










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.34 no.2 Bogotá Apr./June 2014
https://doi.org/10.7705/biomedica.v34i2.1851
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1851
1 Grupo de Investigación en Enfermedades Neurodegenerativas, Departamento de Salud Animal, Facultad de Medicina Veterinaria, Universidad del Tolima, Ibagué, Colombia
2 Grupo de Neurociencias de Antioquia, Área de Neurobiología Celular y Molecular, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
3 Grupo de Modelos Experimentales para las Ciencias Zoohumanas, Facultad de Ciencias Básicas, Universidad del Tolima, Ibagué, Colombia
4 Fundación Valle del Lili, Cali, Colombia
5 Grupo de Investigación Biomédica, Universidad ICESI, Cali, Colombia
Author contributions:
Angélica María Sabogal: conducted the experiments, collected and discussed results, and prepared the manuscript.
César Augusto Arango: consulted on cerebral ischemia procedures and nigrostriatal physiology; discussed results and reviewed the manuscript.
Gloria Patricia Cardona: managed and supervised experiments; reviewed scientific merit; performed input protein analysis; reviewed, revised and approved the manuscript; submitted the manuscript.
Ángel Enrique Céspedes: approved the project; managed and supervised the study; analyzed and interpreted the results, discussions and conclusions; reviewed and approved the manuscript.
Recibido: 14/08/13; aceptado: 20/01/14
Introduction: Cerebral ischemia is the third leading cause of death and the primary cause of permanent disability worldwide. Atorvastatin is a promising drug with neuroprotective effects that may be useful for the treatment of stroke. However, the effects of atorvastatin on specific neuronal populations within the nigrostriatal system following cerebral ischemia are unknown.
Objective: To evaluate the effects of atorvastatin on dopaminergic and GABAergic neuronal populations in exofocal brain regions in a model of transient occlusion of the middle cerebral artery.
Materials and methods: Twenty-eight male eight-week-old Wistar rats were used in this study. Both sham and ischemic rats were treated with atorvastatin (10 mg/kg) or carboxymethylcellulose (placebo) by gavage at 6, 24, 48 and 72 hours post-reperfusion. We analyzed the immunoreactivity of glutamic acid decarboxylase and tyrosine hydroxylase in the globus pallidus, caudate putamen and substantia nigra.
Results: We observed neurological damage and cell loss in the caudate putamen following ischemia. We also found an increase in tyrosine hydroxylase immunoreactivity in the medial globus pallidus and substantia nigra reticulata, as well as a decrease in glutamic acid decarboxylase immunoreactivity in the lateral globus pallidus in ischemic animals treated with a placebo. However, atorvastatin treatment was able to reverse these effects, significantly decreasing tyrosine hydroxylase levels in the medial globus pallidus and substantia nigra reticulata and significantly increasing glutamic acid decarboxylase levels in the lateral globus pallidus.
Conclusion: Our data suggest that post-ischemia treatment with atorvastatin can have neuro-protective effects in exofocal regions far from the ischemic core by modulating the GABAergic and dopaminergic neuronal populations in the nigrostriatal system, which could be useful for preventing neurological disorders.
Key words: GABAergic neurons, dopaminergic neurons, brain ischemia; models, animal; rats.
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1851
La atorvastatina protege las neuronas gabérgicas y dopaminérgicas del sistema nigroestriatal en un modelo experimental de isquemia cerebral focal transitoria en ratas
Introducción. La isquemia cerebral es la tercera causa de muerte y la primera de discapacidad permanente en el mundo. La atorvastatina es un fármaco neuroprotector prometedor para el tratamiento de la apoplejía; sin embargo, su acción sobre las poblaciones neuronales del sistema nigroestriatal después de la isquemia aún se desconoce.
Objetivo. Evaluar el efecto de la atorvastatina sobre poblaciones gabérgicas y dopaminérgicas en regiones exofocales en un modelo de oclusión transitoria de la arteria cerebral media.
Materiales y métodos. Se utilizaron 28 ratas Wistar macho de ocho semanas de edad. Los ejemplares con isquemia simulada y los ejemplares sometidos a isquemia fueron tratados con atorvastatina (10 mg/kg) y carboximetilcelulosa (placebo) administrados por medio de sonda a las 6, 24, 48 y 72 horas después de la reperfusión. Se analizó la inmunorreacción de la descarboxilasa del ácido glutámico y de la tirosina hidroxilasa en el globo pálido, el putamen caudado y la sustancia negra.
Resultados. Los datos confirmaron el daño neurológico y la pérdida celular en el putamen caudado. Se incrementó la inmunorreacción de la tirosina hidroxilasa en el globo pálido medial y la sustancia negra pars reticulata , disminuyendo la inmunorreacción de la descarboxilasa del ácido glutámico en el globo pálido lateral de los animales isquémicos tratados con placebo; sin embargo, el tratamiento con atorvastatina pudo revertirla, lo que logró una disminución significativa de la tirosina hidroxilasa en el globo pálido medial y la sustancia negra pars reticulata y aumentando los niveles de descarboxilasa del ácido glutámico en el globo pálido lateral.
Conclusión. Nuestros datos sugieren que la atorvastatina en el tratamiento posterior a la isquemia ejerce neuroprotección en las zonas exofocales, modulando las poblaciones neuronales gabérgicas y dopaminérgicas del sistema nigroestriatal, lo que podría prevenir trastornos neurológicos.
Palabras clave: neuronas gabérgicas, neuronas dopaminérgicas, isquemia encefálica, modelos animales, ratas .
