogenia es poco conocida (103).
Maduración y ruptura de esquizontes tisulares. Dentro de los hepatocitos, el esporozoíto evoluciona a esquizonte hepático el cual madura y así se forman cientos de merozoítos de primera generación. Durante este período, el hepatocito aumenta varias veces su tamaño, fenómeno que podría inducir la muerte celular, pero ésta es inhibida por el parásito, y sólo hasta el final de esta fase pueden detectarse los primeros signos de muerte celular (102). El proceso de liberación de merozoítos está mediado por la activación de proteasas de cisteína, al parecer de origen parasitario, que inducen la muerte del hepatocito mientras se conserva el citoesqueleto (102). Los merozoítos deben invadir rápidamente su célula blanco, los eritrocitos, ya que, además de poseer una corta vida, están expuestos a la destrucción por las células de Kupffer a su paso por los sinusoides hepáticos. En P. berghei se ha establecido in vitro la formación de estructuras llamadas merosomas que envuelven grupos de merozoítos, mecanismo que permite salir del hepatocito y evitar el ataque por las células de Kupffer en su recorrido por el espacio de Disse hasta la luz del sinusoide hepático (104). Se supone que el paso de los merosomas produce daño por ruptura de las uniones intercelulares del endotelio próximo a las células parasitadas, aunque el mecanismo de muerte en el hepatocito no es completamente conocido, se piensa que las características corresponden a un proceso de autofagia más que de apoptosis o necrosis (102).
Estrés oxidativo en hígado inducido por formas eritrocitarias. Además del daño causado por las formas hepáticas del plasmodio, las formas eritrocitarias inducen estrés oxidativo implicado en la muerte celular del hepatocito. En ratones infectados con P. yoelii se encontró correlación positiva entre la parasitemia con disminución en la concentración hepática de glutatión, peroxidación lipídica y carbonilación de proteínas, esta última una medida de la oxidación de proteínas (103). En estos estudios se observó activación de la vía de apoptosis mitocondrial en hepatocitos por evidencia de activación de la caspasa-3 y regulación en bajo de proteínas Bcl-2 con regulación en alto de proteínas Bax, desequilibrio que conduce a la muerte celular. Se ha establecido que el radical OH está implicado en el estrés oxidativo por su correlación con la generación de peroxidación lipídica y porque desencadena el proceso de apoptosis mitocondrial (104).
Del hepatocito al endotelio pulmonar. Un primer contacto del pulmón con el parásito se presenta con la liberación de merozoítos en sus capilares. En P. yoelii se ha establecido que la desintegración de los merosomas procedentes de los hepatocitos se lleva a cabo en los capilares pulmonares y que este parece ser el mecanismo predominante para la liberación de los merozoítos hacia la circulación sanguínea. Se propone que la detención de los merosomas en los capilares de los tabiques pulmonares permite al plasmodio aprovechar las condiciones particulares de un medio muy oxigenado con las múltiples anastomosis de los vasos pulmonares, lo que permitiría la oclusión de estos capilares sin causar daño por necrosis, proceso que sí se asocia con infartos en otros tejidos. La liberación de los merozoítos de primera generación en los capilares del pulmón, donde la velocidad del flujo sanguíneo es menor que en grandes vasos y donde la densidad de macrófagos es baja, les permite una permanencia transitoria y el tiempo necesario para aumentar su capacidad infecciosa e invadir los eritrocitos (105). No se sabe cuáles son las implicaciones patogénicas de la desintegración de los merosomas en el pulmón, pero lo más probable es que localmente se inicie la respuesta inflamatoria que, aunque no alcance la destrucción de los merozoítos, sí puede desencadenar la secuencia de eventos que expliquen el comienzo de la lesión pulmonar y que sería reforzado por el secuestro posterior de glóbulos rojos parasitados en este órgano.
