










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.33 supl.1 Bogotá Sept. 2013
https://doi.org/10.7705/biomedica.v33i0.706
NOTA TÉCNICA
doi: http://dx.doi.org/10.7705/biomedica.v33i0.706
1 Laboratorio de Virología, Instituto Nacional de Salud, Bogotá, D.C., Colombia
2 Laboratorio de Arbovirus y Enfermedades Víricas Importadas, Centro Nacional de Microbiología, Instituto de Salud Carlos III, Madrid, España
3 Current address: Robert Koch Institut, Berlin, Germany
Authors ' contributions:
María C. Méndez performed most of the experiments and wrote the manuscript. Cristina Domingo participated in the experimental design and provided a critical review of the manuscript.
Antonio Tenorio contributed to the primer design and provided a critical review of the manuscript.
Lissethe C. Pardo performed the experiments to evaluate the specificity of the test. Gloria J. Rey contributed to the interpretation of data and provided a critical review of the manuscript.
Jairo A. Méndez conceived the study, its experimental design, coordinated the job and provided a final review of the manuscript.
All authors read and approved the final version of the manuscript.Recibido: 27/05/12; aceptado: 24/02/13
Introduction. Yellow fever is considered a re-emerging disease and is endemic in tropical regions of Africa and South America. At present, there are no standardized or commercialized kits available for yellow fever virus detection. Therefore, diagnosis must be made by time-consuming routine techniques, and sometimes, the virus or its proteins are not detected. Furthermore, co-circulation with other flaviviruses, including dengue virus, increases the difficulty of diagnosis.
Objective. To develop a specific reverse transcriptase-polymerase chain reaction (RT-PCR) and nested PCR-based assay to improve the detection and diagnosis of yellow fever virus using both serum and fresh tissue samples.
Materials and methods. RT-PCR primers were designed to amplify a short fragment of all yellow fever virus genotypes reported. A second set of primers was used in a nested PCR to increase sensitivity. Thirty-three clinical samples were tested with the standardized reaction.
Results. The expected amplicon was obtained in 25 out of 33 samples analyzed using this approach, and 2 more samples tested positive after a subsequent nested PCR approach.
Conclusion. This improved technique not only ensures the specific detection of a wide range of yellow fever virus genotypes but also may increase the sensitivity of detection by introducing a second round of amplification, allowing a rapid differential diagnosis between dengue and yellow fever infection, which is required for effective surveillance and opportune epidemiologic measures.
Key words: Yellow fever virus, arboviruses, reverse transcription, polymerase chain reaction, diagnosis, epidemiological surveillance.
doi: http://dx.doi.org/10.7705/biomedica.v33i0.1452
Desarrollo de un método de transcripción inversa seguida de reacción en cadena de la polimerasa para la detección del virus de la fiebre amarilla
Introducción. La fiebre amarilla se considera una enfermedad reemergente y endémica en regiones tropicales de África y Suramérica. Actualmente, no existen estuches estandarizados o comerciales disponibles para la detección del virus de la fiebre amarilla y, por lo tanto, el diagnóstico debe hacerse mediante técnicas de rutina que consumen mucho tiempo y algunas veces no garantizan la detección del virus o de sus proteínas. Además, la cocirculación con otros flavivirus, incluyendo el del dengue, hacen el diagnóstico más complicado.
Objetivo. Desarrollar un ensayo específico de amplificación basado en transcripción inversa seguida de reacción en cadena de la polimerasa, con el fin de mejorar la detección y el diagnóstico de la fiebre amarilla, tanto a partir de suero como de tejido fresco.
Materiales y métodos. Se diseñaron iniciadores específicos para amplificar un fragmento conservado del virus de la fiebre amarilla. Un segundo par de iniciadores se usó en una reacción de amplificación anidada para incrementar la sensibilidad. Se probaron 33 muestras clínicas con la técnica estandarizada.
Resultados. El amplímero esperado se obtuvo en 25 de las 33 muestras analizadas usando este método y 2 más resultaron positivas después de la reacción anidada.
Conclusión. Esta técnica mejorada garantiza la detección de todos los genotipos virales de fiebre amarilla y puede incrementar la sensibilidad del ensayo introduciendo una segunda etapa de amplificación, lo cual permite el diagnóstico diferencial con infección por dengue y otros flavivirus, lo cual es de gran importancia para la vigilancia y la toma de medidas epidemiológicas oportunas.