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1851
Cerebrovascular disease is the third leading cause of death worldwide and the leading cause of permanent disability in adults. This disease causes motor and sensory sequelae that affect both the quality of life and longevity of patients and negatively impact the economic and productive sectors (1,2).
Ischemic strokes are caused by arterial occlusions, and the damaged regions are characterized by a central necrotic core and a surrounding penumbral area. The latter tissue can recover from the physiological stress of a nearby ischemia, and these areas are generally the targets for stroke therapies (3). Changes can also occur in tissues removed from the site of physical injury. These are known as exofocal changes, and they may be responsible for some of the clinical manifestations that are not directly related to the ischemic core injury, including behavioral, emotional and cognitive disorders that can appear weeks or months after the ischemic injury (4).
Exofocal changes have been observed in components of the basal ganglia, including the caudate putamen (CPu), globus pallidus (Gp) and substantia nigra (SN), which often show changes near the middle cerebral artery (MCA) following ischemic stroke. The globus pallidus is primarily composed of GABAergic neurons and is located near the putamen, immediately outside the internal capsule; this region can be further divided into the globus pallidus lateral (GpL) and the globus pallidus medial (GpM) (5). The SN is located below the thalamus and is divided into two functionally different structures: the pars reticulata (SNr) and the pars compacta (SNc). The SNr is composed of GABAergic-type fusiform neurons and is innervated by Gp; the SNr sends its axons to the thalamus, where it complements related circuits with sensorimotor and behavioral functions. The SNc is composed of dopaminergic neurons and innervates the caudate putamen (CPu) nucleus, where, among other things, it exerts a modulatory effect on motor functions (6).
With the aim of reducing the consequences of ischemic events, several pharmacological strategies have been elucidated and applied in clinical studies. For example, statins – which were first identified as molecules that inhibit the enzyme 3-hydroxymethylglutaryl coenzyme A (HMG-CoA) – are known to have pleiotropic effects independent of their ability to inhibit cholesterol synthesis. In particular, statins can also enable angiogenesis and synaptogenesis, increase blood flow and inhibit the formation of thrombosis and atheromatous plaques (7-9). One of the most commonly used statins, atorvastatin (ATV), has been shown to reduce infarct volume and improve neurological deficits in a rat cerebral ischemia model (10). In addition, it is known that activation of the pro-survival PI3 kinase pathway decreases excitotoxicity, modulates platelet activity and inhibits the inflammatory response in ischemic settings (11,12). In this study, we focused on the poorly studied exofocal areas, determining the effect of ATV on GABAergic and dopaminergic neuronal populations of the nigrostriatal system in a model of transient focal cerebral ischemia in Wistar rats.
Materials and methods
Animals
Male Wistar albino rats were obtained from an in- house, pathogen-free colony at the vivarium of SIU, University of Antioquia, Medellín, Colombia. Rats were maintained under a 12:12-h dark:light cycle and given food and water ad libitum. Animals were handled in accordance with Colombian standards (Law 84/1989 and Resolution 8430/1993) and European Union guidelines (86/609/EEC). Special care was taken to minimize animal suffering and to reduce the number of animals used in these exper iments. In particular, 2.5-month-old rats weighing between 220-250 g were used in these studies.
Surgical procedure
MCAO and reperfusion protocols were carried out as described previously (13), with minor modifications. Briefly, animals were anesthetized with an i.p. mixture of ketamine (60 mg/kg) and xylazine (5 mg/kg) as well as s.c. atropine (0.01 mg/kg). The right common carotid artery was exposed and dissected through a medial cervical approach. The external and internal carotid arteries (ECA and ICA, respectively) were exposed, and the first arterial branches were cauterized by electrocoagulation (Aaron bipolar cautery). For the following steps, a 4-0 nylon monofilament suture (Ethicon, Johnson & Johnson) was used, which was pretreated with poly-L-lysine (0.1% in de-ionized water), rounded at its tip with a heat treatment and sterilized with UV radiation. The filament was inserted into the ICA from the ECA in order to occlude the right middle cerebral artery. The suture was removed after 60 minutes of occlusion to allow for reperfusion. Finally, the cervical wound was closed along one plane. Sham animals were subjected to the same procedures but the filament was not introduced into the artery. All animals were sacrificed 78 hours after reperfusion.
Administration of atorvastatin
One ischemic group of animals received 10 mg/kg oral doses of calcic atorvastatin (Atorlip®-20, Lafrancol), which were administered using a gastric esophageal catheter. Treatment was repeated for 3 consecutive days following ischemia. The first dose was administered 6 hours post-reperfusion and again after 24, 48 and 72 hours. The other ischemic group received carboxymethylcellulose (CMC) as a placebo, which was delivered at the same time and with the same frequency as the experimental drug. Unoperated rats in the control groups (sham) received either ATV or CMC under the same conditions as the experimental ischemic groups.
Recovery and neurological evaluation
Following surgery, the animals were taken to their home cages to recover from anesthesia. Using the neurological test described by Bederson, et al. , 1986 (14), neurological evaluations were performed six hours after the ischemia/reperfusion procedure and again after 24, 48 and 72 hours. This test is scored on a scale from 0 to 6: 0 = no spontaneous movement; 1 = spontaneous circular movements; 2 = circular movements if held by the tail; 3 = circular movements if lifted and held by the tail; 4 = reduced resistance to lateral pressure; 5 = flexion at one or both forelimbs; 6 = normal extension of both forelimbs when lifted. The animals were also tested for other neurological disorders not accounted for in the above scale, including balance tests along both the vertical and horizontal planes, tests of spatial perception, and tests of corneal, visual, palpebral and acoustic reflexes (13).
