










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.35 no.1 Bogotá jan./mar. 2015
https://doi.org/10.7705/biomedica.v35i1.2401
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v35i1.2401
1 Grupo de Inmunodeficiencias Primarias, Instituto de Investigaciones Médicas, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
2 Grupo de Inmunología Celular e Inmunogenética, Instituto de Investigaciones Médicas, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
3 Grupo de Epidemiología, Facultad Nacional de Salud Pública, Universidad de Antioquia, Medellín, Colombia
Contribución de los autores:
Alejandra Catalina Vélez y Diana María Castaño: revisión de las historias médicas de los pacientes y redacción del manuscrito. Ambas autoras contribuyeron en igual medida al trabajo.
Alejandra Catalina Vélez, Diana María Castaño y Marcela Moncada: pruebas de linfoproliferación, hipersensibilidad retardada y subpoblaciones de linfocitos
Julio César Orrego y José Luis Franco: diagnóstico y seguimiento de los pacientes
Rubén Darío Gómez: análisis estadístico de los resultados
José Luis Franco: dirección del proyecto de investigación y corrección del manuscrito
Recibido: 11/06/14; aceptado: 14/11/14
Introducción. La inmunodeficiencia común variable es un síndrome heterogéneo caracterizado por infecciones recurrentes, hipogammaglobulinemia y producción deficiente de anticuerpos específicos. Las anormalidades en subpoblaciones de linfocitos en sangre periférica, particularmente de linfocitos B, permiten la clasificación de los pacientes en grupos homogéneos.
Objetivo. Caracterizar clínica e inmunológicamente los linfocitos B y tipificar sus subpoblaciones en doce pacientes colombianos con inmunodeficiencia común variable, para clasificarlos en grupos homogéneos.
Materiales y métodos. Se revisaron las historias clínicas de los pacientes y se evaluaron las inmunoglobulinas séricas, la proliferación de linfocitos y la hipersensibilidad retardada, así como las subpoblaciones de linfocitos y de linfocitos B mediante citometría de flujo.
Resultados. Todos los pacientes presentaron infecciones respiratorias o gastrointestinales recurrentes y, algunos, infecciones en otros sistemas. Además, todos presentaban disminución de la IgG, en tanto que la IgA y la IgM fueron bajas en nueve y diez pacientes, respectivamente. En todos hubo disminución de la proliferación de linfocitos inducida por mitógenos, pero fue normal frente a antígenos específicos. La tipificación de subpoblaciones reveló valores elevados de linfocitos T en tres pacientes; siete presentaron disminución en la relación CD4+/CD8+ y, cuatro, linfocitos NK bajos. El conteo de linfocitos B fue normal en once pacientes, ocho de los cuales presentaron linfocitos B de memoria bajos, en tanto que cuatro presentaron aumento de linfocitos B de transición o de linfocitos B CD21 low .
Conclusión. La tipificación de subpoblaciones de linfocitos solo permitió asignar a 11 de los pacientes a grupos homogéneos según los esquemas de clasificación internacionales, lo que indica la necesidad de agregar más criterios hasta lograr una clasificación ideal. Este estudio permitirá establecer mejores seguimientos médicos para pacientes con inmunodeficiencia común variable en grupos con alto riesgo de desarrollar complicaciones clínicas.
Palabras clave: inmunodeficiencia variable común, inmunoglobulinas, proliferación de la célula, linfocitos B, células precursoras de linfocitos B, subgrupos linfocitarios, memoria inmunológica, citometría de flujo.
doi: http://dx.doi.org/10.7705/biomedica.v35i1.2401
Common variable immunodeficiency: Clinical and immunological characterization of patients and homogeneous subgroup definition by means of B lymphocyte subpopulation typing
Introduction: Common variable immunodeficiency is a heterogeneous syndrome characterized by recurrent infections, hypogammaglobulinemia and defective production of specific antibodies. Abnormalities in peripheral blood lymphocyte subpopulations, in particular of B lymphocytes, allow the classification of patients into homogeneous groups.
