










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.34 no.2 Bogotá abr./jun. 2014
https://doi.org/10.7705/biomedica.v34i2.1700
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1700
1 Grupo Malaria, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
2 Laboratorio de Infectología y Parasitología Molecular, Hospital Clementino Fraga Filho, Universidad Federal de Rio de Janeiro, Rio de Janeiro, Brasil
3 Unidad Multidisciplinaria de Genómica, Instituto de Biofísica Carlos Chagas Filho, Universidad Federal de Rio de Janeiro, Rio de Janeiro, Brasil
Author contributions:
Yesid Cuesta-Astroz maintained P. falciparum cultures, prepared protein extracts, carried out 2D electrophoresis and comparative analyses of 2D protein profiles and participated in the preparation of the manuscript.
Wanda Maria de Almeida von Krüger optimized the protocols for 2D electrophoresis and protein extracts preparation, revised and edited the manuscript.
Mariano Zalis provided the infrastructure for parasite cultivation and participated in the preparation of the manuscript.
Paulo Mascarello Bisch optimized the sample preparation protocol for mass spectrometry analysis.
Camila Nunes-Batista optimized the conditions to obtain highly synchronized parasite cultures.
César Segura conceived, designed and coordinated the study, analyzed the results and wrote the manuscript.
All authors read and approved the final manuscript.
Recibido: 11/06/13; aceptado: 05/02/14
Introduction: Despite efforts to control malaria, around 10% of the world population is at risk of acquiring this disease. Plasmodium falciparum accounts for the majority of severe cases and deaths. Malaria control programs have failed due to the therapeutic failure of first-line antimalarials and to parasite resistance. Thus, new and better therapeutic alternatives are required. Proteomic analysis allows determination of protein expression levels under drug pressure, leading to the identification of new therapeutic drug targets and their mechanisms of action.
Objective: The aim of this study was to analyze qualitatively the expression of P.falciparum trophozoite proteins (strain ITG2), after exposure to antimalarial drugs, through a proteomic approach.
Materials and methods: In vitro cultured synchronized parasites were treated with quinine, mefloquine and the natural antiplasmodial diosgenone. Protein extracts were prepared and analyzed by two-dimensional electrophoresis. The differentially expressed proteins were selected and identified by MALDI-TOF mass spectrometry.
Results: The following proteins were identified among those differentially expressed in the parasite in the presence of the drugs tested: enolase (PF10_0155), calcium-binding protein (PF11_0098), chaperonin (PFL0740c), the host cell invasion protein (PF10_0268) and proteins related to redox processes (MAL8P1.17). These findings are consistent with results of previous studies where the parasite was submitted to pressure with other antimalarial drugs.
Conclusion: The observed changes in the P. falciparum trophozoite protein profile induced by antimalarial drugs involved proteins mainly related to the general stress response.
Key words: Plasmodium falciparum , proteome, quinine, mefloquine.
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1700
Caracterización parcial del proteoma del trofozoíto de Plasmodium falciparum bajo tratamiento con quinina, mefloquina y el compuesto natural diosgenona
Introducción. A pesar de los esfuerzos para controlar la malaria, esta sigue siendo un problema de salud pública. Plasmodium falciparum es responsable de la mayoría de los casos graves y de las muertes. Los programas de control de la malaria han sido cuestionados debido al fracaso del tratamiento y a la resistencia del parásito a los antipalúdicos de primera línea, por lo que se requieren nuevas y mejores alternativas. El análisis proteómico permite identificar y determinar los niveles de expresión de las proteínas bajo la presión de los medicamentos, lo que posibilita la identificación de nuevos blancos terapéuticos y mecanismos de acción.
Objetivo. Analizar cualitativamente la expresión diferencial de proteínas del citosol del trofozoíto de P. falciparum bajo tratamiento con quinina, mefloquina y el compuesto natural diosgenona mediante una aproximación proteómica.
Materiales y métodos. Se trataron trofozoítos sincronizados y cultivados in vitro de P. falciparum (cepa ITG2) con quinina, mefloquina y el compuesto natural diosgenona. Los extractos proteicos se prepararon y analizaron por electroforesis bidimensional. Las proteínas con aparente expresión diferencial se seleccionaron e identificaron mediante espectrometría de masas MALDI-TOF.
Resultados. Se encontraron las siguientes proteínas diferencialmente expresadas en el trofozoíto: la enolasa (PF10_0155), la proteína de unión a calcio (PF11_0098), la chaperonina (PFL0740c), la proteína de invasión a la célula del huésped (PF10_0268) y la proteína relacionada con procesos de reducción y oxidación (redox) (MAL8P1.17). Estos hallazgos son congruentes con resultados previos de estudios en los que el parásito fue presionado con otros medicamentos antipalúdicos.