Palabras clave: virus de la fiebre amarilla, arbovirus, transcripción reversa, reacción en cadena de la polimerasa, diagnóstico, vigilancia epidemiológica
doi: http://dx.doi.org/10.7705/biomedica.v33i0.1452
Yellow fever is an arthropod-borne viral disease that is clinically indistinguishable from other viral hemorrhagic fevers or febrile syndromes that are produced by dengue virus, arenavirus, hantavirus, West Nile, hepatitis B Virus and infectious diseases such as leptospirosis and malaria (1-3).
Yellow fever was one of the most feared diseases between the 15 th and 19 th centuries, and it is still a major public health concern in endemic areas, with a case fatality rate ranging from 10% to 50% of confirmed cases, despite an effective and widely available vaccine (3-7). Although asymptomatic in some cases, classic illness consists of three phases, starting with an unspecific infection period, followed by a remission stage in which symptoms disappear (1,3). Severe cases are characterized by the sudden appearance of high fever that may last from hours to days until the intoxication phase, when myocardial, renal and hepatic failure with jaundice present; progression to fulminate disease then occurs, leading to shock and death (1).
Sporadic cases of yellow fever still occur within endemic areas, affecting unvaccinated individuals that get close to or enter into tropical forests (3,7-10). An annual estimate of 200,000 cases and 30,000 deaths makes it a significant cause of morbidity and mortality worldwide (1-3). The official number of cases may be underestimated due to a lack of detection, misdiagnosis and inaccessibility to rural areas and medical facilities. In fact, the real incidence could be as high as 5 to 10 times the official reports (2,10). In addition, the increasing growth of the human population and its forced migration, vector re-infestation and redistribution, difficulties in transport to some areas and the movement of infected travelers may contribute to the spread of the disease (3,10).
The correct and timely diagnosis of yellow fever is an important issue, particularly in countries where it is endemic. Currently, serological tests include IgM antibody capture by enzyme-linked immunosorbent assay (MAC-ELISA), hemagglutination inhibition (HI), complement fixation (CF) and neutralization (N) (1,3). Nevertheless, antibody detection is feasible only 5 to 6 days after the onset of symptoms, when the severity increases, and there is a chance of cross reactivity with other flaviviruses (1). However, diagnosis of yellow fever may also be achieved during the infection period by the isolation of the virus in mosquito cell lines or by genome detection through PCR-based methods.
Fatal cases are confirmed by immunohistochemistry with monoclonal antibodies in well-conserved fixed tissues (1,3,11,12). Virus isolation in mosquito cell lines, although specific, takes at least 7-10 days, and the sensitivity decreases when few virus particles are present in the sample or when toxic biliary salts are present in the serum, inhibiting cell growth (13). Detection of the virus in the acute phase must be specific and sensitive enough to confirm the diagnosis; only then can surveillance and control campaigns begin.
To improve and increase the speed of yellow fever diagnosis, we designed four novel primers for RT-PCR followed by a nested PCR reaction to increase sensitivity. In contrast to previously reported RT-PCR based methods, genetic diversity was considered during the design of these new primers, resulting in a highly sensitive reaction that is able to amplify any yellow fever genotype (3,14). This assay could be easily standardized in developing countries, particularly in small cities or provinces where disease is endemic, increasing the opportunity for diagnosis and improving epidemiological surveillance.
Materials and methods
Virus isolates and clinical samples used in this study were taken during the acute stage of infection and belonged to the sample collection of the Laboratorio de Virología of the Instituto Nacional de Salud, where samples are kept at -20 to -70 ° C. The yellow fever virus strains consisted of 7 lyophilized viral isolates obtained between 1964 and 2000. Those viruses were reconstituted in 1 mL of sterile phosphate buffer saline (PBS) solution prior the extraction of RNA. In addition, we processed 10 yellow fever sera samples collected between 2004 and 2009 that were negative for both IgM and viral isolation and that had been obtained from febrile cases 5 to 7 days after the onset of symptoms. Those samples were compatible with the case definition of yellow fever: illness with a high fever, headache and back pain, including cases that presented with vomiting, bloody diarrhea, abdominal pain, hematemesis, jaundice and epistaxis (1). Travel history to endemic zones or other types of exposure was always considered. To evaluate the performance of the reaction in tissue samples, 13 liver samples collected between 2004 and 2009 from fatal cases were also tested. Ten of these clinical tissue samples were previously confirmed as positive for yellow fever by immunohistochemistry. Finally, the yellow fever-infected brain tissues from 3 mice were used as positive controls.