Histology
Six hours after the final treatment dose, the animals were anesthetized intraperitoneally with a mixture of sodium pentobarbital (60 mg/kg) and xylazine (10 mg/kg) and perfused with 4% paraformaldehyde in saline (0.1 M phosphate buffer [PBS], pH 7.4). The brains were then carefully removed and post-fixed with 4% paraformaldehyde at 4 °C for 48 hours. The brains were divided into right and left hemispheres and sectioned using a vibratome (Leica Microsystems 1000S VT) into 50-micron slices from lateral -0.18 to 4.32 mm, using the Paxinos stereotaxic atlas for rats as a reference (15). Lateral sections (from 2.4 to 2.9 mm) were evaluated by Nissl staining with 1% toluidine blue. The sections were dehydrated in alcohol, treated with xylenes and sealed with Canada balsam.
Immunohistochemistry
Sections were first treated with methanol (50% v/v) and hydrogen peroxide (30% v/v) in 0.1 M PBS (pH 7.4) for 20 minutes to inhibit endogenous peroxidase activity. Next, the sections were washed three times with 0.1 M PBS, and nonspecific binding sites were blocked for 1 hour with a pre-incubation solution consisting of 0.1 M PBS with BSA (1%) and Triton X-100 (0.3% v/v).
The sections were then incubated overnight at 4 °C with the primary antibodies, which were diluted in incubation solution (0.3% BSA and 0.3% v/v Triton X-100 in 0.1 M PBS). Anti-TH (1:1000, mouse monoclonal LNC1-MAB318, Chemicon International, Inc.) was used to detect the enzyme tyrosine hydroxylase, and anti-GAD 65/67 (1:1000, polyclonal rabbit G5163; Sigma) was used to detect glutamic acid decarboxylase. The next day, the sections were washed for 5 minutes in 0.1 M PBS and then incubated with the appropriate secondary antibodies (1:250 biotin-conjugated goat anti-rabbit IgG (H+L) 31822 or 1:250 biotin-conjugated goat anti-mouse IgG (H+L) 31800; Pierce), depending on the host in which primary antibody was prepared, for one hour at room temperature.
After three washes with 0.1 M PBS, the tissues were incubated with avidin-biotin complex (1:250 reagent A and B, ABC Standard Peroxidase Staining Kit, 32020; Pierce) for 1 hour. Once excess complex was removed, three more washes were performed, and the sections were stained with diaminobenzidine (DAB). Subsequently, the sections were dehydrated with alcohol, cleared with xylenes and sealed with Canada balsam. Quantification of immunoreactivity in the areas tested was determined using a 10× objective for GP and SNr. Photomicrographs of the histological and immunohistochemical assays were also analyzed using the ImageJ 1.44 software program (NIH, USA). Controls carried out without primary antibodies did not result in immunoreactivity.
Western blotting analysis
Animals were sacrificed by decapitation and their brains were quickly removed. The globus pallidus and contralateral and ipsilateral substantia nigra were dissected and frozen at -70 °C for later analysis. The tissues were dissected and homogenized in lysis buffer containing 150 mM NaCl, 20 mM Tris (pH 7.4), 10% glycerol, 1 mM EDTA, 1% NP40, 100 µM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin and leupeptin (Sigma) and 100 µM orthovanadate.
Proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% SDS-PAGE) and the Mini-Protean system (Bio-Rad, Hercules, CA) with a wide-range molecular weight standard (Bio-Rad). Each lane was loaded with 10 mg protein and a buffer containing 0.375 M Tris (pH 6.8), 50% glycerol, 10% SDS, 0.5 M DTT and 0.002% bromophenol blue. The samples were heated to 95 °C for 5 minutes before loading on the gel. Following electrophoresis, the proteins were transferred to nitrocellulose membranes (Amersham) using an Electrophoretic Transfer System (Mini Trans-Blot Electrophoretic Transfer Cell) at 300 mA for 1 hour. The membranes were washed with TTBS (20 mM Tris-HCl (pH 7.5), 500 mM NaCl, Tris-buffered saline (pH 7.4) and 0.05% Tween- 20) and 5% lyophilized skimmed milk powder for 1 hour; TTBS was used for all subsequent washes and incubations. The membranes were then incubated overnight with the following primary antibodies: anti-TH (1:1000 mouse monoclonal LNC1-MAB318; Chemicon International Inc.) and anti-GAD 65/67 (1:1000 rabbit polyclonal G5163; Sigma); an anti-actin antibody (1:1000 mouse monoclonal AC-40-A3853; Sigma) was used as a loading control. Next, the membranes were washed and incubated for 1 hour at room temperature with an appropriate secondary antibody (1:250 peroxidase-conjugated anti-rabbit or anti-mouse IgG). Finally, the membranes were washed several more times, and bound antibodies were detected using the Enhanced Chemiluminescence System (ECL) for Western Blotting (34080; Thermo Scientific, Rockford, IL, USA) and exposure on autoradiographic film (Hyperfilm ECL; Amersham; or RP2 plus films; AGFA). The films were analyzed by densitometry using the Bio Rad Quantity One software program, version 4.6 (Umax Powerlook 2100XL Scanner; Bio Rad).
