










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.33 no.2 Bogotá abr./jun. 2013
https://doi.org/10.7705/biomedica.v33i2.830
doi: http://dx.doi.org/10.7705/biomedica.v33i2.830
ARTÍCULO ORIGINAL
1 Área de Neurobiología Celular y Molecular, Grupo de Neurociencias de Antioquia, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
2 Departamento de Salud Animal, Grupo de Investigación en Enfermedades Neurodegenerativas, Facultad de Medicina Veterinaria, Universidad del Tolima, Ibagué, Colombia
3 Fundación Valle del Lili, Cali, Colombia
4 Grupo de Investigación Biomédica, Universidad ICESI, Cali, Colombia
Contribución de los autores:
Ángel Enrique Céspedes: realización de los experimentos, obtención y discusión de los resultados, elaboración del manuscrito.
Gloria Patricia Cardona: aprobación del proyecto y supervisión del estudio; análisis e interpretación de resultados, discusión y conclusiones; revisión y corrección del manuscrito.
César Augusto Arango: capacitación en modelo de isquemia focal, revisión y aporte en el análisis de los resultados, la discusión y redacción del manuscrito.
Recibido: 31/08/12; aceptado:25/02/13
Introducción. Los indicadores espacio-temporales de lesión son esenciales en el estudio neuropatológico y terapéutico de la isquemia cerebral.
Objetivo. Optimizar la técnica de dos modelos de isquemia cerebral (focal y global) y hacer un análisis comparativo de la progresión del daño cerebral, mediante marcadores de neurodegeneración.
Materiales y métodos. Se sometieron ratas Wistar a oclusión temporal de la arteria cerebral media o a oclusión de cuatro vasos, y se evaluaron comparativamente el tiempo quirúrgico, la tasa de supervivencia y la recuperación neurológica. Se utilizó trifenilo de tetrazolio para establecer la distribución del infarto y tinción con Fluoro - Jade B ® como marcador de neurodegeneración. La inmunorreacción de la astroglía se evaluó con el anticuerpo contra la proteína acídica fibrilar de la glía ( Glial Fibrillary Acidic Protein, GFAP) y el anticuerpo AT-8 contra la proteína tau hiperfosforilada, 24, 48 y 72 horas después de la isquemia.
Resultados. Los modelos de isquemia utilizados requirieron menor tiempo quirúrgico y hubo menor riesgo de muerte, respecto a estudios previos. En el modelo focal, las células positivas con Fluoro - Jade B ® y los astrocitos reactivos, se evidenciaron en corteza e hipocampo a las 24 horas después de la isquemia. En el modelo global, se observó tinción Fluoro - Jade B ® positiva a las 24 horas, aumentando significativamente la reacción de la GFAP a las 72 horas en corteza y a las 48 horas en el hipocampo. La reacción contra la proteína tau hiperfosforilada aumentó progresivamente y fue máxima a las 72 horas en ambos modelos.
Conclusiones. Los dos modelos de isquemia cerebral, oclusión temporal de la arteria cerebral media y oclusión de cuatro vasos, fueron optimizados. En estos modelos, los marcadores la tinción Fluoro - Jade B ® y la GFAP permitieron detectar procesos de neurodegeneración 24 horas después de la isquemia, en tanto el marcador de proteína tau hiperfosforilada (AT-8) incrementó progresivamente su reacción hasta las 72 horas, lo cual sugiere la propagación de la excitotoxicidad y la alteración de enzimas implicadas en la fosforilación de proteínas del citoesqueleto.
Palabras clave: accidente cerebrovascular, isquemia, degeneración nerviosa, proteínas tau, ratas Wistar.
doi: http://dx.doi.org/10.7705/biomedica.v33i2.830
Injury markers in two models of cerebral ischemia
Introduction: Spatio-temporal indicators of injury are essential for the study of neuropathological processes and for developing therapeutic approaches for stroke.
Objective: This study sought to optimize the techniques of two cerebral ischemia models (focal and global) and to comparatively evaluate the progression of brain damage by analyzing markers of neurodegeneration.
Materials and methods: Wistar rats were subjected to temporary occlusion of the middle cerebral artery (t-MCAO) or four-vessel occlusion (4-VO), and surgical time, survival rate and neurological recovery were comparatively evaluated. Triphenyl tetrazolium was used to determine the distribution of the infarction, and Fluoro-Jade B was used as a marker of neurodegeneration. Astroglial immunoreactivity was assessed with an anti-glial fibrillary acidic protein (GFAP) antibody, and an anti-AT-8 antibody was used to detect hyperphosphorylated tau protein at 24, 48 and 72 hours post-ischemia.
Results: The cerebral ischemia models employed (t-MCAO and 4-VO) required less surgical time and presented less of a death risk compared to those in previous studies. In the focal model, Fluoro-Jadepositive cells and reactive astrocytes were observed in the cerebral cortex and the hippocampus at 24 hours post-ischemia. In the global model, we observed Fluoro-Jade-positive cells at 24 hours, and a significant increase in the reactivity of GFAP was observed at 72 hours in the cortex and at 48 hours in the hippocampus. The immunoreactivity of hyperphosphorylated tau protein increased progressively, reaching a maximum at 72 hours post-ischemia in both models.
Conclusions: These results suggest that in the t-MCAO and 4-VO ischemia models, the expression of Fluoro-Jade and GFAP indicates early neurodegeneration at 24 hours post-insult. In contrast, the immunoreactivity of the hyperphosphorylated tau protein marker (AT-8) progressively increases until 72 hours post-insult, which suggests that the progression of excitotoxicity and alteration of enzymes involves the phosphorylation of cytoskeletal proteins.
Key words: Stroke, ischemia, nerve degeneration, tau proteins, rats, Wistar.
doi: http://dx.doi.org/10.7705/biomedica.v33i2.830
El accidente cerebrovascular es la primera causa de incapacidad permanente y la tercera de muerte en América Latina (1,2), y ocupó el segundo lugar entre las causas de muerte en el mundo en el 2010 (3). De 70 a 85 % de los accidentes cerebrovasculares son causados por la oclusión de la arteria cerebral media (4,5), lo cual puede ocurrir por la oclusión de pequeños vasos o terminales capilares, daño endotelial y oclusión tromboembólica de una arteria cerebral principal (6,7).
Se han desarrollado diversos modelos experimentales de isquemia cerebral, principalmente en ratas y ratones, como una herramienta indispensable de investigación en el campo del accidente cerebrovascular (7,8), teniendo estos modelos ventajas y desventajas (8). En los últimos años se han desarrollado métodos menos invasivos, seguros y reproducibles, respondiendo a la necesidad de refinar técnicas quirúrgicas enfocadas específicamente a minimizar el riesgo de muerte o las posibles complicaciones que puedan afectar el curso fisiopatológico de la lesión (9,10); sin embargo, la selección de la mayoría de los modelos depende en cada caso de los objetivos particulares del investigador.
La eficacia y la seguridad de un modelo de infarto cerebral, son fundamentales para la comprensión básica de los mecanismos celulares que siguen a la lesión (2,6,9). Muchos modelos de isquemia cerebral van dirigidos a incrementar el conocimiento sobre los mecanismos fisiopatológicos involucrados en el proceso (7,11) o a responder a diversos interrogantes sobre la enfermedad en humanos (8,12,13). Sin embargo, con frecuencia los resultados son variables en este tipo de estudios (6,14), relacionándose la manipulación quirúrgica con el volumen del infarto obtenido. Al momento de seleccionar los dos modelos utilizados en este estudio, se revisaron diferentes modelos de isquemia cerebral, teniendo en cuenta las ventajas relativas de cada uno de ellos.