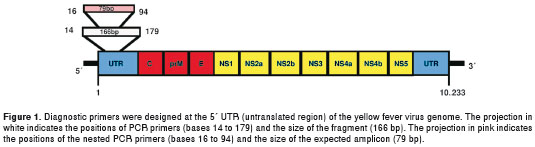
Thirty complete genome sequences, representing all genotypes reported (5 from Africa and 2 from the Americas), were obtained from Gene Bank and aligned using the ClustalW program included in MEGA software (15). Two sets of primers for RT-PCR and nested PCR were designed to target a highly conserved region on the 5´-UTR end of the viral genome (figure 1). Primers were analyzed with the program Gene Runner 3.0 (Hastings Software Inc.) to avoid secondary structures and dimers and to calculate melting temperatures. Primer sequences were as follows: RT-PCR sense primer SYF14, 5´-GCTAATTGAGGTGYATTGGTC-3´, and antisense primer ASYF179, 5´-TTARTTTGRTTG ACAAGGAGCG-3´; nested PCR sense primer NSYF16, 5´-CTAATTGAGGTGYATTGGTCTG-3´ and nested antisense primer NASYF94, 5´- CTGGTCYYTTCTCTGCTAATCG-3´. The name of each primer corresponds to its position on the genome template.
Viral RNA was extracted from 140 µl of serum samples or PBS suspensions of lyophilized virus using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) and eluted with 40 µl of buffer EB. Approximately 1 g of fresh tissue samples was treated with 1 mL of lysis buffer (Tris HCL 1 M, SDS 20%, EDTA 0.5 M pH 8.0, Proteinase K 20 mg/mL, RNAase inhibitor 10 U/µl) at 56°C overnight. RNA was extracted using Trizol, following the manufacturer's instructions. Briefly, 300 µl of processed tissues was mixed with 700 µl of Trizol LS® (Gibco BRL) reagent for 5 min; 200 µl chloroform was added, and the solution was then mixed for 15 sec by vortexing and incubated for 3 min at room temperature. The mixture was subsequently centrifuged for 15 min at 12,000 rpm at 4 °C, and the aqueous phase was carefully separated and precipitated with 500 µl cold isopropanol for 12 hours at 4 °C. Then, the mixture was centrifuged for 10 min at 12,000 rpm at 4 °C, and the supernatant was discarded. The RNA was washed with 500 µl ethanol and resuspended in 40 µl of warm water treated with diethylpyrocarbonate (DEPC, Sigma) and 20 units of RNAse inhibitor (Invitrogen).
RT-PCR and nested PCR optimal conditions were initially estimated to standardize the concentrations of Mg 2+ , dNTPs and primers using yellow fever RNA purified from the culture supernatant. The annealing temperature was determined by a gradient ranging from 50 °C to 60 °C, and serial dilutions of RNA (10 -1 , 10 -2 , 10 -3 , 10 -4 ) were used to test the limit of detection. Once the best setting for the reaction was determined, RT-PCR was performed as follows: 5 µL of purified RNA was added to 45 µL of the RT-PCR one-step reaction buffer (from the Qiagen One-step RT-PCR kit) containing 100 mM of each dNTP, 200 nM of each designed primer (SYF14, ASYF179) and 2 µl of the enzyme mix. RT-PCR was performed in a Perkin Elmer DNA Thermal Cycler 480 (Applied Biosystems, Warrington, England), starting with a 41 °C incubation for 45 min and then 15 min at 94 °C, followed for 40 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 1 min and extension at 72 °C for 1 min, with a final extension at 72 °C for 10 min. PCR products were evaluated in a 1.5% ethidium bromide-stained agarose gel. Nested PCR was performed by adding 1 µl from the original reaction mix of the negative samples to 49 µl of a reaction mix containing 1X PCR buffer, 1.5 mM MgCl 2 , 100 mM of each dNTP, 400 nM of each nested primer (NSYF16, NASYF94) and 2.5 U of DNA recombinant Taq Polymerase (Invitrogen, Life Technologies, Carlsbad). The standardized reaction included denaturation at 94 °C for 30 s, annealing at 57 °C for 4 min, extension at 72 °C for 1 min and a final extension at 72 °C for 10 min. PCR products were then resolved in a 2.0% ethidium bromide-stained agarose gel. Finally, to establish the specificity of the designed primers, RT-PCR was performed using purified dengue RNA (serotypes 1-4) and St. Louis encephalitis virus under the same conditions as described for yellow fever.