Conclusión. Los cambios observados en el perfil de proteínas del trofozoíto de P. falciparum tratado con antipalúdicos involucraron preferencialmente proteínas relacionadas con la respuesta al estrés general.
Palabras clave: Plasmodium falciparum , proteoma, quinina, mefloquina.
doi: http://dx.doi.org/10.7705/biomedica.v34i2.1700
Malaria in humans is caused by five species of Plasmodia: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale , Plasmodium malariae and Plasmodium knowlesi . The disease greatly compromises the health and socioeconomic development of communities living in tropical regions of the world. According to the World Health Organization in 2012 there were about 207 million cases of malaria worldwide and an estimated 627.000 deaths with mortality rates that have fallen by 45% globally since 2000 (1). Malaria control programs in South America have failed in part because of parasite resistance to antimalarial drugs. Present strategies include the search for new drugs, novel therapeutic targets and combination therapies, but their success has been limited.
The genome sequencing of P. falciparum , the description of the transcriptome of the intraerythrocytic developmental cycle, and the proteome analyses have contributed to a systemic understan ding of the disease (2,3). The quantitative analysis of protein levels allows studying the molecular profile of the parasite at different stages of its life cycle and under certain conditions such as drug pressure (3). Proteomic approaches have allowed the understanding of the molecular events occurring in the parasite in response to different antimalarial drugs and environments. However, some are based on different proteomic techniques with contrasting results (4-11).
The drug resistance mechanism to 4- and 8- aminoquinolines in P. falciparum is partially explained by the antimalarial drug efflux from the digestive vacuole and there are evidences of the involvement of transporters and proteases in the process. It is known that under pressure with chloroquine and other antimalarials, P. falciparum differentially expresses proteins related to metabolic processes suggesting that the therapy affects parasite survival and opening the possibility of identifying novel potential therapeutic targets (12).
The aim of this study was to characterize by two- dimensional electrophoresis, followed by MS/MS in tandem, the proteome of P. falciparum trophozoites treated in vitro with quinine, mefloquine and the natural compound diosgenone (Dna). Our working hypothesis was that under treatment with these drugs, P. falciparum modifies protein expression and, therefore, its protein profile. To achieve the objective it was necessary to adapt the parasite in vitro culture conditions to attain high parasitemia.
Materials and methods
Parasite culture and drug treatment
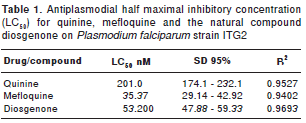
The P. falciparum strain ITG2 (chloroquine resistant) was used in drug treatments. Parasites were cultured in vitro under standard conditions (13), synchronized as described previously (14), with modifications (8) to render 500 m g of protein per 100 ml of culture. Parasites were maintained in human red blood cells type A+ in RPMI 1640 (Sigma, USA), supplemented with 10% human serum at 1% v/v hematocrit, in an atmosphere of 90% N 2 , 5% O 2 and 5% CO 2 , at 37°C. Gas mixture and medium were changed every 8 hours. The 50% inhibitory concentrations (IC 50 ) for quinine, mefloquine and the natural compound diosgenone were determined as described by Smilkstein M, et al ., (15) and calculated using the GraphPad Prism v5.0. For parasite drug treatments, trophozoites in synchronous cultures (18 hours) were treated for three hours with the drugs at their respective IC 50 . Before and after treatment, parasite synchrony and morphology were monitored by light microscopy of Giemsa-stained blood smears. When dead parasites appeared, the sample was discarded. Treated parasites were harvested as described below. All treatments were carried out in duplicate, in three different days, with their respective controls without drug.
Plasmodium falciparum protein extracts
Drug-treated and untreated parasites were collected by centrifugation at 600 g during 5 min at 4°C and the resulting pellet was further washed twice in PBS, pH 7.2, under the same condition. The washed pellet was suspended in 10mM Tris-HCl, 5% sorbitol, pH 7.0, supplemented with 0.15% saponin and protease inhibitors (Protease Inhibitor Cocktail Tablets, Roche), followed by incubation at 4 ° C for 10 min. The suspension was centrifuged at 800 g for 13 min at 4°C and the parasites were freeze-thawed 15 times in liquid nitrogen and in warm water bath at 37°C, respectively, for 5 min each. The lysate was clarified by centrifugation at 800 g for 30 min at 4°C. Proteins in the supernatant were precipitated by 2.5 volumes of acetone at -20°C overnight and then collected by centrifugation at 8,000 g for 30 minutes at 4°C. The supernatants were discarded whereas the pellet was dried at room temperature for 5 - 10 minutes. The resulting pellet was suspended in 9M urea solubilizing buffer, 4% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 1% ampholytes 3-10 (Invitrogen, USA), 40 mM DTT + protease inhibitors (Protease Inhibitor Cocktail Tablets, Roche) by vortexing. The protein suspension was sonicated for 10 cycles of 5 min at 4°C each, and clarified by centrifuging at 12,000 g for 30 min at 4°C. Protein content of extracts was quantified using the 2D Quant Kit (GE Healthcare). Protein integrity was checked in 12% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) (Mini-PROTEAN®, BioRad®), under standard conditions (running buffer Tris/glycine/SDS pH 8.2, at 100V for 60 min).