El modelo de oclusión de los cuatro vasos ( Four-Vessel Occlusion , 4-VO) es uno de los más utilizados para producir lesiones isquémicas globales e infartos hemisféricos en ratas (12,14,15). Consiste en la oclusión temporal de las arterias carótidas, después de cauterizar las arterias vertebrales.
Por otra parte, el modelo más usado para generar isquemia focal transitoria es el de la oclusión temporal de la arteria cerebral media ( Temporal-Middle Cerebral Artery Occlusion , t-MCAO) mediante endarterización de la arteria carótida interna hasta la bifurcación de la arteria cerebral anterior y la cerebral media (13,16,17). Este modelo produce grados variables de isquemia que dependen del tiempo de oclusión y de la permanencia de la oclusión (9); además, se han observado variaciones en el déficit neurológico dependiente del grado de lesión y de la oclusión correcta de la arteria cerebral media (10).
En la mayoría de los modelos de isquemia se replica parcialmente la fisiopatología del accidente cerebrovascular, particularmente en los modelos de isquemia cerebral focal; sin embargo, los modelos de isquemia global también se han utilizado en estudios de aprendizaje y memoria (18). Aún está por esclarecerse si estos modelos pueden servir para identificar marcadores de lesión temprana o de progresión de la enfermedad isquémica.
Se sabe que los ovillos neurofibrilares ( Neurofibrillary Tangles , NFT) son marcadores de enfermedades neurodegenerativas crónicas, como la enfermedad de Alzheimer, la esclerosis lateral amiotrófica y la enfermedad de Pick, entre otras; además, la hiperfosforilación de la proteína tau está relacionada con la demencia después de un accidente cerebrovascular (19). Por otra parte, la expresión de la proteína precursora del ß -amiloide se ha visto incrementada en roedores, tras inducir la isquemia cerebral (20). La proteína tau hiperfosforilada se acumula de manera específica en determinados sitios, en neuronas corticales de humanos con taupatías (19), y puede evidenciarse mediante los filamentos helicoidales pareados ( Paired Helical Filaments , PHF). Bajo condiciones de isquemia, hay falla energética en forma de ATP y desensamblaje del citoesqueleto neuronal, por disociación de la proteína tau de los microtúbulos, como consecuencia de un aumento de la actividad de las enzimas cinasas dirigidas por la prolina ( Proline-Directed Protein Kinases , PDPK) o fosfatasas de serina o treonina (Ser/Thr 2A, 2B), evitando el transporte axonal (21). Sin embargo, la relación directa entre la proteína tau hiperfosforilada y la progresión de la isquemia cerebral aún no está completamente dilucidada.
Se conocen otros marcadores de estrés y muerte celular, como la tinción con Fluoro - Jade B ®, el cual es un derivado de fluoresceína que tiene una gran afinidad por los procesos neuronales acidofílicos, por lo cual la presencia de fluorescencia indica eventos degenerativos intracelulares (22). En la isquemia cerebral, también se han descrito alteraciones morfológicas y bioquímicas en los astrocitos, como respuesta a los cambios fisiopatológicos que se suscitan en etapas tempranas y tardías después del restablecimiento de la perfusión. La reacción astrocitaria que se expresa en forma de hipertrofia, juega un papel importante en la plasticidad neural y en el mantenimiento de la homeostasis, observándose un incremento en la inmunorreacción de GFAP en la isquemia cerebral (22,23).
Con base en lo anterior, se optimizó la técnica en dos modelos distintos de isquemia cerebral (focal y global: t-MCAO y 4-VO) en ratas y se hizo un análisis comparativo de la progresión del daño cerebral a las 24, 48 y 72 horas después de la lesión, utilizando marcadores de neurodegeneración (GFAP, Fluoro - Jade B ® y proteína tau). Los resultados obtenidos tienen utilidad en estudios de neuroprotección, al elegir la ventana temporal de acción, según el modelo y el marcador de degeneración utilizados; también, facilitan la identificación de blancos celulares y su participación espacio-temporal.
Materiales y métodos
Se utilizaron como sujetos experimentales ratas Wistar hembra de cuatro meses de edad y 250 g de peso promedio, obtenidas del bioterio SPF de la Sede de Investigación Universitaria de la Universidad de Antioquia (Medellín, Colombia). Los animales se mantuvieron en ciclos de 12 horas de luz y 12 de oscuridad (luz, 6:00 a.m. a 6:00 p.m.), con alimento y agua ad libitum , siguiendo los lineamientos de la Comunidad Europea (86/609/EEC) y lo normado por las leyes colombianas para tal fin (Ley 84/1989, Resolución 8430/1993 y Ley 576/2000). Se tuvo especial cuidado en evitar el sufrimiento innecesario y se redujo al máximo el número de animales experimentales. Esta investigación contó con la aprobación del Comité de Ética de Animales de Experimentación de la Universidad de Antioquia.
Todos los animales se sometieron a ooferectomía bilateral dos semanas antes de las intervenciones quirúrgicas, para evitar las fluctuaciones hormonales y los efectos de neuroprotección potencialmente atribuibles a la actividad de las hormonas ováricas. La anestesia consistió en ketamina (90 mg/kg) y xilacina (5 mg/kg) por vía intraperitoneal. Se hizo un pequeño corte a través de la piel del dorso a nivel lumbar y se separaron los músculos abdominales de ambos lados; los ovarios se expusieron y resecaron mediante pinzamiento y torsión a nivel de su pedículo, lo cual favoreció la hemostasia.
Modelo de oclusión transitoria de la arteria cerebral media
Se utilizó la técnica de oclusión intravascular de la arteria cerebral media (t-MCAO) mediante endarterización carotídea con monofilamento de poliamida 4/0 durante por 60 minutos, con restablecimiento posterior de la perfusión (13,17). La anestesia se indujo con una mezcla de ketamina (90 mg/kg) y xilacina (5 mg/kg) intraoperitoneal, y se mantuvo con un equipo de anestesia por inhalación usando isoflurano (2-4 %) y oxígeno (96 % v/v) con mascarilla. La temperatura corporal se monitorizó con un termómetro rectal digital durante todo el procedimiento quirúrgico y las ratas se mantuvieron bajo condiciones quirúrgicas de hipotermia controlada sin la influencia del manto de calentamiento corporal.
La arteria carótida común se expuso mediante una incisión cutánea de 2 cm a lo largo de la línea media ventral del cuello, y se disecaron los músculos ventrales (esternohioideo, homohioideo, esternomastoideo) y el nervio vago. Se hizo divulsión de las arterias carótida externa y carótida interna derechas; las primeras ramas de la carótida externa (occipital, tiroidea superior, faríngea ascendente y maxilar externa) se cauterizaron por electrocoagulación (cauterio bipolar Aaron®) y se cortó la carótida externa después de ligarla con poliamida 6/0 de la porción distal craneal creando un muñón. La arteria pterigopalatina fue disecada, cuidadosamente pinzada y electrocoagulada.
La arteria carótida común y la carótida interna derechas se ocluyeron temporalmente mediante pinzamiento (pinzas de aneurisma # 2) y, a través de una microincisión en el muñón de la carótida externa, se introdujo un nailon monofilamento (poliamida 4/0).