Statistical analysis
Each experimental group for neurological evalua-tion consisted of 12 animals, whereas the groups for histological and biochemical analysis consisted of 3 and 4 animals, respectively. The InfoStat program (version 2008) was used to perform tests for normality and homogeneity of the variances. Parametric data were analyzed using ANOVA and Tukey´s test as a post hoc test of multiple comparison between treatment means. For data with non-homogeneous variances, we used a Kruskal-Wallis nonparametric ANOVA test to determine any significant differences between independent groups. Analyses were carried out using the GraphPad Prism 5.0 software program. Differences with p values less than 0.05 were considered to be significant.
Results
ATV protects against motor impairment following cerebral ischemia.
A neurological test was developed for analyzing the recovery of animals treated with ATV following focal cerebral ischemia. We observed sensory and motor deterioration in the ischemic rats 6 hours after injury (2.7/6.0). Slight improvements were observed in the ATV-treated animals by 24 and 48 hours post-ischemia compared with the placebo group. However, by 72 hours post-ischemia, the ATV-treated animals had shown significant recovery (5.6/6.0) compared with the placebo group (4.2/6.0) (P <0.001) (figure 1A). This finding is consistent with previous results from our lab showing that ATV can mitigate the loss of NeuN and MAP2 staining within the cerebral cortex and hippocampus following cerebral ischemia (16,17). It has been demonstrated that focus areas suffer cell death, although is poorly studied exofocal areas that are represented in figure 1B, being the interest of the present study.
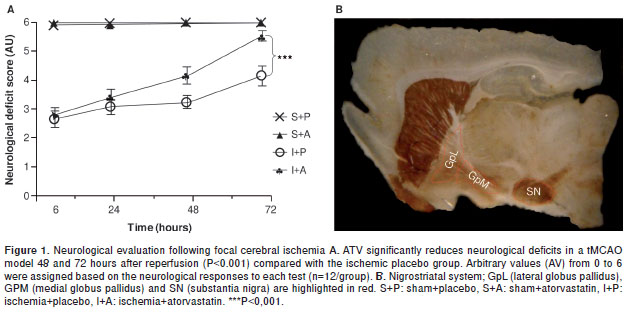
Ischemia-induced injuries near the MCA, such as cellular alterations in the GpM and increased immunoreactivity in the TH, can be reversed by ATV.
Tyrosine hydroxylase catalyzes the rate-limiting step in the synthesis of dopamine – an important neurotransmitter for motor control and reward – motivated behaviors (18) - and this enzyme has been shown to be modulated by ischemia. We did not observe any changes in TH levels in the GpL by immunohistochemistry (figure 2a-A-D, b). Although we did observe that Gp total lysates from ischemic animals had increased TH protein levels compared with controls (P=0.05); furthermore, we found that TH levels were reversed in the ischemic animals by the ATV-treatment similar to those in the controls (figure 2e). These biochemical findings were supported by histological experiments in the GpM that showed increased TH immunoreactivity and altered cytoarchitecture (figure 2a-G,c) in the untreated ischemic animals (P=0.001) compared with the sham+placebo (figure 2a-E,c) and sham+ ATV (figure 2a-F,c) groups. However, the ATV-treated ischemic animals showed decreased TH immunoreactivity (P=0.01) (figure 2a-H,c) and a cellular organization similar to the control animals.
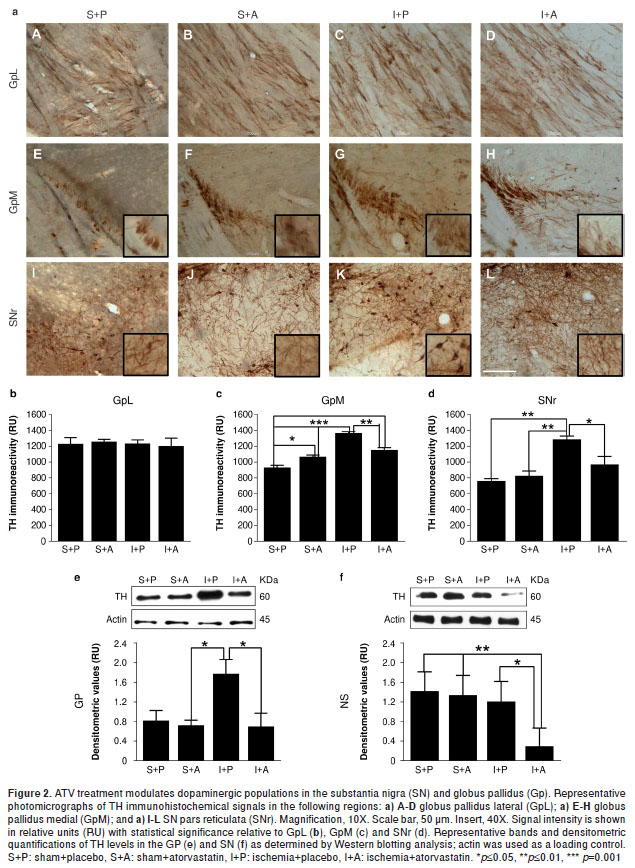
ATV decreases TH immunoreactivity in the SNr and promotes fiber arborization following ischemia in the area of the MCA.
We observed an increase in TH immunoreactivity in the SNr of ischemic animals (P=0.01) (figure 2a-K,d), whereas ATV-treated animals showed a significant decrease in TH staining (P=0.05). Furthermore, we found a more abundant fiber arborization pattern (figure 2a-L,d) compared with the sham-placebo control (figure 2a-I,d) and ATV control groups (figure 2a-J,d). These findings were consistent with TH protein levels in the SN total protein lysates as determined by Western blotting analysis, which showed that ATV treatment could significantly inhibit the upregulation of TH induced by ischemia (P=0.05) (figure 2f).