2D-gel electrophoresis of Plasmodium falciparum proteins
First dimension electrophoresis was performed on 7 cm isoelectrofocusing (IEF) strips, pH 4-7 (Invitrogen, USA), actively hydrated with protein extracts at 50 volts for 12 hours and then focused to accumulate 12,000 V-h at 20°C in an IPGPhor III (GE Healthcare) as described (5). Proteins focused on strips were reduced (DTT 1X NuPAGE LDS buffer, Invitrogen, USA) and alkylated (1X NuPAGE LDS buffer, iodoacetamide, Invitrogen, USA) for 20 min each. Second dimension electrophoresis was run on 4-12% NuPAGE Novex bis-Tris (Invitrogen, USA) and running buffer NuPAGE MES SDS (Invitrogen, USA) for 135 min at 100 V. Gels were silver stained (16).
To improve protein identification scores, gels were fluorescence stained using Flamingo Pink (BioRad®) by 2-hour incubation in 40% ethanol and 10% acetic acid, followed by 3 hours in 1X Flamingo Pink stock solution. Protein spots were visualized under UV light in a Gel Photo documentation system (BioRad®) and images processed using the Gel-Doc EZ System (BioRad®).
2D-gel analysis
Gels were scanned using Image Scanner III (GE Healthcare) and then analyzed and compared with the ImageMaster 2D Platinum software (GE Healthcare). Reference spots were selected (figure 1) and then protein spots in the control and test gels were compared (Gels vs. Match). Protein spots with p < 0.05 were considered differentially expressed. Image Scanner III (GE Healthcare) was previously calibrated as described (10).
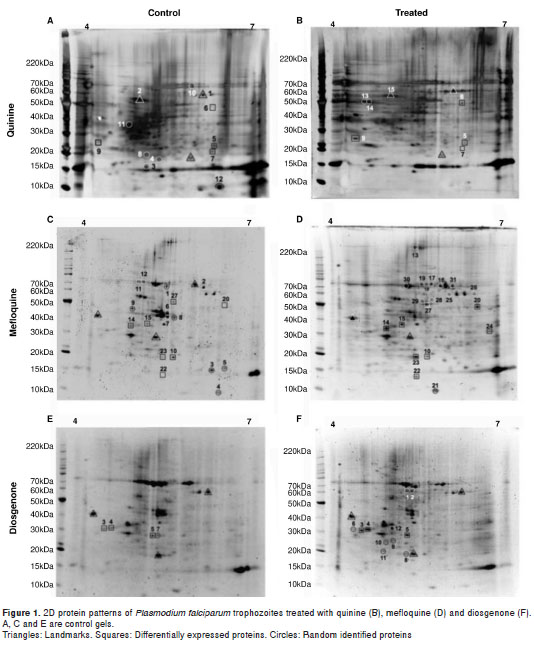
In-gel trypsinization of proteins and preparation of peptide samples for mass spectrometry (MS) analysis
Proteins in differential and reference spots from each gel were identified. Excised gel pieces of about 2 mm of diameter, containing spots, were suspended in 7% acetic acid and distained as described previously (16). Proteins were in-gel trypsinized with 10 ng/µl at 37°C overnight; the peptides were extracted and then suspended in 50% (v/v) methanol and 0.1% (v/v) acetic acid. Peptide containing samples (0.5-0.8 µl) were spotted on a MALDI plate and then mixed with 0.3–0.4 µl of saturated a -cyano-4-hydroxycinnamic acid (4-HCCA) in 50% ACN, and 10mM ammonium citrate. Plates were dried at room temperature.
Mass spectrometry
Protein samples were identified by mass spec trometry using an ABI 4800 MALDI TOF/TOF Analyzer (Applied Biosystems®, USA). MS spectra were executed in a Reflector Positive Ion Mode, mass range 700–4,000 Da and 1,250 total shots/spectrum. MS/MS spectra were executed in a Positive Ion Mode, 2 kV and 1,350 or less total shots/spectrum. The peptides with signal-to-noise ratio above 20 at the MS mode were selected for MS/MS run; a maximum of 45 MS/MS were allowed per spot. The precursor mass window was 200 relative resolution (fwhm).