Los filamentos utilizados en este experimento fueron previamente redondeados en su punta mediante flama y cubiertos con un doble baño de poli-L-lisina (al 0,1 %, Sigma), para incrementar la adhesión al endotelio y prevenir la reperfusión temprana. Se ligó suavemente alrededor del punto de endarterización con polipropileno 8/0 (Prolene®), para evitar hemorragias o la salida del filamento, el cual se avanzó por la arteria carótida interna hasta la bifurcación de la cerebral media y la cerebral anterior, interrumpiendo temporalmente la circulación sanguínea en los territorios cerebrales dependientes de la cerebral media. Se reacomodaron los tejidos y se suturó la incisión con nailon 4/0, dejando protruido 1 cm del extremo distal del nailon de endarterización, el cual fue cuidadosamente retraído y cortado luego de 60 minutos de oclusión, permitiendo el retorno del flujo sanguíneo al cerebro (reperfusión), pero evitando la extracción total del filamento.
Modelo de oclusión de los cuatro vasos
El modelo de oclusión de los cuatro vasos (4-VO) se basó en técnicas previamente descritas, con algunas modificaciones como se describe a continuación. Las ratas se anestesiaron con el mismo protocolo detallado para el modelo de t-MCAO. Se hizo una incisión sobre la piel del dorso del cuello a la altura del occipital y directamente sobre las dos primeras vértebras cervicales. La piel, el músculo cutáneo y los músculos paraespinales se disecaron y retrajeron lateralmente hasta exponer los agujeros alares del primer segmento cervical (atlas). Con un electrocauterio bipolar se cauterizaron las dos arterias vertebrales a través de los respectivos forámenes ovales (derecho e izquierdo), dado que las arterias vertebrales pasan bajo ellos antes de fusionarse en la fosa posterior del occipucio como arteria vértebro-basilar. Los tejidos se reacomodaron y la incisión se cerró en un solo plano con polipropileno 4/0. Las arterias carótidas comunes (derecha e izquierda) se expusieron a través de una incisión en la línea media ventral del cuello e inmediatamente se ocluyeron con pinzas de aneurisma durante 25 minutos, liberándolas posteriormente para permitir la reperfusión. La incisión se suturó con nailon 4/0. La respiración y la temperatura fueron monitorizadas durante todo el procedimiento y en el posoperatorio inmediato.
Recuperación y evaluación neurológica
Los animales fueron retornados a sus cajas luego del procedimiento, hasta su recuperación; posteriormente, se les hizo una valoración neurológica, usando un protocolo previamente descrito (17,24,25) y ligeramente modificado. El déficit neurológico se evaluó después de las primeras seis horas de recuperación de la anestesia y, posteriormente, a 24, 48 y 72 horas después de la isquemia y la reperfusión.
Para complementar la evaluación de la función motora, se hizo una modificación de los métodos previamente descritos para ese fin. La puntuación neurológica se basó en una escala de seis puntos: 0, ausencia de movimientos espontáneos; 1, movimientos espontáneos circulares hacia el lado afectado por la paresia; 2, movimientos en círculos hacia el lado afectado por la paresia, al tirar de la cola; 3, movimientos en círculos hacia el lado afectado por la paresia, al tirar e izar de la cola; 4, reducción de la resistencia a la presión lateral sobre el lado afectado por la paresia; 5, flexión constante del miembro anterior contralateral a la lesión, y 6, extensión normal de ambos miembros anteriores al ser izada de la cola y acercada a una superficie.
También se practicaron otras pruebas neurológicas que no están incluidas en la escala, como por ejemplo, el balanceo o pérdida del equilibrio sobre barra vertical y horizontal, la percepción espacial, y los reflejos de enderezamiento, motor ocular, óptico, palpebral y acústico (no se presentan los datos).
Determinación del volumen del infarto
Para establecer la magnitud de la lesión en ambos modelos (t-MCAO y 4-VO), se utilizó el método de tinción con el cloruro de tetrazolio ( Triphenyl Tetrazolium Chloride , TTC), descrito previamente en otros estudios (17,26-28). Las ratas se anestesiaron con una mezcla de ketamina/xilacina (100/15 mg/kg) a las 24 horas después de la isquemia y la reperfusión, y por punción cardiaca y avance aórtico, se perfundieron 200 ml de solución salina normal al 0,9 % mediante una bomba peristáltica.
Los cerebros se resecaron y almacenaron durante 10 minutos en solución tampón salina de fosfato ( Phosphate Buffered Saline, PBS ) y luego se cortaron en secciones coronales a intervalos de 2 mm, usando una matriz acrílica para rata (Stoelting Co., Ref #51382). Los cortes se incubaron en el colorante cloruro de tetrazolio al 0,5 % (Sigma, St. Louis, MO) durante 30 minutos a 37 °C, protegidos de la luz. Las secciones se fijaron con paraformaldehído en solución tampón al 4 % y luego se tomaron las fotografías digitales.
Los volúmenes de infarto cerebral se determinaron usando el software Image-J (versión 1.0), mediante integración de las áreas de infarto en cada segmento y por cada cerebro. El volumen del infarto se calculó usando la siguiente fórmula: volumen del infarto = [volumen del hemisferio izquierdo – (volumen del hemisferio derecho – volumen del infarto medido)] / volumen del hemisferio izquierdo. El volumen del infarto se corrigió en cada cerebro (29) para compensar el edema cerebral en el hemisferio isquémico, mediante la siguiente fórmula: volumen del infarto corregido = volumen del hemisferio izquierdo – (volumen del hemisferio derecho–volumen del infarto medido) . El volumen del infarto se expresó en mm3 y en porcentaje del hemisferio izquierdo.
Células en proceso neurodegenerativo
Se utilizó la tinción de Fluoro - Jade B ® para marcar las células en proceso de neurodegeneración. Las ratas se anestesiaron como se describió anteriormente y se les perfundieron 200 ml de solución salina 0,9 %N seguidos de 200 ml de PFA al 4 % en PBS 0,1M.
Los cerebros se extrajeron y posfijaron en paraformaldehído al 4 % durante 48 horas, y luego se lavaron en solución tampón 0,1M. Se cortaron secciones de 50 µm con un micrótomo Leica de hoja vibrante (VT 1000S-Leica Microsystems). Las secciones se incubaron inicialmente en una solución de NaOH al 1 % en etanol (80 %) durante cinco minutos y luego se hidrataron en alcohol con gradientes descendientes (75, 50 y 25 %) durante cinco minutos en cada uno, seguido de agua destilada.
Los cortes se incubaron en una solución de permanganato de potasio (KMnO4 al 0,6 %) durante 10 minutos, se lavaron con agua destilada e se incubaron en una solución de Fluoro - Jade B ® al 0,001 % (Chemicon International AG310) en ácido acético al 0,1 % durante 30 minutos.
Las secciones se montaron en láminas y se cubrieron con laminillas bajo un medio de montaje acuoso (Vecta-shield Mounting Medium). Todos los cortes se observados, analizaron y fotografiaron en un microscopio de fluorescencia (Olympus IX-81) a 450-490 nm.
Inmunohistoquímica
Los estudios inmunohistoquímicos se desarrollaron con cortes coronales de 50 µm, correspondientes a las coordenadas -2,30 a -3,60 de Bregma, usando el siguiente protocolo. Las secciones se colocaron en metanol: PBS (1:1) más H2O2 al 1 % durante 20 minutos para inhibir la peroxidasa endógena y luego se lavaron tres veces consecutivas con PBS 0,1 M (5 minutos cada uno). Las secciones cerebrales se pre-incubaron (PBS 0,1 M, Tritón 100X-3 % y BSA 1 %) durante una hora en agitación suave y a temperatura ambiente; posteriormente, los cortes se incubaron (PBS 0,1M, Tritón 100X-0,3 % y BSA 0,3 %) en el anticuerpo primario contra proteína tau hiperfosforilada (AT8 monoclonal mouse, 1:250, Pierce) y se dejaron en agitación constante a 4 °C toda la noche.