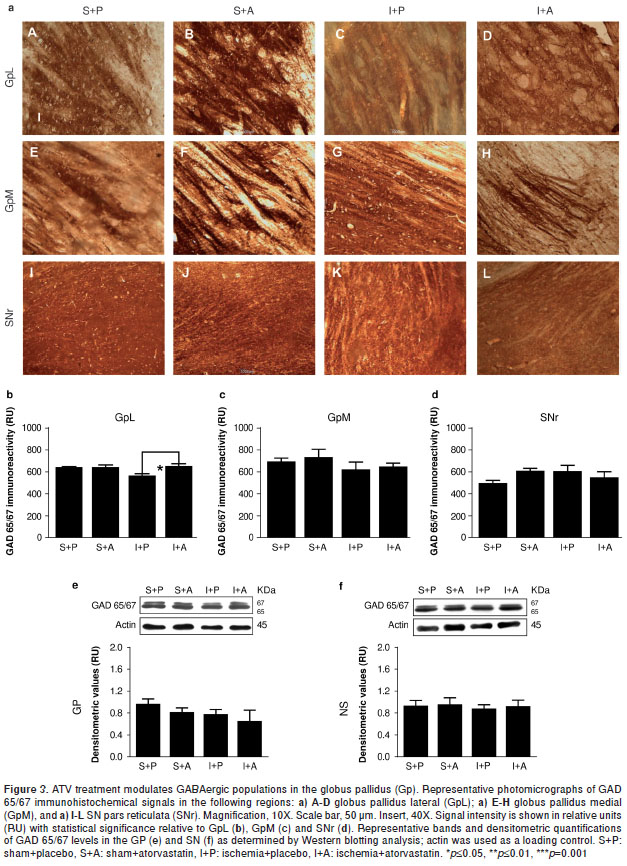
ATV rescues the loss of GAD 65/67 immunoreactivity in the GpL near the MCA following ischemia.
It is well known that glutamic acid decarboxylase (GAD) is one of the rate-limiting enzymes in GABA synthesis and that it can be used as a reliable marker for inhibitory interneurons (19). GAD65 plays an important role in the local control of GABA synthesis at synapses, whereas GAD67 is responsible for maintaining GABA at baseline levels for use as both a neurotransmitter and a metabolite (20). GAD 65/67 immunoreactivity was significantly decreased within the GpL by 78 hours post-injury (P =0.05) (figure 3a-C,b), a phenomenon that could be rescued with ATV treatment (P =0.05) (figure 3a-D,b) to levels similar to those observed in the controls (figure 3a-A-B,b). No changes were observed in the GpM (figure 3-E-H, c). We also note that no significant differences in GAD 65/67 protein levels could be detected between Gp total lysates from injured and control samples (figure 3e), although it is likely that such differences were lost when the GpL and GpM proteins were pooled. In addition, no changes in GAD 65/67 immunoreactivity were detected within the SNr by immunohistochemistry (figure 3a-I-L,d) or by Western blotting analysis (figure 3-f).
Discussion
Our findings demonstrate that the expression levels of specific enzymes involved in the synthesis of the neurotransmitters dopamine and GABA within certain exofocal brain regions, as well as in the nigrostriatal system, are affected by focal cerebral ischemia, and they also suggest that these changes can be reversed by post-ischemia ATV treatment. It appears that excitotoxic conditions resulted in increased immunoreactivity in TH-positive neurons within the GpM and SNr, as well as a decrease in the GABAergic cell population (GAD 65/67+) within the GpL. Strikingly, ATV treatment could successfully block the negative effects of this cellular environment, leading to an increase in the GABAergic neuronal population within the GpL and blocking the increases in TH immunoreactivity within the GpM and SNr. These observations are supported by previous reports where TH staining increased in the SNc and striatum in injured rats (21). Also, considering that SNr and SNc are functionally depending and these project to the striatum (22), could mean an up-regulation of dopaminergic neurotransmission in a tissue-specific manner at the nigrostriatal system depending of the brain injury. Also, these findings suggest that ATV helps restore equilibrium in tissues affected by ischemia and that ATV reduces the expression of TH in the dopaminergic neurons of the SNr while maintaining projections to the striatum. These results support a protective role for statins following stroke, and they are consistent with our previous description of the induction of survival pathways and synaptic connectivity under post-ischemic conditions by ATV (7,9). Furthermore, these data suggest that statins can block excitotoxic events in a tissue-specific manner. In particular, ATV may act to silence an increase in inhibitory signals coming from the GpL that control the excitatory efferents to the SNr, which are a product of exofocal phenomena following ischemia, generating a protective modulation of protein levels within the nigrostriatal system, which may be related to neurological recovery. It is important to emphasize that these types of neurological deficits caused by cerebral ischemia have been correlated with injury severity and size in experimental models (13,23,24).
Previous studies from our group and others demonstrated that ATV treatment (10 mg/kg) can significantly improve neurological scores at 48 and 72 hours post-ischemia (9). This was also found to be true for pravastatin (1 mg/kg) 5 days after the induction of cerebral ischemia in the vicinity of the MCA (8), as well as with other statins and neuroprotective agents (25,26).