En resumen, durante las infecciones por P. falciparum, la génesis de la lesión en distintos órganos se da a partir de los procesos generados por la obstrucción vascular y procesos inflamatorios asociados al ciclo eritrocityario del parásito, entre los cuales han sido bien caracterizados los mecanismos relacionados con hipoxia, hemólisis y daño endotelial, con los efectos resultantes de acidosis y aumento de la permeabilidad capilar, y que explican complicaciones como la malaria cerebral, el edema pulmonar y la anemia. Procesos menos caracterizados se relacionan con el daño hepático y la lesión pulmonar durante la fase preeritrocitaria. Cada vez son más frecuentes los informes de casos graves debidos a P. vivax, motivo para intensificar el estudio de su patogenia.
Los autores declaramos ausencia de conflicto de intereses en la publicación de este manuscrito.
Este documento es producto de un proyecto de investigación realizado por Colciencias y la Universidad de Antioquia (CT-489-2009; RC-111549326146).
Correspondencia: Ana María Vásquez, Sede Investigación Universitaria, Universidad de Antioquia, Calle 62 N° 52-59, laboratorio 610, Medellín, Colombia Telefax: (574) 219 6487 amvc.ana@gmail.com
1. Newton CR, Krishna S. Severe falciparum malaria in children: Current understanding of pathophysiology and supportive treatment. Pharmacol Ther. 1998;79:1-53. [ Links ]
2. Baird JK. Neglect of Plasmodium vivax malaria. Trends Parasitol. 2007;23:533-9. [ Links ]
3. Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673-9. [ Links ]
4. Riganti M, Pongponratn E, Tegoshi T, Looareesuwan S, Punpoowong B, Aikawa M. Human cerebral malaria in Thailand: A clinico-pathological correlation. Immunol Lett. 1990;25:199-205. [ Links ]
5. Haldar K, Murphy SC, Milner DA, Taylor TE. Malaria: Mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol. 2007;2:217-49. [ Links ]
6. Sherman IW, Eda S, Winograd E. Cytoadherence and sequestration in Plasmodium falciparum: Defining the ties that bind. Microbes Infect. 2003;5:897-909. [ Links ]
7. Urban BC, Roberts DJ. Malaria, monocytes, macrophages and myeloid dendritic cells: Sticking of infected erythrocytes switches off host cells. Curr Opin Immunol. 2002;14:458-65. [ Links ]
8. Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev Mol Med. 2009;11:e16. [ Links ]
9. Heddini A, Pettersson F, Kai O, Shafi J, Obiero J, Chen Q, et al. Fresh isolates from children with severe Plasmodium falciparum malaria bind to multiple receptors. Infect Immun. 2001;69:5849-56. [ Links ]
10. Afonso Nogueira P, Wunderlich G, Shugiro Tada M, d´Arc Neves Costa J, Jose Menezes M, Scherf A, et al. Plasmodium falciparum: Analysis of transcribed var gene sequences in natural isolates from the Brazilian Amazon region. Exp Parasitol. 2002;101:111-20. [ Links ]
11. Newbold C, Warn P, Black G, Berendt A, Craig A, Snow B, et al. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. Am J Trop Med Hyg. 1997;57:389-98. [ Links ]
12. Rogerson SJ, Tembenu R, Dobano C, Plitt S, Taylor TE, Molyneux ME. Cytoadherence characteristics of Plasmodium falciparum-infected erythrocytes from Malawian children with severe and uncomplicated malaria. Am J Trop Med Hyg. 1999;61:467-72. [ Links ]
13. Traore B, Muanza K, Looareesuwan S, Supavej S, Khusmith S, Danis M, et al. Cytoadherence characteristics of Plasmodium falciparum isolates in Thailand using an in vitro human lung endothelial cells model. Am J Trop Med Hyg. 2000;62:38-44. [ Links ]
14. Udomsangpetch R, Reinhardt PH, Schollaardt T, Elliott JF, Kubes P, Ho M. Promiscuity of clinical Plasmodium falciparum isolates for multiple adhesion molecules under flow conditions. J Immunol. 1997;158:4358-64. [ Links ]