MS data analysis
All spectra were searched against Rat taxonomy, NCBInr database, using GPS Explorer Software Version 3.6 (ABI) and Mascot (Matrix Science) search engine. Variable modifi34n2a10cations included oxidation (M) and acetylation (K). Mass tolerance was 80 ppm for precursor ions and 0.5 Da for fragment ions and two-missed cleavage were allowed. Identified peptides from P. falciparum proteins were confirmed in PlasmoDB (www.plasmodb.org), Uniprot (http://www.uniprot.org) and their access numbers obtained. Database access numbers for each protein were obtained from NCBI (www.ncbi.nlm.nih.gov). Protein ontology was obtained from the Protein Information Resource, PIR (http://pir.georgetown.edu).
Results
Antiplasmodial activity
Table 1 summarizes the antiplasmodial half maximal inhibitory concentration IC 50 (µM) for quinine, mefloquine and the natural compound diosgenone.
Proteins from quinine-treated parasites
The two-dimensional protein profile of trophozoites (18 hours post culture) treated at a 201 nM final concentration of quinine for 3 hours is shown in figure 1B. Proteins stained with Flamingo Pink were marked as follows: Differentially expressed (squares), landmarks (triangles) and randomly identified proteins (circles). The identified proteins are organized in table 2, and their corresponding peptide sequences are in table 3. Seven out of 15 proteins were identified as belonging to human red blood cells ( Homo sapiens ), as observed in other P. falciparum proteome analyses (7).
Proteins from mefloquine-treated parasites
The two-dimensional protein profile of trophozoites (18 hours post culture) treated with 35.4 nM mefloquine for 3 hours is shown in figure 1D. Proteins were stained with Flamingo Pink (BioRad®) and the spots in the gel were marked as follows: Differentially expressed (squares), landmarks (triangles) and others (arrows). The identified proteins are listed in table 4 and their corresponding peptide sequences in table 5. Eight out of 31 proteins were identified as belonging to the human red blood cells ( H. sapiens ) and 23 are parasite proteins (figure 1D, table 4).
Proteins from diosgenone-treated parasites
The two-dimensional protein profile of trophozoites (18 hours post culture) treated with 53.2 nM diosgenone for 3 hours is shown in figure 1F. Proteins in the gel were stained with Flamingo Pink (BioRad®).The identified proteins are listed in table 6 and the corresponding peptide sequences of those differentially expressed are shown in table 7. Three out of 12 were identified as belonging to human red blood cells ( H. sapiens ) and 9 were parasite proteins (figure 1F, table 6).
Discussion
We found very few proteins differentially expressed by the parasite under quinine, mefloquine and diosgenone treatments relative to the control at the resolution of the 2D-gel electrophoresis. In contrast to previous reports, we did not observe alterations in the levels of human proteins in the parasite extracts upon drug treatment (6,7,17). All three drugs affected the expression of parasite proteins involved in the stress response relative to the control. Quinine and diosgenone induced differential expression of the merozoite surface protein 7 precursor (PF13_0197) and the endoplasmic reticulum-resident calcium binding protein (PF11_0098) (tables 3 and 7). It is well known that aryl amino alcohols, such as quinine and mefloquine, interact with hemozoin and inhibit its crystallization in vitro (18). Interestingly, in the present work we showed that these drugs appar ently affect expression of parasite proteins that are not biochemically related to that process. Such was the case of the enolase (PF10_0155) (spot 6, figure 1B and spot 20, figure 1D), a highly active metalloenzyme during the intraerythrocytic stage (19) that mediates the conversion of 2-phosphoglycericacid into phosphoenolpyruvate in glycolysis, the main ener getic process of the intraerythrocytic form of the parasite (20). Enolase has been identified as an abundant protein in two-dimensional gels (6). The differential expression of enolase has been reported in the parasite under pressure with artemether-lumefatrine (6) and doxycycline (10). However, this was not observed when the parasite was treated with pyrimethamine (7). The diverse localization of P. falciparum enolase suggests that, apart from its catalytic activity, it might play other biological roles. For instance, enolase localized on the merozoite surface may be involved in red blood cell invasion, vacuolar enolase may be involved in food vacuole formation and/or development, while nuclear enolase may play a role in transcription (21). Therefore, it seems that many drugs can induce over-expression of enolase in the parasite.
Effect of quinine on Plasmodium falciparum protein profile
Quinine treated parasites produced a reduced amount of heat shock protein 70 (HSP-70) (Pf10875w) (spot 7, figure 1, A and B) relative to the untreated cells. This protein is localized in the endoplasmic reticulum (ER) at high levels in the exoerythrocytic stages and trophozoites, and it has also been shown that it is involved in the develop-ment and pathogenesis of malaria (22). In vitro P. falciparum produces 87% of its total protein as new proteins in 48 hours (12). Thus, Hsp70 might assist protein folding through the parasite life cycle. On the other hand, fevers caused by the parasite add a further stress to its protein-folding machinery (22).