In the present study, GAD 65/67 immunoreactivity was dramatically decreased in the GpL following focal ischemia, which was indicative of a loss in either the activity or number of GABAergic neurons within the GpL. This specific effect has not been reported before, although it has been reported that deficiencies in GABA production can induce neuronal death (19). Furthermore, changes in GAD 67 enzyme levels have been described in the cerebral cortex following ischemia; in particular, GAD 67 levels were shown to increase after 12 hours of reperfusion and then progressively decrease over the next 4 days (19). Nishino, et al., (27) reported a 50% decrease in pallidal GABA in ischemic animals compared with control animals, as determined using microdialysis analysis. It is important to emphasize that during an ischemic stroke, glutamatergic activity increases, which decreases the endogenous synthesis and release of GABA, leading to reduced GABAergic transmission. As glutamatergic and GABAergic transmissions have antagonistic effects, increases in GABAergic activity in an ischemic setting can compensate for the excess glutamatergic signaling, leading to decreased cell death; conversely, decreased GABAergic signaling in this setting promotes cell death (28,29). The findings in this study were also consistent with our previous observations of increased neuronal death and decreased Bcl-2 levels in the SNr following an ischemic event (20). In addition, our data show that post-ischemia ATV treatment increases GAD 65/67 immunoreactivity, which may suggest a potential mechanism for the observed neuroprotective effects by enhancing GABAergic transmission, which is consistent with other studies demonstrating that inducing certain inhibitory conditions can be used to control cell death following ischemia (28,30-33).
The observed reduction in TH immunoreactivity in the exofocal area of the GpM following ATV treatment has not been previously described. Existing data point to increased levels of dopa-mine during ischemia (34,35), particularly in the neostriatum (18,36-39). For example, substantial increases in dopamine levels in this brain region were reported following 10 minutes of arterial occlusion, which then returned to baseline levels after 60 minutes (40). However, other studies have reported that dopamine protects neurons against glutamate-induced excitotoxicity (37) and that dopamine levels are reduced following ischemia (38-40). In addition, it has been shown that some experimental conditions can lead to increased TH levels in the striatum (21), suggesting the existence of tissue-specific modulation. The observed increase in dopamine levels in the SNr following MCA ischemia was similar to the modulation observed in the GpM, which could be due to the structural proximity and similar functions of these two brain regions. It is important to stress that changes in dopamine production and release during ischemia are potentially harmful to cells. However, ATV treatment was able to restore basal dopamine levels, suggesting a specific function in dopamine synthesis, which is supported by the neurological responses and behavioral recovery (20,36). On the other hand, we also recorded increased TH immunostaining in the GpM of sham animals treated with ATV, but these changes were not accompanied by neurological or behavioral defects. It is possible that these changes were balanced out by the weak increase in GAD65/67 immunoreactivity observed in the GpL and GpM, suggesting that neurotransmission may actively stimulated by atorvastatin in these areas. It should be noted, however, that a recent report indicates that ATV has variable effects on membrane transporters in the liver (41). Taken together, the ATV-treated ischemic animals had better neurological recovery and reversed altered neurotransmission signals, indicating that ATV can exert neuroprotective effects in these areas, even in late ischemic injuries.
Finally, our findings suggest a relationship between behavioral deficits and the impairment of GABAergic and dopaminergic neuronal populations in specific exofocal regions. These regions are involved in processing sensorimotor, cognitive and affective information, which is consistent with the types of pathophysiological conditions that often occur following ischemia, such as depression (42,43), anxiety, cognitive disorders and other behavioral changes (44-46).
In conclusion, atorvastatin plays a neuroprotective role in specific exofocal areas affected by ische mia through the tissue-specific modulation of dopaminergic and GABAergic neuronal populations in the nigrostriatal system. These findings will be useful for developing novel preclinical therapies related to the pathophysiology of exofocal regions and help determine therapeutic uses for ATV following cerebral ischemia.
We would like to thank Catalina Lapuente from the Grupo de Investigación en Enfermedades Neurodegenerativas at the Universidad del Tolima for her valuable technical support in conducting this research. We also thank the Grupo de Modelos Experimentales para las Ciencias Zoohumanas and the Grupo de Neurociencias de Antioquia for their scientific and technical support during our experiments.
The authors certify that they have no conflicts of interest with the institution that financed this project or with any other public or private entity.
This research was funded by Colciencias (GP C-G, project #111545921467), the Universidad de Antioquia CODI and the Office of Research at the Universidad del Tolima , Colombia (110220).