15. Al-Yaman F, Genton B, Mokela D, Raiko A, Kati S, Rogerson S, et al. Human cerebral malaria: Lack of significant association between erythrocyte rosetting and disease severity. Trans R Soc Trop Med Hyg. 1995;89:55-8. [ Links ]
16. Doumbo OK, Thera MA, Kone AK, Raza A, Tempest LJ, Lyke KE, et al. High levels of Plasmodium falciparum rosetting in all clinical forms of severe malaria in African children. Am J Trop Med Hyg. 2009;81:987-93. [ Links ]
17. Kun JF, Schmidt-Ott RJ, Lehman LG, Lell B, Luckner D, Greve B, et al. Merozoite surface antigen 1 and 2 genotypes and rosetting of Plasmodium falciparum in severe and mild malaria in Lambarene, Gabon. Trans R Soc Trop Med Hyg. 1998;92:110-4. [ Links ]
18. Normark J, Nilsson D, Ribacke U, Winter G, Moll K, Wheelock CE, et al. PfEMP1-DBL1alpha amino acid motifs in severe disease states of Plasmodium falciparum malaria. Proc Natl Acad Sci USA. 2007;104:15835-40. [ Links ]
19. Rowe A, Obeiro J, Newbold CI, Marsh K. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect Immun. 1995;63:2323-6. [ Links ]
20. Treutiger CJ, Hedlund I, Helmby H, Carlson J, Jepson A, Twumasi P, et al. Rosette formation in Plasmodium falciparum isolates and anti-rosette activity of sera from Gambians with cerebral or uncomplicated malaria. Am J Trop Med Hyg. 1992;46:503-10. [ Links ]
21. Pain A, Ferguson DJ, Kai O, Urban BC, Lowe B, Marsh K, et al. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc Natl Acad Sci USA. 2001;98:1805-10. [ Links ]
22. Clark IA, Cowden WB. The pathophysiology of falciparum malaria. Pharmacol Ther. 2003;99:221-60. [ Links ]
23. Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262-70. [ Links ]
24. Kyes SA, Kraemer SM, Smith JD. Antigenic variation in Plasmodium falciparum: Gene organization and regulation of the var multigene family. Eukaryot Cell. 2007;6:1511-20. [ Links ]
25. Roberts DJ, Craig AG, Berendt AR, Pinches R, Nash G, Marsh K, et al. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357:689-92. [ Links ]
26. Falk N, Kaestli M, Qi W, Ott M, Baea K, Cortes A, et al. Analysis of Plasmodium falciparum var genes expressed in children from Papua New Guinea. J Infect Dis. 2009;200:347-56. [ Links ]
27. Kyriacou HM, Stone GN, Challis RJ, Raza A, Lyke KE, Thera MA, et al. Differential var gene transcription in Plasmodium falciparum isolates from patients with cerebral malaria compared to hyperparasitaemia. Mol Biochem Parasitol. 2006;150:211-8. [ Links ]
28. Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen AT, Muller D, et al. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun. 2006;74:3904-11. [ Links ]
29. Kaestli M, Cockburn IA, Cortes A, Baea K, Rowe JA, Beck HP. Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. J Infect Dis. 2006;193:1567-74. [ Links ]
30. Kirchgatter K, Portillo Hdel A. Association of severe noncerebral Plasmodium falciparum malaria in Brazil with expressed PfEMP1 DBL1 alpha sequences lacking cysteine residues. Mol Med. 2002;8:16-23. [ Links ]
31. Bull PC, Kortok M, Kai O, Ndungu F, Ross A, Lowe BS, et al. Plasmodium falciparum-infected erythrocytes: Agglutination by diverse Kenyan plasma is associated with severe disease and young host age. J Infect Dis. 2000;182:252-9. [ Links ]
32. García F, Cebrian M, Dgedge M, Casademont J, Bedini JL, Neves O, et al. Endothelial cell activation in muscle biopsy samples is related to clinical severity in human cerebral malaria. J Infect Dis. 1999;179:475-83. [ Links ]