Differential expression of heat shock proteins is a typical stress-response of the parasites to the toxic effects of drugs (i.e., quinine). However, variations in the expression level of heat shock proteins are needed to allow the parasite to adapt to environmental changes and for its maturation in the host. HSP-70 has been found upregulated in the parasite under treatment with artemether-lumefantrine (6) and an oxidized form of Hsp70 has also been found upon CQ treatment (8). Our observations, and those from other studies, clearly suggest that drugs induce differential expression of this particular HSP-70 in P. falciparum (Pf10875w).
It has been observed that antimalarials have contrasting effects on the expression of enolase (4,6,7). Glycolytic enzymes seem to have reduced expression in a chloroquine resistant strain of parasites treated with artemisinin, but over-expression when treated with lumefantrine (3 fold) (6). The apparent change in expression of enolase, also a glycolytic enzyme, induced by quinine here reported, is consistent with these previous findings (6). Quinine and lumefantrine belong to the same amino-alcohol group, whereas artemisinin is a sesquiterpenlactone. Thus, specific effects of drugs on the expression of aldolase seem to be apparent.
The parasite endoplasmic reticulum-resident calcium binding protein (PF11_0098) has been proposed to play a role in protein trafficking in the malaria parasite (23) . Here we showed that quinine and diosgenone increased the level of this protein (spot 9, figure 1B, and spot 3, figure 1F). Calcium storage is critical for parasite cell mobility and invasion (24) .Therefore, overexpression of the calcium binding protein (PF11_0098) might be an important strategy for the maintenance of calcium homeostasis in the parasite cells. Similar increase in the calcium binding protein level has also been observed in P . falciparum under the artemether/lumefantrine combination (6) and other experimental antiplasmodials (4). Thus, the increased expression of this protein in quinine and diosgenone treated cells suggests that they interfere with P . falciparum calcium homeostasis.
Interestingly, quinine downregulated and diosgenone upregulated the merozoite surface protein 7 (MSP7) (PF13_0197) (spot 5, figure 1, A and F), which is localized in the cytoplasm of trophozoites . MSP7, a peripheral membrane protein, is the precursor of the 22 kDa protein (MSP722) and of the 19 kDa protein (MSP719) found on the merozoite surface and non-covalently associated to the merozoite surface protein 1 (MSP1) c omplex that is shed upon erythrocyte invasion (25). MSP7 and its orthologous probably have a specific function either in the development of the merozoite or in parasite release from the erythrocyte, most likely via interaction with MSP1. MSP7 is translocated to the merozoite surface via interaction with MSP1 , suggesting a possible role in invasion (25) . This particular observation deserves further investigation. In the case of quinine, a recognized antimalarial drug, the downregulation of MSP7 might have effects on the parasite maturation and red blood cell release, on merozoite binding, as well as in invasion. In the case of diosgenone, the over-expression of MSP7 is difficult to frame in the light of its apoptotic activity in the parasite, but it might occur as part of the apoptotic effects induced by the drug (26). MSP-7 has been reported as differentially expressed in the trophozoite stage under different treatments (4,6).
Effect of mefloquine on Plasmodium falciparum protein profile
Mefloquine downregulated the expression of the eukaryotic translation initiation factor 5a, putative EIF-5A (PFL0210c) (spot 10, figure 1C and D) localized in the cytoplasm. Factor EIF-5A in P. falciparum is a highly conserved essential protein, which contains the unique amino acid hypusine (27). The aminoacid hypusine and its deoxyhypusine hydroxylase have been proposed as putative, novel antimalarial-drug targets (28). Factor EIF-5A has been identified in P. falciparum trophozoites (6), but it has not been previously reported as differentially expressed under pressure with antimalarials. Thus, it is of special interest to further study the effect of mefloquine on the parasite and its hypothetical target.
Circumsporozoite related antigen EXP-1 (PF11_ 0224) (spot 23 fig 1D) was upregulated by mefloquine. EXP-1 is a negatively charged integral protein located at the endoplasmic reticulum (ER) in the intraerythrocytic stages (29,30). EXP-1 has been identified by mass spectrometry (31) and found differentially expressed in chloroquine resistant/sensitive strains relative to the wild-type strain (17). It has been demonstrated that chloroquine significantly induces the expression of EXP-1 in resistant P. falciparum strains (K1 – Dd2), but downregulates it in sensitive ones (3D7 – HB3) (17). Findings here reported agree with the above observations. In our hands, mefloquine upregulated EXP-1 expression in the chloroquine resistant P. falciparum ITG2. EXP-1 has, in fact, been considered a chloroquine resistance marker (17) and we propose that it could also be used as a marker of mefloquine resistance. Further investigation is needed to establish the role of EXP-1 in the parasite and its relation with the chloroquine resistance phenotype and mefloquine treatment.