Luego de cinco lavados de 10 minutos cada uno, los cortes se incubaron en anticuerpo secundario (mouse biotinilado' 1:250) durante dos horas a temperatura ambiente y a continuación se lavaron con PBS (5 veces durante 5 minutos cada una). Se incubaron en el complejo avidina/biotina (ABC) 1:250 (de cada uno) durante dos horas a temperatura ambiente y en la oscuridad. Luego de cinco lavados consecutivos (10 minutos cada uno) con PBS, se revelaron con diaminobenzidina, tableta de 11 mg (Merck), en 15 ml de PBS 0,1M protegido de la luz. Se adicionó H2O2 al 0,02 % (30 % de la stock solution ), una vez filtrado. Las secciones se montaron en láminas portaobjeto, cubiertas con laminillas y selladas con medio de montaje (Permount Fisher).
Todos los cortes se fotografiaron con cámara digital bajo microscopio óptico, analizando al menos seis cortes por cerebro; las cuantificaciones se hicieron mediante densitometría (unidades relativas) como la relación de células marcadas, empleando el programa Image-J, versión 1.0.
Inmunofluorescencia
Las secciones almacenadas en criopreservador, se seleccionaron, se lavaron con PBS 0,1M y se incubaron con cloruro de amonio 50 mM durante 10 minutos para evitar la autofluorescencia. Los cortes se preincubaron en solución TXTBS ( Tritón X-Tween Buffer Saline ) con adición de BSA al 3 % en agitación constante durante 60 minutos y a temperatura ambiente; posteriormente, se incubaron en el anticuerpo primario (Anti-GFAP rabbit, 1:2.000) en solución de incubación TXTBS que contenía BSA al 1 %, durante toda la noche a 4 °C en agitación constante.
Las secciones se lavaron cuatro veces con PBS 0,1 M y se incubaron en el anticuerpo secundario acoplado al fluoróforo (Alexa-Fluor 594 Goat antirabbit IgG) 1:2.000, durante dos horas a temperatura ambiente y en agitación constante. Los cortes se lavaron cuatro veces con PBS 0,1 M, se montaron en láminas y se sellaron con laminillas, usando medio acuoso (Gel Mount).
Las láminas se analizaron bajo microscopio de fluorescencia (Olympus IX 81). Para los controles negativos, se omitió el anticuerpo primario durante el procedimiento. El análisis densitométrico fue similar al descrito en la inmunohistoquímica convencional (Image-J 1.0 software ).
Análisis estadístico
Al menos cinco ratas fueron evaluadas por cada grupo experimental, excepto para la evaluación de puntaje neurológico, en la cual el n (número de animales por grupo) fue de 15. Se aplicó estadística no paramétrica mediante la prueba de Kruskal-Wallis y las diferencias se analizaron mediante la prueba de Dunns para la comparación múltiple entre medias de tratamientos. Los datos se expresaron como la media ± EEM (error estándar de la media). Para el análisis estadístico se usó el programa Graph Pad Prism 4.0 (Software Inc., San Diego, CA, 2003).
Resultados
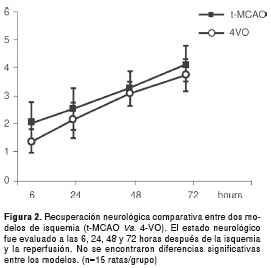
Ventajas comparativas de t-MCAO Vs. 4VO como modelos de isquemia El volumen del infarto se determinó mediante la tinción de cloruro de tetrazolio a las 24 horas después de la isquemia y la reperfusión y las áreas afectadas fueron claramente delimitadas en el modelo t-MCAO, con un volumen de infarto de 43,59±9,3 mm3; en tanto, en el modelo de 4-VO, la tinción fue difusa y variable en todos los casos y en ambos hemisferios, lo cual no permitió cuantificar el volumen del infarto ni determinar las áreas específicas afectadas (figura 1). El déficit neurológico fue evaluado en ambos modelos a las seis horas después de la isquemia y la reperfusión, y luego, a las 24, 48 y 72 horas. La escala de evaluación neurológica se basó en lo descrito previamente en otros estudios (24,25); además, se practicó un examen neurológico complementario, evaluando el reflejo palpebral, corneal, acústico, de retirada (pedal), de enderezamiento y de percepción espacial (propiocepción), así como el equilibrio en barra horizontal y en plataforma inclinada (no se presentan los datos). En ambos modelos fue evidente el déficit neurológico a las 24 horas después de la isquemia y la reperfusión, con una tendencia a la recuperación a las 48 y 72 horas, sin diferencias significativas entre ellos (figura 2). El tiempo quirúrgico, el porcentaje de supervivencia y la reproducibilidad en cuanto a las áreas afectadas, también se evaluaron en ambos modelos (cuadro 1).
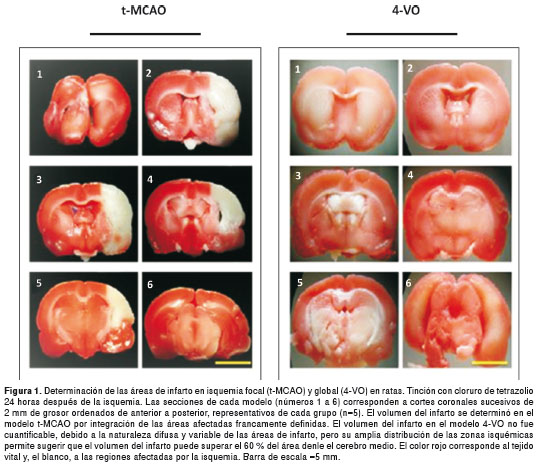
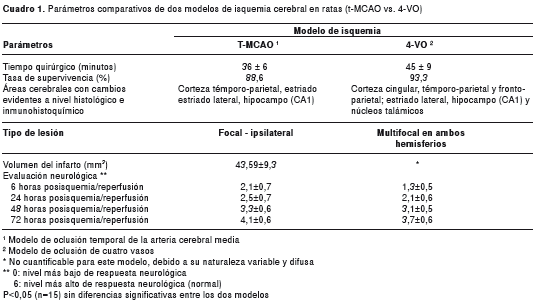
El tiempo de cirugía fue de 36±6 minutos en el modelo focal (t-MCAO) y de 45±9 minutos en el modelo multifocal (4-VO), sin diferencias importantes entre ellos, considerando que el segundo se llevó a cabo en un solo tiempo quirúrgico, contrario a lo descrito en otros trabajos, en los cuales la técnica se ejecuta en dos tiempos con un intervalo de 24 horas (12,15,30). En cuanto a la tasa de supervivencia, no se observaron diferencias significativas entre los dos modelos de isquemia (86,6 % para t-MCAO Vs . 93,3 % para 4-VO). El modelo t-MCAO aventaja al modelo 4-VO, en cuanto a la reproducibilidad en las zonas afectadas y su delimitación.