33. Turner GD, Ly VC, Nguyen TH, Tran TH, Nguyen HP, Bethell D, et al. Systemic endothelial activation occurs in both mild and severe malaria. Correlating dermal microvascular endothelial cell phenotype and soluble cell adhesion molecules with disease severity. Am J Pathol. 1998;152:1477-87. [ Links ]
34. Combes V, Taylor TE, Juhan-Vague I, Mege JL, Mwenechanya J, Tembo M, et al. Circulating endothelial microparticles in Malawian children with severe falciparum malaria complicated with coma. JAMA. 2004;291:2542-4. [ Links ]
35. Conroy AL, Phiri H, Hawkes M, Glover S, Mallewa M, Seydel KB, et al. Endothelium-based biomarkers are associated with cerebral malaria in Malawian children: A retrospective case-control study. PloS One. 2010;5:e15291. [ Links ]
36. Dietmann A, Lackner P, Helbok R, Spora K, Issifou S, Lell B, et al. Opposed circulating plasma levels of endothelin-1 and C-type natriuretic peptide in children with Plasmodium falciparum malaria. Malar J. 2008;7:253. [ Links ]
37. Hollestelle MJ, Donkor C, Mantey EA, Chakravorty SJ, Craig A, Akoto AO, et al. von Willebrand factor propeptide in malaria: Evidence of acute endothelial cell activation. Br J Haematol. 2006;133:562-9. [ Links ]
38. Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, Piera K, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci USA. 2008;105:17097-102. [ Links ]
39. Viebig NK, Wulbrand U, Forster R, Andrews KT, Lanzer M, Knolle PA. Direct activation of human endothelial cells by Plasmodium falciparum-infected erythrocytes. Infect Immun. 2005;73:3271-7. [ Links ]
40. Taoufiq Z, Gay F, Balvanyos J, Ciceron L, Tefit M, Lechat P, et al. Rho kinase inhibition in severe malaria: Thwarting parasite-induced collateral damage to endothelia. J Infect Dis. 2008;197:1062-73. [ Links ]
41. Susomboon P, Maneerat Y, Dekumyoy P, Kalambaheti T, Iwagami M, Komaki-Yasuda K, et al. Down-regulation of tight junction mRNAs in human endothelial cells co-cultured with Plasmodium falciparum-infected erythrocytes. Parasitol Int. 2006;55:107-12. [ Links ]
42. Pino P, Vouldoukis I, Kolb JP, Mahmoudi N, Desportes-Livage I, Bricaire F, et al. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003;187:1283-90. [ Links ]
43. Toure FS, Ouwe-Missi-Oukem-Boyer O, Bisvigou U, Moussa O, Rogier C, Pino P, et al. Apoptosis: A potential triggering mechanism of neurological manifestation in Plasmodium falciparum malaria. Parasite Immunol. 2008;30:47-51. [ Links ]
44. Jain V, Armah HB, Tongren JE, Ned RM, Wilson NO, Crawford S, et al. Plasma IP-10, apoptotic and angiogenic factors associated with fatal cerebral malaria in India. Malar J. 2008;7:83. [ Links ]
45. Kern P, Dietrich M, Hemmer C, Wellinghausen N. Increased levels of soluble Fas ligand in serum in Plasmodium falciparum malaria. Infect Immun. 2000;68:3061-3. [ Links ]
46. Pino P, Vouldoukis I, Dugas N, Hassani-Loppion G, Dugas B, Mazier D. Redox-dependent apoptosis in human endothelial cells after adhesion of Plasmodium falciparum-infected erythrocytes. Ann N Y Acad Sci. 2003;1010:582-6. [ Links ]
47. Clough B, Atilola FA, Pasvoi G. The role of rosetting in the multiplication of Plasmodium falciparum: Rosette formation neither enhances nor targets parasite invasion into uninfected red cells. Br J Haematol. 1998;100:99-104. [ Links ]
48. Deans AM, Rowe JA. Plasmodium falciparum: Rosettes do not protect merozoites from invasion-inhibitory antibodies. Exp Parasitol. 2006;112:269-73. [ Links ]
49. Cockburn IA, Mackinnon MJ, O´Donnell A, Allen SJ, Moulds JM, Baisor M, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc Natl Acad Sci USA. 2004;101:272-7. [ Links ]