The merozoite capping protein 1 (MCP-1) (PF10_0268) (spot 14, figure 1D), a protein related to the entry into the host cell, was upregulated by mefloquine. MCP-1 is a highly conserved protein that is one of the components of the invasion machinery of the parasite. Plasmepsin III, a histo-aspartic protease (PF14_0078) (spot 15, figure 1D) also was upregulated by mefloquine in the parasite. Hemoglobin degradation is a metabolic process central to the growth and maturation of the malaria parasite P. falciparum . Plasmepsins (PMs) initiate hemoglobin degradation and have been identified and extensively characterized (26). Plasmepsin III is one of the 10 plasmepsins coded in the P. falciparum genome and the protein is located in the food vacuole. Thus, it has been proposed as a potential anti-malarial therapeutic target (32). Upregulation of this protein by mefloquine suggests increased hemoglobin degradation, which agrees with the known mode of action of the drug. This protein has also been found differentially expressed in the parasite treated with doxycycline, relative to the control (10).
The 10 kDa chaperonin (PFL0740c) (spot 22, figure 1D) was upregulated by mefloquine. This co-chaperonin localizes in the mitochondria of P. falciparum and, with chaperonin 60 (cpn60 or GroEL), an ATPase, assists the folding and assembly of proteins in the mitochondrial matrix (33). The apparent differential expression of 10 kDa chaperonin in response to mefloquine and its localization deserve further investigation. The differential expression of the chaperonin proteins under different drug treatments in P. falciparum has been reported previously (4,10).
The conserved Plasmodium protein of unknown function (PF14_0046) (spot 24, figure 1D ) was upregulated by mefloquine. This protein is localized in the endoplasmic reticulum (ER) and shares highly amino-acidic identity with a protein of unknown function from P. vivax (PVX _086190) (65%) and another from P. knowlesi (PKH_134200) (63%). This protein does not align with proteins from other parasites suggesting that it is unique to the genus Plasmodium . Our results suggest a possible role for PF14_0046 in the parasite response to mefloquine.
The disulfide isomerase (MAL8P1.17) (spot 27, figure 1D) is an enzyme of the endoplasmic reticulum and displays typical biochemical functions of disulphide isomerase, namely oxidase/isomerase, reductase and a chaperone-like activity. Pf52 contains domains of thiol: disulfide oxidoreductases, and it has been proposed as a potential parasite therapeutic target (33,34).
Effect of diosgenone on Plasmodium falciparum protein profile
Diosgenone, a triterpene of 30 carbons, induces parasites death in the intraerythrocytic stages (35). Treatment of P. falciparum trophozoites with diosgenone caused overexpression of the following proteins: The merozoite capping protein 1 (MCP-1) (PF10_0268) localized in the nucleus and related to the entry into the host cell; the endoplasmic reticulum-resident calcium binding protein (PF11_ 0098), and the merozoite surface protein 7 (MSP7) (P F13_0197) . Interestingly, expression of the latter two proteins appears to be controlled also by quinine, which caused upregulation of the endoplasmic reticulum-resident calcium binding protein and downregulation of the merozoite surface protein 7. This observation suggests that diosgenone, quinine and mefloquine regulate the expression of similar proteins in P. falciparum trophozoites. More sensitive techniques will be required to confirm this interesting observation.
Four out of five proteins upregulated by the drugs under study are localized in the endoplasmic reticulum suggesting increased protein packing and trafficking due to the synthesis of proteins for detoxification and oxidative stress. The upregulation of disulfide isomerase was evidenced by mefloquine treatment. It is expected that minor parasite proteins, differentially expressed in the presence of the antimalarial drugs, could also be identified if high-sensitivity techniques were used. In general, our results have shown that in the presence of the antimalarials quinine, mefloquine and the natural compound diosgenone, parasite proteins related to redox and energy metabolism, synthesis and transport had their expression levels altered.
Two-dimensional gel electrophoresis-based proteome of humans and rodents in different experimental settings revealed similar protein expression patterns (36). It has been reported that the glycolytic enzyme enolase is the most frequently identified among proteins expressed differentially in nearly every third experiment. Similarly, the heat-shock proteins 27 (HSP27) and 60 (HSP60) are also induced under many conditions and have been found expressed in about 30 percent of human and rodent samples (36). These findings suggest that they are typical cellular stress response proteins (33).
By using a proteomics approach, we detected changes in the P. falciparum protein expression profile, following treatment with quinine, mefloquine and diosgenone, relative to the control. A similar protein pattern emerged: Enolase, chaperonins, disulphide isomerases and the endoplasmic reticulum-resident calcium binding protein were the major differentially expressed proteins in P. falciparum treated cells under the experimental conditions used in this work. However, highly sensitive techniques such as SILAC (stable isotope labeling) and MudPIT (Multi-Dimensional Protein Identification) have made it possible to identify over 1,000 proteins in drug treated parasites in one experiment, from which approximately 50 were regulated relative to the control (9). About 40% of the identified and regulated proteins were of unknown functions (9), showing that a high number of P. falciparum proteins remain to be characterized for a better understanding of the mechanisms by which drugs affect the parasite physiology.