Análisis comparativo de Fluoro-Jade B® y reacción astrocitaria en dos modelos de isquemia cerebral
La tinción con Fluoro-Jade B® como método de histofluorescencia, permitió detectar células en proceso de neurodegeneración en ambos modelos. El modelo de isquemia focal t-MCAO mostró un incremento de las células Fluoro-Jade B® positivas a las 24 horas después de la isquemia y la reperfusión en la corteza cerebral témporo-parietal (figuras 3, A-1), disminuyendo significativamente (p<0,05) a las 48 horas (figuras 3, A-3 y E), mientras que, en la zona CA1 del hipocampo, la inmunorreacción fue mayor hacia las 48 horas de inducido el infarto (figuras 3A-4), sin diferencias significativas en relación con los otros tiempos de evaluación (figuras 3E). La tinción de células Fluoro-Jade B® positivas fue igualmente evidente en el modelo 4-VO y en ambos territorios cerebrales (corteza e hipocampo) a las 24 horas después de la isquemia y la reperfusión (figuras 3B-1,2), con un incremento muy significativo en la corteza a las 24 horas en relación con las 48 horas (p<0,001) y, significativo, en relación con las 72 horas (p<0,05) después de la isquemia y la reperfusión (figura 3B-3, 3B-5 y 3F). En la zona CA1 del hipocampo, hubo menos marcación que en la corteza, sin diferencias significativas entre los distintos tiempos después de la isquemia y la reperfusión (figuras 3B-2, 4, 6 y 3F).
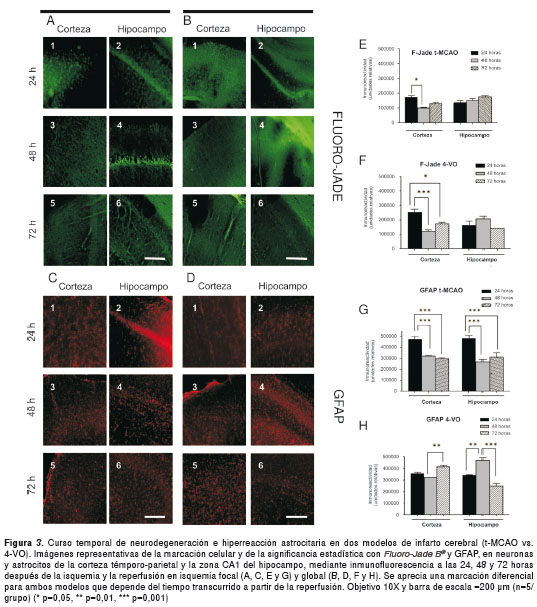
Por otra parte, en el modelo t-MCAO la hiperreacción astrocitaria se detectó a las 24 horas después de la isquemia y la reperfusión, tanto en la corteza somatosensorial (figuras 3C-1) como en la zona CA1 del hipocampo (figuras 3C-2), con diferencias muy significativas (p<0,001) en comparación con lo observado a las 48 y 72 horas en las mismas regiones cerebrales (figuras 3C-3 a 6 y 3G).
Respecto al modelo 4-VO, la hiperreacción astrocitaria en la corteza somatosensorial (figuras 3D-1) y la zona CA1 del hipocampo (figuras 3D-2) fue ligeramente menor y difusa a las 24 horas, con un incremento significativo a las 72 horas en la corteza (p<0,01) (figuras 3D-5 y 3H), mientras que, en el hipocampo, el incremento en la reacción fue significativo (p<0,01) a las 48 horas (figuras 3D-4 y 3H) y disminuyó a las 72 horas (p<0,001) (figuras 3, D-6 y H).
Progresión de la inmunorreacción de la proteína tau hiperfosforilada en dos modelos de isquemia cerebral
La inmunorreacción de células AT-8 (positivas para filamentos helicoidales pareados) como marcador de la proteína tau hiperfosforilada, se ha descrito en diversas enfermedades neurodegenerativas (21). En el presente estudio, se evidenció inmunorreacción de las células AT-8 (proteína tau hiperfosforilada) en la corteza témporo-parietal (figuras 4A-1 y 4-B1) y en la zona CA1 del hipocampo (figuras 4A-2 y 4B-2), tanto en el modelo t-MCAO (focal) como en el modelo 4-VO (global), desde las primeras 24 horas después de la isquemia y la reperfusión.
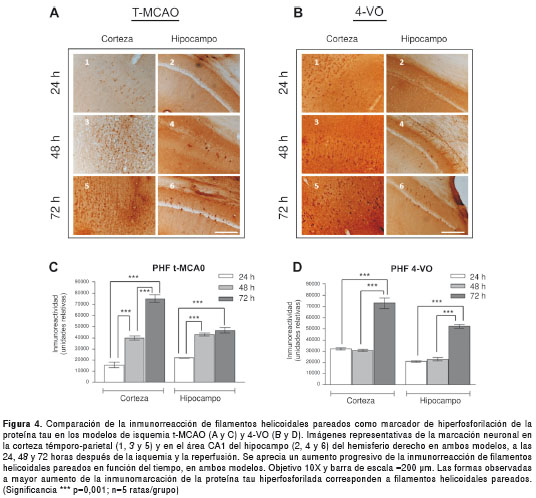
La marcación positiva de filamentos helicoidales pareados se presentó en el hemisferio ipsilateral (derecho) en el modelo focal (t-MCAO), mientras que en el global (4-VO) se observó una inmunorreacción similar en los dos hemisferios.
En ambos modelos (t-MCAO y 4-VO) fue muy evidente la progresión en la inmunorreacción de la proteína tau hiperfosforilada a 48 y 72 horas después de la isquemia y la reperfusión, tanto en la corteza (figuras 4A-3,5 y 4B-3,5) como en la zona CA1 del hipocampo (figuras 4A-4,6 y 4B-4,6), con diferencias muy significativas (p<0,001) (figuras 4C y 4D).
Discusión
En el presente estudio, se optimizaron los dos modelos de isquemia cerebral utilizados, t-MCAO y 4-VO, al requerir menor tiempo quirúrgico y generar menor riesgo de muerte, respecto a estudios previos. En estos modelos de isquemia, los marcadores Fluoro-Jade B® y GFAP permitieron detectar procesos de neurodegeneración a las 24 horas después de la lesión, en tanto el marcador de proteína tau hiperfosforilada (AT-8) incrementó progresivamente su reacción hasta las 72 horas, lo cual sugiere propagación de la excitotoxicidad y alteración de enzimas implicadas en la fosforilación de proteínas del citoesqueleto. Estos hallazgos son importantes porque permitirán planear estudios de neuroprotección en una ventana temporal específica, según el modelo y el marcador de degeneración utilizados; también, facilitará la identificación dirigida de blancos celulares y su participación espacio-temporal.
Los modelos de isquemia cerebral (tMCAO y 4-VO), usados de manera frecuente en investigación biomédica, fueron estandarizados y optimizados en la presente investigación, haciéndolos más prácticos, seguros y ágiles, lo cual permitió reducir significativamente el tiempo efectivo del procedimiento quirúrgico e incrementar la tasa de supervivencia transoperatoria y posoperatoria, en relación con lo descrito en otros trabajos previos (2). Es decir, se logró optimizar dos protocolos confiables y reproducibles de modelos animales adecuados para estudiar los eventos fisiopatológicos dependientes del tiempo después de un evento isquémico, e ideales para estudios preclínicos de tratamiento protectores.
La precisión en el abordaje quirúrgico, el acceso vascular directo y la endarterización, sumados al uso del electrocauterio y las condiciones de temperatura controlada, reducen sustancialmente el tiempo quirúrgico, disminuyen significativamente el riesgo de muerte intraoperatoria, aumentan las probabilidades de supervivencia posoperatoria y reducen el tiempo de recuperación neurológica, tal como se evidenció en el presente estudio (cuadro 1).