Corresponding author: Ángel Enrique Céspedes, Grupo de Investigación en Enfermedades Neurodegenerativas, Laboratorio 33-L101, Departamento de Salud Animal, Facultad de Medicina Veterinaria, Universidad del Tolima, Barrio Santa Helena, Parte Alta, Ibagué, Colombia. Teléfono: (578) 277 1212, extensión 9220; fax: (578) 277 2042 aecesped@ut.edu.co, biomedicineresearch@yahoo.es
1. Bousser MG. Stroke prevention: An update. Front Med. 2012;6:22-34. http://dx.doi.org/10.1007/s11684-012-0178-6 [ Links ]
2. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al . Heart disease and stroke statistics –2010 update: A report from the American Heart Association. Circulation. 2010;121:e46-e215. http://dx.doi.org/10.1161/CIRCULATIONAHA.109.192667 [ Links ]
3. A rango C, Escobar M, Cardona GP, Pimienta H. Fisio -patología de la isquemia cerebral focal: aspectos básicos y proyección a la clínica. Neurología. 2004;39:156-65. [ Links ]
4. Kronenberg G, Balkaya M, Prinz V, Gertz K, Ji S, Kirste I, et al . Exofocal dopaminergic degeneration as antide-pressant target in mouse model of poststroke depression. Biol Psychiatry. 2012;72:273-81. http://dx.doi.org/10.1016/j.biopsych.2012.02.026 [ Links ]
5. Boyes J, Bolam JP. Localization of GABA receptors in the basal ganglia. Prog Brain Res. 2007;160:229-43. http://dx.doi.org/10.1016/S0079-6123(06)60013-76 [ Links ]
6. Purves D, Augustine G, Fitzpatrick D, Hall W, LaMantia A, McNara J, et al . Neuroscience. Third edition. Sunderland (MA): Sinauer Associates; 2004. p. 832. [ Links ]
7. Fisher M, Moonis M. Neuroprotective effects of statins: Evidence from preclinical and clinical studies. Curr Treat Options Cardiovasc Med. 2012;14:252-9. http://dx.doi.org/10.1007/s11936-012-0174-9 [ Links ]
8. Berger C, Xia F, Maurer MH, Schwab S. Neuroprotection by pravastatin in acute ischemic stroke in rats. Brain Res Rev. 2008;58:48-56. http://dx.doi.org/10.1016/j.brainresrev.2007.10.010 [ Links ]
9. Céspedes A, Wandosell F, Cardona G. p120 Catenin/N- catenin are molecular targets in the neuroprotection and neuronal plasticity mediated by atorvastatin after focal cerebral ischemia. J Neurosci Res. 2010;88:3621-34. http://dx.doi.org/10.1002/jnr.22511 [ Links ]
10. Paciaroni M, Hennerici M, Agnelli G, Bogousslavsky J. Statins and stroke prevention. Cerebrovasc Dis. 2007; 24:170-82. http://dx.doi.org/10.1586/erc.09.106 [ Links ]
11. Sacco RL, Liao JK. Drug insight: Statins and stroke. Nat Clin Pract Cardiovasc Med. 2005;2:576-84. [ Links ]
12. Bösel J, Gandor F, Harms C, Synowitz M, Harms U, Djoufack PC, et al . Neuroprotective effects of atorvastatin against glutamate-induced excitotoxicity in primary cortical neurones. J Neurochem. 2005;92:1386-98. http://dx.doi.org/10.1111/j.1471-4159.2004.02980 [ Links ]
13. Belayev L, Busto R, Zhao W, Fernández G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-L-lysine: Neurological and histological validation. Brain Res. 1999;833:181-90. http://dx.doi.org/10.1016/S0006-8993(99)01528-0 [ Links ]
14. Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472-6. http://dx.doi.org/10.1161/01.STR.17.3.472 [ Links ]
15. Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Fourth edition. San Diego: Academic Press; 1998. [ Links ]
16. Deguchi K, Takaishi M, Hayashi T, Oohira A, Nagotani S, Li F, et al . Expression of neurocan after transient middle cerebral artery occlusion in adult rat brain. Brain Res. 2005;1037:194-9. http://dx.doi.org/10.1016/j.brainres.2004.12.016 [ Links ]
17. Sehara Y, Hayashi T, Deguchi K, Nagotani S, Zhang H, Shoji M, et al . Distribution of inducible nitric oxide synthase and cell proliferation in rat brain after transient middle cerebral artery occlusion. Brain Res. 2006;1093:190-7. http://dx.doi.org/10.1016/j.brainres.2006.03.092 [ Links ]
18. Globus MY, Busto R, Dietrich WD, Martínez E, Valdés I, Ginsberg MD. Effect of ischemia on the in vivo release of striatal dopamine, glutamate, and gamma-aminobutyric acid studied by intracerebral microdialysis. J Neurochem. 1988;51:1455-64. [ Links ]
19. Hwang IK, Yoo KY, Li H, Park OK, Lee CH, Choi JH, et al . Transient increases of glutamic acid decarboxylase 67 immunoreactivity and its protein levels in the somatosensory cortex after transient cerebral ischemia in gerbils. J Vet Med Sci. 2008;70:1005-10. [ Links ]
20. Tsunekawa N, Yanagawa Y, Obata K. Development of GABAergic neurons from the ventricular zone in the superior colliculus of the mouse. Neurosci Res. 2005;51:243-51. http://dx.doi.org/10.1016/j.neures.2004.11.011 [ Links ]
21. O´Byrne MB, Bolam JP, Hanley JJ, Tipton KF. Tyrosine- hydroxylase immunoreactive cells in the rat striatum following treatment with MPP+. Adv Exp Med Biol. 2000;483:369-74. http://dx.doi.org/10.1007/0-306-46838-7_41 [ Links ]
22. Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20:91-127. http://dx.doi.org/10.1016/0165-0173(94)00007-C [ Links ]
23. Wakayama K, Shimamura M, Sata M, Sato N, Kawakami K, Fukuda H, et al . Quantitative measurement of neurological deficit after mild (30 min) transient middle cerebral artery occlusion in rats. Brain Res. 2007;1130:181-7. http://dx.doi.org/10.1016/S0022-510X(00)00268-9 [ Links ]