Authors thank the Systems-Proteomics Core Facility, University of North Carolina at Chapel Hill School of Medicine, for the mass spectrometry identification services.
The authors declare that they have no competing interests.
This work was supported by Grant 1115-408-20500 from the Departamento Administrativo de Ciencia, Tecnología e Innovación , Colciencias, Colombia, the Estrategia para la Sostenibilidad de los Grupos de Investigación , Universidad de Antioquia, 2013-2014, Medellín, Colombia, the Carlos Chagas Filho Institute of Biophysics, Federal University of Rio de Janeiro, Brazil, and the National Research Council (Cap), Brazil.
Corresponding author: César Segura, Grupo Malaria, Facultad de Medicina, Universidad de Antioquia, Carrera 62 N° 52-59, Medellín, Colombia Telephone: (574) 219 6490; fax: (574) 219 6487 cesar.segura@siu.udea.edu.co
1. World Health Organization. World Malaria Report 2013. Geneva: WHO; 2013. [ Links ]
2. Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum . Biol.2003;1:85-100. http://dx.doi.org/10.1371/journal.pbio.0000005 [ Links ]
3. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al . Genome sequence of the human malaria parasite Plasmodium falciparum . Nature. 2002;419:498-511. http://dx.doi.org/10.1038/nature01097 [ Links ]
4. Aly NS, Hiramoto A, Sanai H, Hiraoka O, Hiramoto K, Kataoka H, et al . Proteome analysis of new antimalarial endoperoxide against Plasmodium falciparum . Parasitol Res.2007;100:1119-24. http://dx.doi.org/10.1007/s00436-007-0460-8 [ Links ]
5. Gelhaus C, Fritsch J, Krause E, Leippe M. Fractionation and identification of proteins by 2-DE and MS: Towards a proteomic analysis of Plasmodium falciparum . Proteomics. 2005;5 : 4213-22. http://dx.doi.org/10.1002/pmic.200401285 [ Links ]
6. Makanga M, Bray PG, Horrocks P, Ward SA. Towards a proteomic definition of CoArtem action in Plasmodium falciparum malaria. Proteomics . 2005;5 : 1849-58. http://dx.doi.org/10.1002/pmic.200401076 [ Links ]
7. Nirmalan N, Sims PF, Hyde JE. Quantitative proteomics of the human malaria parasite Plasmodium falciparum and its application to studies of development and inhibition. Mol Microbiol.2004;52:1187-99. http://dx.doi.org/10.1111/j.1365-2958.2004.04049.x [ Links ]
8. Radfar A, Díez A, Bautista JM. Chloroquine mediates specific proteome oxidative damage across the erythrocytic cycle of resistant Plasmodium falciparum . Free Radic Biol Med. 2008;44:2034-42. http://dx.doi.org /10.1016/j.freeradbiomed. 2008.03.010 [ Links ]
9. Prieto JH, Koncarevic S, Park SK, Yates J 3rd, Becker K. Large-scale differential proteome analysis in Plasmodium falciparum under drug treatment. PLoS One. 2008;3:e4098. http://dx.doi.org/10.1371/journal.pone.0004098 [ Links ]
10. Briolant S, Almeras L, Belghazi M, Boucomont- Chapeaublanc E, Wurtz N, Fontaine A, et al . Plasmodium falciparum proteome changes in response to doxycycline treatment. Malar J. 2010;9:141. http://dx.doi.org/10.1186/1475-2875-9-141 [ Links ]
11. Le Roch K, Johnson J, Ahiboh H, Chung D, Prudhomme J, Plouffe D, et al . A systematic approach to understand the mechanism of action of the bisthiazolium compound T4 on the human malaria parasite, Plasmodium falciparum . BMC Genomics. 2008;9:513. http://dx.doi.org/10.1186/1471- 2164-9-513 [ Links ]
12. Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol. 2002;62:1364-72. http://dx.doi.org/10.1124/mol.62.6.1364 [ Links ]
13. Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673-5. http://dx.doi.org/10.1126/science.781840 [ Links ]
14. Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979; 65:418-20. [ Links ]
15. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803-6. http://dx.doi.org/10.1128/AAC.48.5.1803-1806.2004 [ Links ]
16. Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850-8. http://dx.doi.org/10.1021/ac950914h [ Links ]
17. Koncarevic S, Bogumil R, Becker K. SELDI-TOF-MS analysis of chloroquine resistant and sensitive Plasmodium falciparum strains. Proteomics.2007;7:711-21. http://dx.doi.org/10.1002/pmic.200600552 [ Links ]