El modelo de isquemia t-MCAO demostró ser reproducible y poco variable, ocasionó lesiones focales específicas, delimitadas y francamente definidas, mientras que el modelo 4-VO causó lesiones multifocales, difícilmente definidas y con alteraciones celulares en ambos hemisferios cerebrales; en este modelo no se pudo delimitar claramente las áreas rescatables de penumbra ni establecer el volumen del infarto. Sin embargo,
la amplia distribución de las zonas isquémicas descrita en el cuadro 1 y la figura 1, permite sugerir que el volumen del infarto puede superar el 60 % del área denle el cerebro medio.
El modelo focal (t-MCAO) se basó en el protocolo propuesto por Longa, et al . (13), y adaptado posteriormente en otros trabajos (31,32), con pequeñas modificaciones según las experiencias de otros autores (6-8,10,14,16,17,25,33-41). La fórmula para la corrección del factor edema cerebral, se usó para calcular el volumen del infarto en este modelo (40); sin embargo, en el modelo 4-VO no fue posible calcular el volumen del infarto dadas las características difusas y multifocales de la lesión. El volumen del infarto en t-MCAO se estimó en 43,59±9,3 mm3 y las variaciones pueden haber obedecido al tipo de sutura, la longitud de la misma introducida en la arteria carótida interna, el diámetro de la esfera en la punta del filamento, la profundidad de introducción del mismo en la arteria carótida interna, el tiempo de permanencia o la reperfusión temprana (7,32,40,42).
El monofilamento utilizado en este estudio fue poliamida 4/0 recubierto con poli-L-lisina para aumentar la adhesividad dentro de la luz; sin embargo, en pocos casos no se evidenció una lesión focal, lo cual pudo haber obedecido a una oclusión incompleta de la arteria cerebral media o a una reperfusión temprana por desplazamiento de la sutura. En ninguno de los casos se presentó ruptura vascular o hemorragia subaracnoidea; solamente en tres de ellos se observó hipertermia espontánea posquirúrgica y muerte consecuente, lo cual puede asociarse con daño hipotalámico por lesión cerebral posterior, tal como se ha descrito en otros trabajos (6).
A pesar de que el t-MCAO ha sido el modelo de isquemia focal más comúnmente utilizado en ratas para simular los efectos fisiopatológicos del infarto en humano, su anatomía vascular cerebral difiere de la del hombre, lo cual genera en las ratas infartos focales relativamente extensos que no corresponden con la mayoría de los infartos focales comúnmente evidenciados y tratados en la práctica clínica (6,7).
Por otra parte, en el modelo de 4-VO, la electrocoagulación de las arterias vertebrales solo produjo déficit neurológico en 20 a 30 % de las ratas; sin embargo, la técnica tuvo éxito en 50 a 75 % de los animales intervenidos, por lo tanto, es muy variable, tal como lo describen otros autores (7,43). Los resultados obtenidos en el presente estudio fueron similarmente variables, en particular con relación a la difusión de la lesión. Los animales se sometieron a oclusión temporal (25 minutos) de ambas arterias carótidas y enseguida a electrocoagulación de las arterias vertebrales, por lo cual se mantuvieron bajo anestesia durante todo el procedimiento y solo recuperaron parcialmente el movimiento y el comportamiento exploratorio cuatro horas después de culminada la cirugía. Aunque los animales se desplazaban con dificultad y los reflejos estuvieron disminuidos, la disfunción neurológica fue recuperándose gradualmente en el transcurso de las siguientes 24 horas después de la isquemia y la reperfusión, sin que la actividad motora llegara a normalizarse totalmente, similar a lo descrito en otros trabajos (12,15).
El porcentaje de supervivencia con el modelo 4-VO fue ligeramente mayor que con el del modelo t-MCAO, lo que evidencia que el procedimiento es seguro. Además, el modelo de isquemia por 4-VO es relativamente sencillo, tiene una alta incidencia de daño neuronal isquémico (43), pero con gran variabilidad multiinfarto de difícil medición. La cauterización de las arterias vertebrales es fundamental en este modelo (4-VO) debido a la circulación colateral de la rata (15), que puede suplir el déficit que sigue a la simple oclusión de las arterias carótidas.
Además, nuestros datos muestran que las células positivas con Fluoro-Jade B® (marcador de neurodegeneración), estuvieron presentes en el área de penumbra del modelo t-MCAO a 24 horas después de la isquemia y la reperfusión, pero no hacia las 48 y 72 horas; mientras que, la inmunorreacción de GFAP fue mayor en la corteza témporo-parietal y el hipocampo (zona CA1) ipsilaterales, a las 24 horas. Los filamentos helicoidales pareados como indicadores de la hiperfosforilación de la proteína tau, fueron evidentes en las mismas zonas desde las 24 horas después de la isquemia, incrementándose de manera significativa la inmunorreacción a las 48 horas y más aún hacia las 72 horas.
La reacción de Fluoro-Jade B® y de GFAP en el modelo 4-VO a las 24 horas después de la isquemia y la reperfusión, fue mucho más extensa en la corteza temporo-parietal de ambos hemisferios, pero menos notable en la zona CA1 del hipocampo; en un trabajo similar, de observaron células positivas con Fluoro-Jade B® en el estrato piramidal de la zona CA1 (22). A las 48 horas, la reacción astrocitaria fue mayor en este modelo, con incremento hacia las 72 horas en corteza y hacia las 48 horas en el hipocampo. Concordante con nuestros resultados, un posible mecanismo de plasticidad que podría contribuir a la recuperación neuronal después del evento isquémico, es la activación de los astrocitos, al recaptar el exceso de neurotransmisor excitador y producir factores tróficos (22). Se ha descrito que la activación de GFAP se ve incrementada a partir de las 48 horas después de la isquemia y la reperfusión, acompañando el daño neuronal con una mayor intensidad en la reacción astrocitaria en las zonas de penumbra (modelo focal); mientras que, a las 24 horas, los astrocitos reactivos pueden aún apreciarse en el foco isquémico (23).
En estudios previos se describe que el número de células apoptóticas es bajo durante las primeras horas, se incrementa hacia las 48 y 72 horas al igual que la astroglía, y la apoptosis declina hacia el séptimo día de la lesión (44). Estos resultados relacionados con la muerte celular coinciden con nuestros hallazgos, pues encontramos un incremento de la acidosis a las 24 horas y mayor número de células positivas con GFAP a las 48 y 72 horas después de la isquemia y la reperfusión, lo que refleja la progresión del deterioro tisular, el cual se propaga a otra regiones exofocales en tiempo más tardíos (2).
Las neuronas que sobrevivieron inicialmente a la isquemia, pueden morir dentro de las primeras horas posteriores a la reperfusión, después de que el flujo sanguíneo cerebral es restablecido. Esta muerte se debe a la falla energética, al incremento de los niveles de calcio intracelular, que conllevan a la excitotoxicidad, la acidosis y a acumulación de especies reactivas de oxígeno, lo cual, a su vez, produce activación de células gliales, las señales apoptóticas y respuesta inflamatoria, con la concomitante alteración de la barrera hematoencefálica (43,45). Las neuronas en la zona de penumbra isquémica pueden entrar en reposo en donde la perfusión sanguínea se ha preservado; sin embargo, muchas de ellas pueden sufrir sobreexcitación y una serie de eventos bioquímicos, los cuales se han registrado al menos durante las primeras seis a ocho horas después de la isquemia y la reperfusión, aumentando el área del infarto (44). La penumbra isquémica comprende la mitad del volumen total de la lesión durante las primeras etapas de la isquemia y representa la región en la cual existe posibilidad de recuperación (43). El reestablecimiento funcional después de la isquemia depende, por lo tanto, del grado de pérdida tisular y de la preservación o rescate de las redes neuronales que fueron afectadas (46).