Objective: To perform a clinical and immunological characterization and to evaluate lymphocyte subpopulations of twelve Colombian patients with common variable immunodeficiency in order to define homogeneous groups.
Materials and methods: We reviewed medical records and evaluated serum immunoglobulins (Ig), lymphoproliferation, delayed hypersensitivity and used flow cytometry to quantify peripheral blood total lymphocyte and B cell populations.
Results: All patients had recurrent respiratory and/or gastrointestinal infections, while some also had infections affecting other systems. All patients had abnormally low serum IgG levels, while IgA and IgM levels were reduced in nine and ten patients, respectively. Lymphoproliferation to mitogen was lower in patients than in healthy controls but lymphoproliferation to specific antigen was normal in all. Flow cytometry revealed high numbers of T cells in three patients, while seven had a low CD4+/CD8+ ratio and four had reduced NK cells . Eleven patients had normal B cell counts, and eight of them also showed decreased memory B lymphocytes, and four had increased transitional or CD21 low B lymphocytes.
Conclusion: Lymphocyte typing allowed assigning all but one patient to homogeneous groups according to international classification schemes, indicating the necessity of including more criteria until an ideal classification is achieved. This study will lead to a better medical monitoring of common variable immunodeficiency patients in groups at high risk of developing clinical complications.
Key words: common variable immunodeficiency, immunoglobulins, cell proliferation, B-lymphocytes; precursor cells, B-lymphoid; lymphocyte subsets; immunologic memory; flow cytometry.
doi: http://dx.doi.org/10.7705/biomedica.v35i1.2401
La inmunodeficiencia común variable ( MIM#240500) es la inmunodeficiencia primaria sintomática más frecuente en humanos, con prevalencia de 1 por cada 10.000-50.000 nacidos vivos, y afecta por igual a ambos sexos, generalmente durante las tres primeras décadas de la vida (1,2). Mientras la mayoría de casos son esporádicos, 20 a 25 % tiene presentación familiar con patrones de herencia autosómica dominante o recesiva (1). Esta condi- ción se caracteriza por infecciones bacterianas recurrentes de las vías respiratorias superior e inferior y del tubo digestivo, mayor predisposición a manifestaciones de tipo autoinmunitario, inflamación crónica del tubo digestivo, lesiones granulomatosas del tipo de la sarcoidosis, y mayor incidencia de neoplasias hematológicas y cáncer (1,3,4).
El principal defecto inmunitario en la inmunodeficien cia común variable es la hipogammaglobulinemia con disminución de los niveles séricos de IgG, IgA y, ocasionalmente, de IgM (1). No obstante, se han descrito múltiples anormalidades inmunitarias tales como hipertrofia del tejido linfoide, esplenomegalia (1,3,4), respuesta linfoproliferativa e hipersensibilidad retardada normales o ausentes, ausencia de isohemaglutininas séricas y respuesta deficiente de los anticuerpos a las vacunas proteicas y de polisacáridos (5,6). Aunque en la mayoría de los pacientes el número total de linfocitos en sangre periférica suele ser normal o estar levemente disminuido, entre 5 y 10 % de ellos presentan linfocitos B CD19+ <1 % (7,8).
En la inmunodeficiencia común variable se han descrito múltiples alteraciones en la respuesta inmunitaria que comprometen la función, la activación y la diferenciación de los linfocitos (9-1 1). Específicamente en los linfocitos B, se han descrito defectos en el cambio de isotipo, la maduración de la afinidad, la generación de memoria y la diferenciación terminal de células plasmáticas, entre otros. De forma similar, en los linfocitos T se han descrito alteraciones en las subpoblaciones y la activación celular, la señalización del receptor del linfocito T, la producción de ciertas citocinas, mayor número de linfocitos T CD8+CD28- y aumento de su actividad supresora, menor número de los linfocitos T reguladores CD4+CD25hiFoxp3+ y disminución en la relación CD4+/CD8+ (<1). Más aún, se ha descrito aumento de la apoptosis en los linfocitos B y T, así como defectos en la expresión y función de algunas moléculas coestimuladoras, entre otros (9-11).