Results
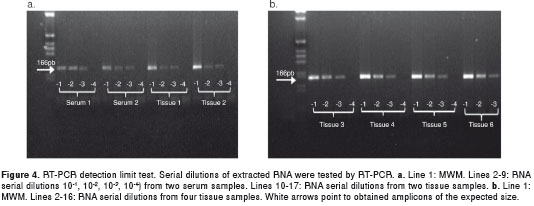
The degenerated primers SYF14 and ASYF179 were designed to target a highly conserved region of the non-coding 5' end. The amplicon corresponded to a 166-bp fragment (from base 14 to base 179) of the yellow fever virus genome. This amplicon was of the expected size and was obtained from the three positive controls (mouse brain tissue), the 7 lyophilized viruses and all fresh tissue samples, including those previously confirmed as infected with yellow fever by immunohistochemistry (figure 2). However, the amplicon was obtained from only 2 out of 10 sera within a clinical frame compatible with yellow fever (figure 3).
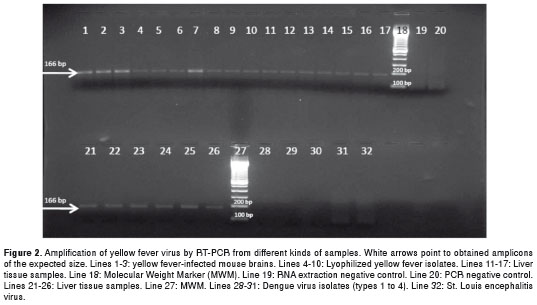
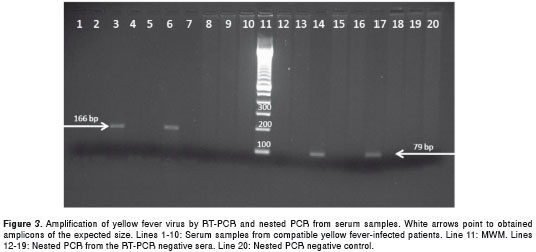
The primers NSYF16 and NASYF94 were designed to improve the sensitivity of the PCR reaction and targeted an internal region of the 166-bp amplicon. Nested PCR may increase the chance of virus detection in samples with few viral copies and can also be used to confirm the first reaction. Nested PCR was performed using the first PCR reaction from the 8 RT-PCR-negative sera as templates. A 79-bp amplification product was obtained from 2 samples (figure 3). The remaining 6 samples were taken approximately 5 to 7 days after symptom onset, when the virus might not be present. Finally, reactions using dengue virus and St. Louis encephalitis virus RNA as a template were negative, even under nested PCR conditions (figure 3).
To infer the detection limit of the PCR reaction, serial dilutions of extracted RNA (10 -1 , 10 -2 , 10 -3 , 10 -4 ) from positive sera (2 samples) and tissue samples (13 samples) were tested. Both the tissue samples and sera were positive up to a -3 dilution of the first RT-PCR amplification (figure 4). All dilutions tested positive by nested PCR.
Discussion
Colombia is an endemic yellow fever country and is surrounded by endemic yellow fever areas. In Colombia, there is an enormous risk of receiving viruses from infected incoming travelers and a high probability of the co-circulation of flavivirus and yellow fever genotypes that are not detectable by routinely used molecular probes (3,8,10,16-19). No molecular epidemiology study of yellow fever has been performed in Colombia whose aim was to determine the genotype diversity that may result in misdiagnosis; therefore, the true number of cases may be higher than reported. Dengue fever is also endemic, as it is one of the most recurrent febrile syndromes in Colombia (3,8,10,20). Co-circulation of the four dengue virus serotypes in yellow fever-endemic regions makes both syndromes easy to misdiagnose, especially in cases for which symptoms are nonspecific (3,8,10,20). Furthermore, co-circulation is a high risk of urbanization because both viruses share a common urban vector (7,21). Urbanization of yellow fever is a public health threat because vaccination is only given to people who are travelling to sylvatic areas. Although yellow fever vaccination campaigns in Colombia have been successful, war conditions and forced displacements in endemic regions near forests where viral activity has been confirmed have increased the risk of transmission and eventual urbanization (3,8,10).