Uno de los hallazgos novedosos obtenidos en la presente investigación, fue el incremento de la inmunorreacción de la proteína tau hiperfosforilada de un modo dependiente del tiempo en ambos modelos (t-MCAO y 4-VO), lo que sugiere la detección de proteína tau hiperfosforilada, como marcador de la progresión del daño isquémico cerebral, similar a lo que acontece con otras taupatías en el humano, pero en un curso temporal corto tras el daño agudo.
Estos resultados, a su vez, son apoyados por hallazgos previos de nuestro laboratorio, donde se describe la correlación entre la taupatía y el déficit cognitivo a largo plazo a 30 días después de la isquemia en un modelo de 2-VO. En dicha lesión cerebral tardía, se observó, además, acompañado de apoptosis, alteración de los microtúbulos y del citoesqueleto de actina, altos niveles de la proteína Rac1 inactiva la cual es una RhoGTPasa involucrada en supervivencia celular (47) así como un incremento en la actividad de CDK5 y de la proteína RhoA (RhoGTPasa implicada en muerte celular) (48).
Otros estudios han descrito la presencia de proteína tau hiperfosforilada en cerebros isquémicos de rata y ratón. La demencia se ha correlacionado con la presencia de taupatía en humanos (19,49), incluyendo la enfermedad de Alzheimer, la enfermedad de Parkinson y la demencia frontotemporal, lo cual sugiere que la proteína tau patológica es un marcador de progresión de la lesión cerebral (19,21).
De acuerdo con los resultados obtenidos en el presente estudio, actualmente hay investigaciones de laboratorio encaminadas a bloquear el progreso de la hiperfosforilación de la proteína tau, controlando la expresión de la cinasa dependiente de ciclina 5 ( Cyclin-Dependent Kinase 5, CDK5 ), desde la reperfusión y en los tres primeros días de mayor inmunorreacción de la proteína tau. Una estrategia análoga se utilizó en el modelo triple transgénico de Alzheimer, lográndose reversión de los filamentos helicoidales pareados y ovillos neurofibrilares (50).
En conclusión, tras la utilización de modelos de isquemia cerebral focal o global, optimizadas y reproducibles, en estos modelos de isquemia, los marcadores Fluoro-Jade B® y GFAP permitieron detectar procesos de neurodegeneración a las 24 horas después de la lesión, mientras que el marcador de proteína tau hiperfosforilada (AT-8) incrementó progresivamente su reacción hasta las 72 horas, lo cual sugiere la propagación de la excitotoxicidad y la alteración de enzimas implicadas en la fosforilación de proteínas del citoesqueleto. Estos hallazgos proponen el análisis de la proteína tau hiperfosforilada como indicador de la magnitud de la lesión, su repercusión en el deterioro funcional y como sensor de respuesta a estrategias neuroprotectoras y restauradoras.
Agradecimientos
Queremos expresar nuestro agradecimiento a Johanna Gutiérrez Vargas, por su apoyo técnico y el mantenimiento de la colonia de ratas SPF en el bioterio de la Universidad de Antioquia.
Conflicto de intereses
Los autores de la presente investigación declaran no tener de manera directa o indirecta, ningún tipo de conflicto de intereses que puedan poner en riesgo la validez de este estudio.
Financiación
Este trabajo fue realizado gracias a la financiación otorgada por COLCIENCIAS (proyectos 11150416372, 111545921467) y CODI de la Universidad de Antioquia - Colombia.
Correspondencia:
Gloria Patricia Cardona, Sede de Investigación Universitaria, Universidad de Antioquia, Calle 62 N°52-59, torre 1, laboratorio 412, Medellín, Colombia
Telefax: (574) 219 6444
patricia.cardona@neurociencias.udea.edu.co
Referencias
1. Dearborn JL, McCullough LD. Perception of risk and knowledge of risk factors in women at high risk for stroke. Stroke. 2009;40:1181-6. http://dx.doi.org/10.1161/STROKEAHA.108.543272 [ Links ]
2. Arango CA, Escobar M, Cardona GP, Pimienta H. Fisiopatología de la isquemia cerebral focal: aspectos básicos y proyección a la clínica. Rev Neurol. 2004;39:156-65. [ Links ]
3. Myers GL, Christenson RH, Cushman M, Ballantyne CM, Cooper GR, Pfeiffer CM, et al . Biomarcadores emergentes para la prevención primaria de la enfermedad cardiovascular y del accidente cerebrovascular. Acta Bioquím Clín Latinoam. 2010;44:75-100. [ Links ]
4. Rodrigo J, Fernández AP, Serrano J, Peinado MA, Martínez A. The role of free radicals in cerebral hypoxia and ischemia. Free Radical Biol Med. 2005;39:26-50. http://dx.doi.org/10.1016/j.freeradbiomed.2005.02.010 [ Links ]
5. Wu D. Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRX. 2005;2:120-8. [ Links ]
6. Carmichael ST. Rodent models of focal stroke: Size, mechanism, and purpose. NeuroRX. 2005;2:396-409. [ Links ]
7. Durukan A, Tatlisumak T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav.2007;87:179-97. http://dx.doi.org/10.1016/j.pbb.2007.04.015 [ Links ]
8. Graham SM, McCullough LD, Murphy SJ. Animal models of ischemic stroke: Balancing experimental aims and animal care. Comp Med. 2004;54:486-96. [ Links ]
9. Sicard KM, Fisher M. Animal models of focal brain ischemia. Exp Transl Stroke Med. 2009;1:7. http://dx.doi.org/10.1186/2040-7378-1-7 [ Links ]
10. Gerriets T, Stolz E, Walberer M, Müller C, Rottger C, Kluge A, et al . Complications and pitfalls in rat stroke models for middle cerebral artery occlusion: A comparison between the suture and the macrosphere model using magnetic resonance angiography. Stroke. 2004;35:2372-7. [ Links ]
11. Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006;26:1055-81. http://dx.doi.org/10.1007/s10571-006-9008-1 [ Links ]
12. Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267-72. http://dx.doi.org/10.1161/01.STR.10.3.267 [ Links ]
13. Longa ZE, Weinstein PR, Carlson S, Cummins R. reversible middle cerebral artery occlusion without craniectomy in rats. Stroke.1989;20:84-91. http://dx.doi.org/10.1161/01.STR.20.1.84 [ Links ]
14. Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85-95. [ Links ]
15. Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: Method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke.1988;19:913-4. http://dx.doi.org/10.1161/01.STR.19.7.913 [ Links ]
16. Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627-42. http://dx.doi.org/10.1161/01.STR.20 . 12.162 [ Links ]
17. Belayev L, Busto R, Zhao W, Fernández G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-L-lysine: Neurological and histological validation. Brain Res. 1999;833:181-90. http://dx.doi.org/10.1016/S0006-8993(99)01528-0 [ Links ]
18. Wang J, Shen J, Gao Q, Ye Z, Shang S, Liang H, et al . Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke.2008;39:983-90. http://dx.doi.org/10.1161/STROKEAHA.107.499079 [ Links ]
19. Wen Y, Yang S, Liu R, Simpkins JW. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004;1022:30-8. http://dx.doi.org/10.1016/j.brainres.2004.05.106 [ Links ]