Toda esta variabilidad en los hallazgos clínicos y de laboratorio, reflejan una amplia heterogeneidad en estos pacientes (6), lo que ha dificultado la identificación de los defectos genéticos subyacen tes en este síndrome. De todas maneras, hasta ahora se han descrito mutaciones asociadas a la inmunodeficiencia común variable, en los siguientes genes: ICOS ( Inducible CO-Stimulator ) (12,13), TACI ( Transmembrane Activator and Calcium-modulating cyclophilin ligand –CAML– Interactor ) (14-18), CD19 (Artac H, Reisli I, Kara R, Adin Cinar S, Pico-Knijnenburg I, Pekcan S, et al . Consequences of heterozygous loss of CD19 expression. XIIIth Meeting of the European Society for Immunodeficiencies. Abstract #P135. Clin Exp Immunol. 2008;154(S1):95) (19-21), y BAFF-R ( B-cell activating factor receptor ) (22). Recientemente, se reportaron pacientes con cuadros clínicos similares a los de esta condición, con mutaciones en los genes CD81 , CD21 , CD20 , CD27 y LRBA ( Lipopolysaccharide Responsive Beige-like Anchor protein ) (23-27). Sin embargo, las mutaciones en estos genes explican menos del 15 % de los casos.
En el proceso de desarrollo, maduración, respuesta específica de antígeno y diferenciación terminal, los linfocitos B se caracterizan por la expresión de diversas moléculas de membrana que acompañan la expresión del receptor específico para el antígeno ( B-cell Receptor, BCR). En sangre periférica, los linfocitos B vírgenes convencionales son los CD19+CD10 low CD21-IgM++IgD+, los cuales, al comenzar a circular de nuevo por los tejidos linfoides (principalmente el bazo), adquieren un fenotipo intermedio (linfocitos B de transición, CD 19+CD21 low/+ IgM++IgD low/+ CD24++CD38 ++) antes de convertirse en linfocitos B maduros: CD19+ CD21+ IgM+IgD++ (28,29). Durante la respuesta específica de antígeno, los linfocitos B pueden diferenciarse en células plasmáticas de larga vida productoras de anticuerpos (inmunoglobulinas, Ig) o en linfocitos B de memoria que, en su mayoría, expresan el antígeno CD27 (30,31). De esta manera, los linfocitos B IgM+IgD+CD27- se consideran primordialmente vírgenes, mientras que los CD27+ se denominan ´de memoria´ y se subdividen en linfocitos B CD27+IgM+IgD+ (de tipo zona marginal que no hacen cambio de isotipo) y los linfocitos B CD27+IgM-IgD- (que hacen cambio de isotipo) (32).
En la inmunodeficiencia común variable se han descrito anormalidades en la distribución de las subpoblaciones de linfocitos B (33-37), lo que ha llevado a proponer su evaluación sistemática en sangre periférica mediante citometría de flujo, herramienta que permite una rápida tipificación y clasificación de los pacientes en subgrupos homogéneos y su correlación con el riesgo de ciertas complicaciones (33,38-40). En el 2008, en ocho centros europeos se evaluaron 303 pacientes con esta condición y se propuso una nueva clasificación (EUROclass), con base en las subpoblaciones de linfocitos B, inmaduros, de transición y de memoria (40).
Así, según el número total de linfocitos B, los pacientes se dividen en dos grupos: grupo B- , es decir, pacientes con <1 % de linfocitos B, y grupo B+, es decir, aquellos con >1 %. El grupo B+ se divide según el porcentaje de linfocitos B que han hecho cambio de isotipo: <2 % ( switched memory , smB-) o >2 % (smB+). A su vez, estos subgrupos se dividen según si el porcentaje de los linfocitos B CD21- es elevado (>10 %, grupos smB- 21 lo o smB+ 21 lo ), o si es normal (<10 %, grupos smB- 21 norm o smB+ 21 norm ); también, se pueden clasificar en smB- Tr hi , cuando hay aumento de los linfocitos B de transición (>9 %) o smB- Tr norm , cuando están en el nivel normal (<9 %). Este esquema de clasificación permite evaluar el riesgo de algunas complicaciones en la inmunodeficiencia común variable (por ejemplo, mayor incidencia de esplenomegalia y enfermedad granulomatosa en los smB-, smB-21 lo y smB+21 lo ) (40), y podría facilitar la identificación de los defectos genéticos y moleculares asociados a este síndrome.