18. Bray PG, Ward SA, O´Neill PM. Quinolines and artemisinin: Chemistry, biology and history. Curr Top Microbiol Immunol. 2005;295:3-38. http://dx.doi.org/10.1007/3-540-29088-5_1 [ Links ]
19. Roth EF, Calvin MC, Max-Audit I, Rosa J, Rosa R. The enzymes of the glycolytic pathway in erythrocytes infected with Plasmodium falciparum malaria parasites. Blood. 1988;72:1922-5. [ Links ]
20. Vivas L, Easton A, Kendrick H, Cameron A, Lavandera JL, Barros D, et al. Plasmodium falciparum : Stage specific effects of a selective inhibitor of lactate dehydrogenase. Exp Parasitol. 2005;111:105-14. http://dx.doi.org/10.1016/j.exppara.2005.06.007 [ Links ]
21. Bhowmick IP, Kumar N, Sharma S, Coppens I, Jarori GK. Plasmodium falciparum enolase: Stage-specific expression and sub-cellular localization. Malar J. 2009;8:179. http://dx.doi.org/10.1186/1475-2875-8-179 [ Links ]
22. Shonhai A, Boshoff A, Blatch GL. Plasmodium falciparum heat shock protein 70 is able to suppress the thermosensitivity of an Escherichia coli DnaK mutant strain. Mol Genet Genomics. 2005;274:70-8. http://dx.doi.org/10.1007/s00438-005-1150-9 [ Links ]
23. La Greca N, Hibbs AR, Riffkin C, Foley M, Tilley L. Identification of an endoplasmic reticulum-resident calcium- binding protein with multiple EF-hand motifs in asexual stages of Plasmodium falciparum . Mol Biochem Parasitol. 1997;89:283-93. http://dx.doi.org/10.1016/S0166-6851(97)00134-5. [ Links ]
24. Koch GL. The endoplasmic reticulum and calcium storage. Bioessays. 1990;12:527-31. http://dx.doi.org/10.1002/bies.950121105 [ Links ]
25. Pachebat JA, Ling IT, Grainger M, Trucco C, Howell S, Fernández-Reyes D, et al . The 22 kDa component of the protein complex on the surface of Plasmodium falciparum merozoites is derived from a larger precursor, merozoite surface protein 7. Mol Biochem Parasitol. 2001;117:83-9. http://dx.doi.org/10.1016/S0166-6851(01)00336-X [ Links ]
26. Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci USA. 2002;99 : 990-5. http://dx.doi.org/10.1073/pnas.022630099 [ Links ]
27. Molitor IM, Knobel S, Dang C, Spielmann T, Allera A, Konig GM. Translation initiation factor eIF-5A from Plasmodium falciparum . Mol Biochem Parasitol. 2004;137:65-74. http://dx.doi.org/10.1016/j.molbiopara.2004.04.013 [ Links ]
28. Kersher B, Nzukou E, Kaiser A. Assessment of deoxyhypusine hydroxylase as a putative, novel drug target. Amino Acids. 2010;38:471-7. http://dx.doi.org/10.1007/s00726-009-0406-9 [ Links ]
29. Ansorge I, Paprotka K, Bhakdi S, Lingelbach K. Permeabilization of the erythrocyte membrane with streptolysin O allows access to the vacuolar membrane of Plasmodium falciparum and a molecular analysis of membrane topology. Mol Biochem Parasitol. 1997;84:259-61. http://dx.doi.org/10.1016/S0166-6851(96)02806-X [ Links ]
30. Spielmann T, Gardiner DL, Beck HP, Trenholme KR, Kemp DJ. Organization of ETRAMPs and EXP-1 at the parasite-host cell interface of malaria parasites. Mol Microbiol. 2006;59:779-94. http://dx.doi.org/10.1111/j.1365-2958.2005.04983.x [ Links ]
31. Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520-6. http://dx.doi.org/10.1038/nature01107 [ Links ]
32. Ersmark K, Samuelsson B, Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med Res Rev. 2006;26:626-66. http://dx.doi.org/10.1002/med.20082 [ Links ]
33. Sato S, Wilson RJ. Organelle-specific cochaperonins in apicomplexan parasites. Mol Biochem Parasitol. 2005;141: 133-43. http://dx.doi.org/10.1016/j.molbiopara.2005.01.010 [ Links ]
34. Mouray E, Moutiez M, Girault S, Sergheraert C, Florent I, Grellier P. Biochemical properties and cellular localization of Plasmodium falciparum protein disulfide isomerase. Biochimie. 2007;89:337-46. http://dx.doi.org/10.1016/j.biochi.2006.11.001 [ Links ]
35. López ML, Vommaro R, Zalis M, de Souza W, Blair S, Segura C. Induction of cell death on Plasmodium falciparum asexual blood stages by Solanum nudum steroids. Parasitol Int. 2010;59:217-25. http://dx.doi.org/10.1016/j.parint.2010.02.002 [ Links ]
36. Petrak J, Ivanek R, Toman O, Cmejla R, Cmejlova J, Vyoral D, et al. Déjà vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics. 2008;8:1744-9. http://dx.doi.org/10.1002/pmic.200700919 [ Links ]