20. Popa-Wagner A, Schr ? der E, Walker LC, Kessler C. B-amyloid precursor protein and b-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion. Stroke. 1998;29:2196-202. http://dx.doi.org/10.1161/01.STR.29.10.2196 [ Links ]
21. Buée L, Bussiére T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95-130. http://dx.doi.org/10.1016/S0165-0173(00)00019-9 [ Links ]
22. Briones TL, Woods J, Wadowska M, Rogozinska M, Nguyen M. Astrocytic changes in the hippocampus and functional recovery after cerebral ischemia are facilitated by rehabilitation training. Behav Brain Res. 2006;171:17-25. http://dx.doi.org/10.1016/j.bbr.2006.03.01 [ Links ]
23. Li Y, Chopp M, Zhang ZG, Zhang RL. Expression of glial fibrillary acidic protein in areas of focal cerebral ischemia accompanies neuronal expression of 72-kDa heat shock protein. J Neurol Sci. 1995;128:134-42. http://dx.doi.org/10.1016/0022-510X(94)00228-G [ Links ]
24. Yrjänheikki J, Koistinaho J, Kettunen M, Kauppinen RA, Appel K, Hull M, et al . Long-term protective effect of atorvastatin in permanent focal cerebral ischemia. Brain Res. 2005;1052:174-9. http://dx.doi.org/10.1016/j.brainres.2005.06.004 [ Links ]
25. Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472-6. http://dx.doi.org/10.1161/01.STR.17.3.472 [ Links ]
26. Yang Y, Shuaib A, Li O, Siddiqui MM. Neuroprotection by delayed administration of topiramate in a rat model of middle cerebral artery embolization. Brain Res. 1998;804:169-76. http://dx.doi.org/10.1016/S0006-8993(98)00410-7 [ Links ]
27. Joshi CN, Jain SK, Murthy PSR. An optimized triphenyltetrazolium chloride method for identification of cerebral infarcts. Brain Res Brain Res Protoc. 2004;13:11-7. http://dx.doi.org/10.1016/j.brainresprot.2003. 12.001 [ Links ]
28. Céspedes-Rubio A, Wandosell F, Cardona-Gómez GP. p120 catenin/an-catenin are molecular targets in the neuroprotection and neuronal plasticity mediated by atorvastatin after focal cerebral ischemia. J Neurosci Res. 2010;88:3621-34. http://dx.doi.org/10.1002/jnr.22511 [ Links ]
29. Roulston CL, Callaway JK, Jarrott B, Woodman OL, Dusting GJ. Using behaviour to predict stroke severity in conscious rats: Post-stroke treatment with 3 ',4'-d ihydroxyflavonol improves recovery. Eur J Pharmacol. 2008;584:100-10. http://dx.doi.org/10.1016/j.ejphar.2008.01.046 [ Links ]
30. Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM. Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav. 2006;49:246-60. http://dx.doi.org/10.1016/j.yhbeh.2005.07.010 [ Links ]
31. Kuge Y, Minematsu K, Yamaguchi T, Miyake Y. Nylon monofilament for intraluminal middle cerebral artery occlusion in rats. Stroke. 1995;26:1655-8. http://dx.doi.org/10.1161/01.STR.26.9.1655 [ Links ]
32. Schmid-Elsaesser R, Zausinger S, Hungerhuber E, Baethmann A, Reulen HJ, García JH. A critical reevaluation of the intraluminal thread model of focal cerebra ischemia: Evidence of inadvertent premature reperfusion and subarachnoid hemorrhage in rats by laser-Doppler flowmetry. Stroke. 1998;29:2162-70. http://dx.doi.org/10.1161/01.STR.29.10.2162 [ Links ]
33. Braeuninger S, Kleinschnitz C. Rodent models of focal cerebral ischemia: Procedural pitfalls and translational problems. Exp Transl Stroke Med. 2009;1:8. http://dx.doi.org/10.1186/2040-7378-1-8 [ Links ]
34. Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: Technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res. 2005;1042:29-36. http://dx.doi.org/10.1016/j.brainres.2005.02.021 [ Links ]
35. Gupta YK, Briyal S. Animal models of cerebral ischemia for evaluation of drugs. Indian J Physiol Pharmacol. 2004;48:379-94. [ Links ]
36. Herz RCG, Kasbergen CM, Hillen B, Versteeg DHG, Wildt DJ. Rat middle cerebral artery occlusion by an intraluminal thread compromises collateral blood flow. Brain Res. 1998;791:223-8. http://dx.doi.org/10.1016/S0006-8993(98)00106-1 [ Links ]
37. Hossmann KA. Cerebral ischemia: Models, methods and outcomes. Neuropharmacology. 2008;55:257-70. http://dx.doi.org/10.1016/j.neuropharm.2007.12.004 [ Links ]
38. Peña-Tapia PG, Díaz AH, Torres JL. Oclusión endovascular permanente de la arteria cerebral media en ratas Wistar: descripción del abordaje a través de la arteria carótida interna. Rev Neurol. 2004;39:1011-6. [ Links ]
39. Smr c ka M, Otev r el F, Kucht íc kov á , Hork ý M, Jur án V, Duba M, et al. Experimental model of reversible focal ischaemia in the rat. Scripta Medica. 2001;74:391-8. [ Links ]
40. Woitzik J, Schneider UC, Thome C, Schroeck H, Schilling L. Comparison of different intravascular thread occlusion models for experimental stroke in rats. J Neurosci Methods.2006;151:224-31. http://dx.doi.org/10.1016/j.jneumeth.2005.07.007 [ Links ]
41. Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, et al . AKT contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci.2005;25:9794-806. http://dx.doi.org/10.1523/JNEUROSCI.3163-05.2005 [ Links ]
42. Sicard KM, Fisher M. Emerging drugs for acute ischemic stroke. Expert Opin Emerg Drugs. 2009;14:33-42. http://dx.doi.org/10.1517/14728210902748365 [ Links ]
43. Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6:11. http://dx.doi.org/10.1186/1750-1326-6-11 [ Links ]
44. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999;22:391-7. http://dx.doi.org/10.1016/S0166-2236(99)01401-0 [ Links ]
45. Culmsee C, Krieglstein J. Mechanisms of neuronal degeneration after ischemic stroke –Emerging targets for novel therapeutic strategies. Drug Discov Today. 2005;2:463-70. http://dx.doi.org/10.1016/j.ddmec.2005.11.006 [ Links ]
46. Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. 2006;16:258-64. http://dx.doi.org/10.1016/j.conb.2006.05.011 [ Links ]
47. Gutiérrez-Vargas J, Castro-Álvarez JF, Velásquez-Carvajal D, Montañez-Velásquez MN, Céspedes-Rubio A, Cardona-Gómez GP. Rac1 activity changes are associated with neuronal pathology and spatial memory long-term recovery after global cerebral ischemia. Neurochem Int. 2010;57:762-73. http://dx.doi.org/10.1016/j.neuint.2010.08.014 [ Links ]
48. Castro-Álvarez JF, Gutiérrez-Vargas J, Darnaudery M, Cardona-Gómez GP. ROCK inhibition prevents tau hyperphosphorylation and p25/CDK5 increase after global cerebral ischemia. Behav Neurosci. 2011;125:465-72. http://dx.doi.org/10.1037/a0023167 [ Links ]
49. Wen Y, Yang SH, Liu R, Pérez EJ, Brun-Zinkernagel AM, Koulen P, et al . Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats Biochim Biophys Acta. 2007;1772:473-83. http://dx.doi.org/10.1016/j.bbadis.2006.10.011 [ Links ]
50. Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, et al . Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer's mice. J Neurosci. 2010;30:13966-76. http://dx.doi.org/10.1523/JNEUROSCI.3637-10.2010 [ Links ]