Due to the lack of commercially available kits, specific diagnosis of yellow fever may be difficult and is often limited to specialized and reference labs (1). Thus, serologic diagnostic techniques are useful after the acute phase when virus is no longer present and IgM antibodies become detectable. Nevertheless, these antibodies may cross-react with other flaviviruses that are genetically and antigenically similar to yellow fever, including dengue virus, meaning that correct diagnosis is even more difficult (1,3).
Viral isolation in mosquito cell lines is a useful technique for acute phase samples. However, this technique is time consuming, and it is not easy to perform in small laboratories. In addition, samples for yellow fever virus detection often suffer from a lack of proper transportation conditions from endemic areas to diagnostic laboratories. Tissue or serum samples may pass through several freeze-thaw cycles, decreasing the probability of virus isolation, causing genome degradation or fragmentation and resulting in false-negative results, especially in those samples with low viral copy numbers (1,3,10). For these reasons, we have developed a specific RT-PCR assay with novel sets of primers that match a highly conserved region of all yellow fever genomes reported to date; this assay is thus capable of detecting a wide range of genotypes (6,19,21). This conserved genome region has a low selective pressure and no role in the immune response or the virus-environment relationships. Thus, if this sequence is present in yellow fever genotypes that have not yet been discovered, the use of these two sets of novel primers would enable the identification of new yellow fever variants. Another advantage of our approach is that the amplicon size is small enough to appear even in low copy number or degraded samples, and when combined with a nested PCR reaction, this technique generates a strong detectable amplification signal that may increase the chance of detection and perhaps the sensitivity of the method. Some of the previously published primers for yellow fever diagnosis are located in variable genome regions that may be exposed to selective pressure from the host's immune response, leading to mutation that may reduce the chance of detection (3,10).
In conclusion, we propose a novel specific RT-PCR-based assay designed to detect all yellow fever genotypes circulating worldwide. Although further experiments involving the titration of the virus or the quantification of RNA should be performed to determine the exact sensitivity of this method, these preliminary results allow us to infer a good limit of detection and; therefore, the assay offers an increased chance of identifying the viral genome. Conventional PCR is easy to perform, even in small laboratories where complex techniques such as viral isolation or real time-PCR are not feasible. Finally, this technique can be used as a complementary tool to improve yellow fever surveillance and control strategies because fast and specific detection is critical for proper control measures – not just to reduce the number of cases but also to prevent urbanization of the disease.
We thank the Red Nacional de Laboratorios, Instituto Nacional de Salud, Colombia; Noelia Reyes for technical assistance in amplifying at the ISCIII; Martha Gracia for technical assistance at Instituto Nacional de Salud; and Jose Usme-Ciro for helping in primer design. RIVE /CYTED (Red Iberoamericana de Virosis Emergentes) allowed the authors to meet with several other researchers in the field.
The authors declare that they do not have any conflicts of interest.
The Instituto Nacional de Salud government resources supported this research.
Corresponding author:
Jairo A. Méndez, Laboratorio de Virología, Instituto Nacional de Salud, Avenida calle 26 N° 51-20, CAN Zona 6, Bogotá, D.C., Colombia. Tel.: (571) 220 7700, extensión 1426; fax: (571) 220 7700 jmendez@ins.gov.co
1. Monath TP. Yellow fever: An update. Lancet Inf Dis. 2001;1:11-20. http://dx.doi.org/10.1016/S1473-3099(01)00016-0 [ Links ]