50. Biswas AK, Hafiz A, Banerjee B, Kim KS, Datta K, Chitnis CE. Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathog. 2007;3:1271-80. [ Links ]
51. Das BS, Nanda NK. Evidence for erythrocyte lipid peroxidation in acute falciparum malaria. Trans R Soc Trop Med Hyg. 1999;93:58-62. [ Links ]
52. Greve B, Lehman LG, Lell B, Luckner D, Schmidt-Ott R, Kremsner PG. High oxygen radical production is associated with fast parasite clearance in children with Plasmodium falciparum malaria. J Infect Dis. 1999;179:1584-6. [ Links ]
53. Schwarzer E, Kuhn H, Valente E, Arese P. Malaria-parasitized erythrocytes and hemozoin nonenzymatically generate large amounts of hydroxy fatty acids that inhibit monocyte functions. Blood. 2003;101:722-8. [ Links ]
54. Pabón A, Carmona J, Burgos LC, Blair S. Oxidative stress in patients with non-complicated malaria. Clin Biochem. 2003;36:71-8. [ Links ]
55. Das BS, Patnaik JK, Mohanty S, Mishra SK, Mohanty D, Satpathy SK, et al. Plasma antioxidants and lipid peroxidation products in falciparum malaria. Am J Trop Med Hyg. 1993;49:720-5. [ Links ]
56. Griffiths MJ, Ndungu F, Baird KL, Muller DP, Marsh K, Newton CR. Oxidative stress and erythrocyte damage in Kenyan children with severe Plasmodium falciparum malaria. Br J Haematol. 2001;113:486-91. [ Links ]
57. Nanda NK, Das BS. Presence of pro-oxidants in plasma of patients suffering from falciparum malaria. Trans R Soc Trop Med Hyg. 2000;94:684-8. [ Links ]
58. Gyan B, Troyeblomberg M, Perlmann P, Bjorkman A. Human monocytes cultured with and without interferon-gamma inhibit Plasmodium falciparum parasite growth in vitro via secretion of reactive nitrogen intermediates. Parasitol Immunol. 1994;16:371-75. [ Links ]
59. Mellouk S, Hoffman SL, Liu ZZ, de la Vega P, Billiar TR, Nussler AK. Nitric oxide-mediated antiplasmodial activity in human and murine hepatocytes induced by gamma interferon and the parasite itself: Enhancement by exogenous tetrahydrobiopterin. Infect Immun. 1994;62:4043-6. [ Links ]
60. Skorokhod OA, Schwarzer E, Ceretto M, Arese P. Malarial pigment haemozoin, IFN-gamma, TNF-alpha, IL-1beta and LPS do not stimulate expression of inducible nitric oxide synthase and production of nitric oxide in immuno-purified human monocytes. Malar J. 2007;6:73. [ Links ]
61. Al Yaman FM, Mokela D, Genton B, Rockett KA, Alpers MP, Clark IA. Association between serum levels of reactive nitrogen intermediates and coma in children with cerebral malaria in Papua New Guinea. Trans R Soc Trop Med Hyg 1996;90:270-3. [ Links ]
62. Maneerat Y, Viriyavejakul P, Punpoowong B, Jones M, Wilairatana P, Pongponratn E, et al. Inducible nitric oxide synthase expression is increased in the brain in fatal cerebral malaria. Histopathology. 2000;37:269-77. [ Links ]
63. Gyan B, Kurtzhals JA, Akanmori BD, Ofori M, Goka BQ, Hviid L, et al. Elevated levels of nitric oxide and low levels of haptoglobin are associated with severe malarial anaemia in African children. Acta Trop. 2002;83:133-40. [ Links ]
64. Keller CC, Kremsner PG, Hittner JB, Misukonis MA, Weinberg JB, Perkins DJ. Elevated nitric oxide production in children with malarial anemia: hemozoin-induced nitric oxide synthase type 2 transcripts and nitric oxide in blood mononuclear cells. Infect Immun. 2004;72:4868-73. [ Links ]
65. Agbenyega T, Angus B, Bedu-Addo G, Baffoe-Bonnie B, Griffin G, Vallance P, et al. Plasma nitrogen oxides and blood lactate concentrations in Ghanaian children with malaria. Trans R Soc Trop Med Hyg. 1997;91:298-302. [ Links ]