24. Reglodi D, Tamas A, Lengvari I. Examination of sensorimotor performance following middle cerebral artery occlusion in rats. Brain Res Bull. 2003;59:459-66. http://dx.doi.org/10.1016/S0361-9230(02)00962-0 [ Links ]
25. Xing Y, Hua Y, Keep RF, Xi G. Effects of deferoxamine on brain injury after transient focal cerebral ischemia in rats with hyperglycemia. Brain Res. 2009;1291:113-21. http://dx.doi.org/10.1016/j.brainres.2009.07.032 [ Links ]
26. Liu XF, Fawcett JR, Thorne RG, Frey WH. Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci Lett. 2001;308:91-4. http://dx.doi.org/10.1016/S0304-3940(01)01982-6 [ Links ]
27. Nishino H, Aihara N, Czurko A, Hashitani T, Isobe Y, Ichikawa O, et al . Reconstruction of GABAergic transmission and behavior by striatal cell grafts in rats with ischemic infarcts in the middle cerebral artery. J Neural Transplant Plast. 1993;4:147-55. http://dx.doi.org/10.1155/NP.1993.147 [ Links ]
28. Costa C, Leone G, Saulle E, Pisani F, Bernardi G, Calabresi P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke. 2004;35:596-600. http://dx.doi.org/10.1161/01.STR.0000113691.32026.06 [ Links ]
29. Pisani A, Bonsi P, Calabresi P. Calcium signaling and neuronal vulnerability to ischemia in the striatum. Cell Calcium. 2004;36:277-84. http://dx.doi.org/10.1016/j.ceca.2004.02.010 [ Links ]
30. Allen NJ, Rossi DJ, Attwell D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J Neurosci. 2004;24:3837-49. http://dx.doi.org/10.1523/JNEUROSCI.5539-03.2004 [ Links ]
31. Liang R, Pang ZP, Deng P, Xu ZC. Transient enhancement of inhibitory synaptic transmission in hippocampal CA1 pyramidal neurons after cerebral ischemia. Neuroscience. 2009;160:412-8. http://dx.doi.org/10.1016/j.neuroscience.2009.02.046 [ Links ]
32. Haberg A, Qu H, Saether O, Unsgard G, Haraldseth O, Sonnewald U. Differences in neurotransmitter synthesis and intermediary metabolism between glutamatergic and GABAergic neurons during 4 hours of middle cerebral artery occlusion in the rat: The role of astrocytes in neuronal survival. J Cereb Blood Flow Metab. 2001;21:1451-63. http://dx.doi.org/10.1097/00004647-200112000-00010 [ Links ]
33. Joh HD, Searles RV, Selmanoff M, Alkayed NJ, Koehler RC, Hurn PD, et al . Estradiol alters only GAD67 mRNA levels in ischemic rat brain with no consequent effects on GABA. J Cereb Blood Flow Metab. 2006;26:518-26. http://dx.doi.org/10.1038/sj.jcbfm.9600211 [ Links ]
34. Saulle E, Centonze D, Martin AB, Moratalla R, Bernardi G, Calabresi P. Endogenous dopamine amplifies ischemic long- term potentiation via D1 receptors. Stroke. 2002;33:2978-84. http://dx.doi.org/10.1161/01.STR.0000038093.42512.0F [ Links ]
35. Toner CC, Stamford JA. ´Real time´ measurement of dopamine release in an in vitro model of neostriatal ischaemia. J Neurosci Methods. 1996;67:133-40. http://dx. doi.org/10.1016/0165-0270(96)00030-1 [ Links ]
36. Inoue H, Ochi M, Shibata S, Watanabe S. Effects of transient forebrain ischemia on long-term enhancement of dopamine release in rat striatal slices. Brain Res. 1995;671:95-9. http://dx.doi.org/10.1016/0006-8993(94)01325-C. [ Links ]
37. Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633-43. [ Links ]
38. Slivka A, Brannan TS, Weinberger J, Knott PJ, Cohen G. Increase in extracellular dopamine in the striatum during cerebral ischemia: A study utilizing cerebral microdialysis. J Neurochem. 1988;50:1714-8. http://dx.doi.org/10.1111/j.1471-4159.1988.tb02468 [ Links ]
39. Zhang Y, Deng P, Ruan Y, Xu ZC. Dopamine D1 -like receptors depress excitatory synaptic transmissions in striatal neurons after transient forebrain ischemia. Stroke. 2008;39:2370-6 . http://dx.doi.org/10.1161/STROKEAHA.107.506824 [ Links ]
40. Furuwaka N, Arai N, Goshima Y, Miyamae T, Ohshima E, Fujita K, et al . Endogenously released DOPA is a causal factor for glutamate release and resultant delayed neuronal cell death by transient ischemia in rat striata. Neurochemistry. 2001;76:815-24. http://dx.doi.org/10.1046/j.1471-4159.2001.00068 [ Links ]
41. Bjorkhem-Bergman L, Bergstrom H, Johansson M, Parini P, Eriksson M, Rane A, et al. Atorvastatin treatment induces uptake and efflux transporters in human liver. Drug Metab Dispos. 2013;41:1610-5. http://dx.doi.org/10.1124/dmd.113.051698 [ Links ]
42. Angeleri F, Angeleri VA, Foschi N, Giaquinto S, Nolfe G. The influence of depression, social activity, and family stress on functional outcome after stroke. Stroke.1993;24:1478-83. http://dx.doi.org/10.1161/01.STR.24.10.1478 [ Links ]
43. Kotila M, Numminen H, Waltimo O, Kaste M. Depression after stroke: Results of the FINNSTROKE Study. Stroke. 1998;29:368-72. http://dx.doi.org/10.1191/0269215502cr487oa [ Links ]
44. Huh Y, Jung JW, Park C, Ryu JR, Shin CY, Kim WK, et al . Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neurosci Lett. 2003; 349:63-7. [ Links ]
45. Wang Q, Yan J, Chen X, Li J, Yang Y, Weng J, et al . Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol. 2011;230:27-34. http://dx.doi.org/10.1016/j.expneurol.2010.04.006 [ Links ]
46. Robinson RG, Starkstein SE. Current research in affective disorders following stroke. J Neuropsychiatry Clin Neurosci. 1990;2:1-14. [ Links ]