2. Tesh R. Viral hemorrhagic fevers of South America. Biomédica. 2002;22:178-93. [ Links ]
3. Méndez JA, Rodríguez G, Bernal MP, Calvache D, Boshell J. Detección molecular del virus de la fiebre amarilla en muestras de suero de casos fatales humanos y en cerebros de ratón. Biomédica. 2003;23:232-8. [ Links ]
4. Weir E, Haider S. Yellow fever: Readily prevented but difficult to treat. CMAJ. 2004;170:1909-10. http://dx.doi.org/10.1503/cmaj.1040826 [ Links ]
5. Monath TP. Dengue and yellow fever: Challenges for the development and use of vaccines. N Engl J Med. 2007;357:2222-5. http://dx.doi.org/10.1056/NEJMp 0707161 [ Links ]
6. Bryant JE, Holmes E, Barrett ADT. Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Path. 2007;3:668-73. http://dx.doi.org/10.1371/journal.ppat.0030075 [ Links ]
7. Mutebi JP, Gianella A, Travassos A, Tesh RB, Barret A, Higgs S. Yellow fever virus infectivity for Bolivian Aedes aegypti mosquitoes. Emerg Infect Dis. 2004;10:1657-60. http://dx.doi.org/10.3201/eid1009.031124 [ Links ]
8. Groot H, Morales A, Romero M, Ferro C, Prías E, Vidales H, et al . Estudios de arbovirus en Colombia en la década de 1970. Biomédica. 1996;16:331-44. [ Links ]
9. Filippis AM, Schatzmayr HG, Nicolai C, Baran M, Miagostovich MP, Sequeira PC, et al . Jungle yellow fever, Rio de Janeiro. Emerg Infect Dis. 2001; 7:484-5. http://dx.doi.org/10.3201/eid0703.017331 [ Links ]
10. Méndez JA, Parra E, Neira M, Rey GJ. Detección por reacción en cadena de la polimerasa de transcriptasa inversa del virus de la fiebre amarilla en monos silvestres: una herramienta sensible para la vigilancia epidemiológica. Biomédica. 2007;27:461-7. [ Links ]
11. Monath TP, Nystrom RR. Detection of yellow fever virus in serum by enzyme immunoassay. Am J Trop Med Hyg. 1984;33:151-7. [ Links ]
12. Ricaurte O, Sarmiento L, Caldas ML, Rodríguez G. Evaluación de un método inmunohistoquímico para el diagnóstico de la fiebre amarilla. Biomédica. 1993;13:15-9. [ Links ]
13. Deubel V, Huerre M, Cathomas G, Drouet MT, Wuscher N, LeGuenno B, et al . Molecular detection and characterization of yellow fever virus in blood and liver specimens of a non-vaccinated fatal human case. J Med Virol. 1997; 53:212-7. http://dx.doi.org/10.1002(SICI)1096-9071(199711)53:3<212::AID-JMV5>3.0.CO;2-B [ Links ]
14. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007; 24:1596-9. http://dx.doi.org/10.1093/molbev/msm092 [ Links ]
15. Vidales H, Buitrago B, Sanín LH, Morales A, Groot H. Estudio de un brote epidémico de fiebre amarilla selvática en el piedemonte de la Sierra Nevada de Santa Marta, 1979. Biomédica. 1981;1:171-8. [ Links ]
16. García I, Velandia MP, Olano VA, Molina J, Salas T, Bernal MP, et al . Sistema de vigilancia centinela sobre enfermedades febriles transmitidas por vectores con énfasis en fiebre amarilla, dengue y malaria en los departamentos de Caquetá, Nariño, Putumayo, La Guajira y Valle, 2000. Inf Quinc Epid Nac. 2001;6:1-8. [ Links ]
17. Vera MJ, Velandia MP, Rodríguez G, Neira M, Bernal MP, Méndez JA, et al . Fiebre amarilla selvática en la región del Catatumbo, Colombia 2003. Inf Quinc Epid Nac. 2004;9:49-53. [ Links ]
18. Vasconcelos PF, Bryant JE, Da Rosa TP, Tesh RB, Rodrigues SG, Barrett AD. Genetic divergence and dispersal of yellow fever virus, Brazil. Emer Infect Dis. 2004;10:1578-84. http://dx.doi.org/10.3201/eid1009.040197 [ Links ]
19. Méndez JA, Usme-Ciro JA, Domingo C, Rey GJ, Sánchez JA, Tenorio A, Gallego-Gómez JC. Phylogenetic reconstruction of dengue virus type 2 in Colombia. Virol J. 2012;9:64. http://dx.doi.org/10.1186/1743-422X-9-64 [ Links ]
20. van der Stuyft AG, Pirad M, Céspedes J, Lora J, Peredo C, Pelegrino J, et al . Urbanization of yellow fever in Santa Cruz, Bolivia. Lancet. 1999;353:1558-62. http://dx.doi.org/10.1016/S0140-6736(99)03291-2, [ Links ]
21. Bryant JE, Barret ADT. Comparative phylogenies of yellow fever isolates from Peru and Brazil. FEMS Immunol Med Microbiol. 2003;39:103-18. http://dx.doi.org/10.1016/S0928-8244(03)00238-4 [ Links ]