66. Dondorp AM, Planche T, de Bel EE, Angus BJ, Chotivanich KT, Silamut K, et al. Nitric oxides in plasma, urine, and cerebrospinal fluid in patients with severe falciparum malaria. Am J Trop Med Hyg. 1998;59:497-502. [ Links ]
67. Taylor AM, Day NP, Sinh DX, Loc PP, Mai TT, Chau TT, et al. Reactive nitrogen intermediates and outcome in severe adult malaria. Trans R Soc Trop Med Hyg. 1998;92:170-5. [ Links ]
68. Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA, et al. Nitric oxide in Tanzanian children with malaria: Inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J Exp Med. 1996;184:557-67. [ Links ]
69. Chiwakata CB, Hemmer CJ, Dietrich M. High levels of inducible nitric oxide synthase mRNA are associated with increased monocyte counts in blood and have a beneficial role in Plasmodium falciparum malaria. Infect Immun. 2000;68:394-9. [ Links ]
70. Lopansri BK, Anstey NM, Weinberg JB, Stoddard GJ, Hobbs MR, Levesque MC, et al. Low plasma arginine concentrations in children with cerebral malaria and decreased nitric oxide production. Lancet. 2003;361:676-8. [ Links ]
71. Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, McNeil YR, et al. Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693-704. [ Links ]
72. Coban C, Ishii KJ, Horii T, Akira S. Manipulation of host innate immune responses by the malaria parasite. Trends Microbiol. 2007;15:271-8. [ Links ]
73. Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60:1623-35. [ Links ]
74. Odeh M. The role of tumour necrosis factor-alpha in the pathogenesis of complicated falciparum malaria. Cytokine. 2001;14:11-8. [ Links ]
75. Kern P, Hemmer CJ, van Damme J, Gruss HJ, Dietrich M. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med. 1989 87:139-43. [ Links ]
76. Kwiatkowski D, Hill AV, Sambou I, Twumasi P, Castracane J, Manogue KR, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336:1201-4. [ Links ]
77. Nyakundi JN, Warn P, Newton C, Mumo J, Jephthah-Ochola J. Serum tumour necrosis factor in children suffering from Plasmodium falciparum infection in Kilifi District, Kenya. Trans R Soc Trop Med Hyg. 1994;88:667-70. [ Links ]
78. John CC, Opika-Opoka R, Byarugaba J, Idro R, Boivin MJ. Low levels of RANTES are associated with mortality in children with cerebral malaria. J Infect Dis. 2006;194:837-45. [ Links ]
79. D´Ombrain MC, Robinson LJ, Stanisic DI, Taraika J, Bernard N, Michon P, et al. Association of early interferon-gamma production with immunity to clinical malaria: A longitudinal study among Papua New Guinean children. Clin Infect Dis. 2008;47:1380-7. [ Links ]
80. Ho M, Schollaardt T, Snape S, Looareesuwan S, Suntharasamai P, White NJ. Endogenous interleukin-10 modulates proinflammatory response in Plasmodium falciparum malaria. J Infect Dis. 1998;178:520-5. [ Links ]
81. Othoro C, Lal AA, Nahlen B, Koech D, Orago AS, Udhayakumar V. A low interleukin-10 tumor necrosis factor-alpha ratio is associated with malaria anemia in children residing in a holoendemic malaria region in western Kenya. J Infect Dis. 1999;179:279-82. [ Links ]
82. Chaiyaroj SC, Rutta AS, Muenthaisong K, Watkins P, Na Ubol M, Looareesuwan S. Reduced levels of transforming growth factor-beta1, interleukin-12 and increased migration inhibitory factor are associated with severe malaria. Acta Trop. 2004;89:319-27. [ Links ]
83. Ochiel DO, Awandare GA, Keller CC, Hittner JB, Kremsner PG, Weinberg JB, et al. Differential regulation of beta-chemokines in children with Plasmodium falciparum malaria. Infect Immun. 2005;73:4190-7. [ Links ]
84. Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20:597-603. [ Links ]
85. Maguire GP, Handojo T, Pain MC, Kenangalem E, Price RN, Tjitra E, et al. Lung injury in uncomplicated and severe falciparum malaria: A longitudinal study in Papua, Indonesia. J Infect Dis. 2005;192:1966-74. [ Links ]
86. Giraldo C, Blair S, Tobón A. Complicaciones pulmonares en malaria. Infectio. 2004;8:279-92. [ Links ]
87. Martínez O. Sindrome de dificultad respiratoria aguda en malaria por P. vivax. Acta Médica Colombiana. 1996;21:146-50. [ Links ]
88. Lang F, Lang PA, Lang KS, Brand V, Tanneur V, Duranton C, et al. Channel-induced apoptosis of infected host cells-the case of malaria. Pflugers Arch. 2004;448:319-24. [ Links ]
89. Angus BJ, Chotivanich K, Udomsangpetch R, White NJ. In vivo removal of malaria parasites from red blood cells without their destruction in acute falciparum malaria. Blood. 1997; 90:2037-40. [ Links ]
90. Dondorp AM, Angus BJ, Chotivanich K, Silamut K, Ruangveerayuth R, Hardeman MR, et al. Red blood cell deformability as a predictor of anemia in severe falciparum malaria. Am J Trop Med Hyg. 1999;60:733-7. [ Links ]
91. Groux H, Gysin J. Opsonization as an effector mechanism in human protection against asexual blood stages of Plasmodium falciparum: Functional role of IgG subclasses. Res Immunol. 1990;141:529-42. [ Links ]
92. Waitumbi JN, Opollo MO, Muga RO, Misore AO, Stoute JA. Red cell surface changes and erythrophagocytosis in children with severe Plasmodium falciparum anemia. Blood. 2000;95:1481-6. [ Links ]
93. Ekvall H. Malaria and anemia. Curr Opin Hematol. 2003;10:108-14. [ Links ]
94. Wickramasinghe SN, Abdalla SH. Blood and bone marrow changes in malaria. Baillieres Best Pract Res Clin Haematol. 2000;13:277-99. [ Links ]
95. Stoute JA, Odindo AO, Owuor BO, Mibei EK, Opollo MO, Waitumbi JN. Loss of red blood cell-complement regulatory proteins and increased levels of circulating immune complexes are associated with severe malarial anemia. J Infect Dis. 2003;187:522-5. [ Links ]
96. Weatherall DJ, Miller LH, Baruch DI, Marsh K, Doumbo OK, Casals-Pascual C, et al. Malaria and the red cell. Hematology Am Soc Hematol Educ Program. 2002:35-57. [ Links ]
97. Abdalla SH. Hematopoiesis in human malaria. Blood Cells. 1990;16:401-16. [ Links ]
98. Bachmann A, Esser C, Petter M, Predehl S, von Kalckreuth V, Schmiedel S, et al. Absence of erythrocyte sequestration and lack of multicopy gene family expression in Plasmodium falciparum from a splenectomized malaria patient. PloS One. 2009;4:e7459. [ Links ]
99. Munasinghe A, Ileperuma M, Premawansa G, Handunnetti S, Premawansa S. Spleen modulation of cytoadherence properties of Plasmodium falciparum. Scand J Infect Dis. 2009;41:538-9. [ Links ]
100. Safeukui I, Correas JM, Brousse V, Hirt D, Deplaine G, Mule S, et al. Retention of Plasmodium falciparum ring-infected erythrocytes in the slow, open microcirculation of the human spleen. Blood. 2008;112:2520-8. [ Links ]
101. Ejigiri I, Sinnis P. Plasmodium sporozoite-host interactions from the dermis to the hepatocyte. Curr Opin Microbiol. 2009;12:401-7. [ Links ]
102. Sturm A, Heussler V. Live and let die: Manipulation of host hepatocytes by exoerythrocytic Plasmodium parasites. Med Microbiol Immunol. 2007;196:127-33. [ Links ]
103. Guha M, Kumar S, Choubey V, Maity P, Bandyopadhyay U. Apoptosis in liver during malaria: Role of oxidative stress and implication of mitochondrial pathway. Faseb J. 2006;20:1224-6. [ Links ]
104. Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287-90. [ Links ]
105. Baer K, Klotz C, Kappe SH, Schnieder T, Frevert U. Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog. 2007;3:e171. [ Links ]