El objetivo del presente estudio fue establecer las características clínicas e inmunológicas de doce pacientes colombianos con inmunodeficiencia común variable, así como la tipificación de subpo blaciones de linfocitos B en sangre periférica mediante citometría de flujo, con el fin de asignarlos a los diferentes grupos de acuerdo con la clasifica ción europea propuesta en el 2008.
Materiales y métodos
Tipo de estudio y población
En el presente estudio se describieron 12 pacientes con inmunodeficiencia común variable y 24 individuos sanos que participaron voluntariamente y de conformidad con las normas del Comité de Bioética de la Sede de Investigación Universitaria de la Universidad de Antioquia en Medellín, Colombia.
El diagnóstico de la inmunodeficiencia común variable se estableció con base en los criterios de la European Society for Immunodeficiencies (ESID) (www.esid.org) (41). Se excluyeron otras causas conocidas de hipogammaglobulinemia y de inmunodeficiencia secundaria en todos los pacientes (42). Además, en los pacientes 1 a 8 se descartaron previamente mutaciones en los genes ICOS y TACI mediante secuenciación del ADN genómico (13,16).
En el momento del estudio, todos los pacientes recibían tratamiento de reemplazo con gammaglobulina humana intravenosa (400-600 mg/kg al mes). Los controles incluyeron individuos sanos con edad y sexo similares a los de los pacientes y sin evidencia clínica o antecedentes de infección recurrente, alergia, enfermedad autoinmunitaria o cáncer. La mayoría de los pacientes y controles habían recibido la vacuna de DPT en la niñez y presentaban cicatriz por inmunización con BCG. Esta información se obtuvo de los registros de vacu nación o por el propio reporte de los participantes.
Los niveles de las inmunoglobulinas séricas en el momento del diagnóstico, y los datos clínicos y epidemiológicos obtenidos de las historias clínicas, incluyeron el sexo, la edad de inicio del síndrome, la edad de diagnóstico, el tratamiento, la historia de infecciones, el tipo de caso (esporádico o familiar), la clase y la localización de las infecciones, las enfermedades asociadas y la supervivencia desde el diagnóstico.
Aislamiento de células mononucleares de sangre periférica
La separación de las células mononucleares de sangre periférica se hizo a partir de sangre venosa periférica extraída en heparina con medio de separación de linfocitos (Sigma-Aldrich, St Louis, MO) a 500 g durante 30 minutos y a temperatura ambiente. Posteriormente, las células se lavaron con RPMI 1640 (Sigma) a 325 g durante 10 minutos a temperatura ambiente y se evaluó su viabilidad (>97 %) y su número mediante exclusión con azul de tripano (Sigma-Aldrich).
Respuesta de proliferación de las células mononucleares de sangre periférica inducida por mitógenos y antígenos específicos
Las células mononucleares de sangre periférica de pacientes y controles, se cultivaron por triplicado a 2 x 10 5 células por pozo en RPMI 1640 con suple mento de L-glutamina (Sigma-Aldrich) y 10 % de plasma autólogo (inactivado a 56 °C durante 60 minutos) en platos de cultivo de 96 pozos con fondo en "U" (BD Bioscience, San Diego, CA).
Para las estimulaciones, se empleó candidina en diluciones de 1:1000 y 1:2000 (Greer Laboratories, Lenoir, NC), toxoide tetánico en diluciones de 1:200 y 1:400 (Aventis Pasteur, Toronto, Canadá), o 5 µg/ml y 10 µg/ml de fitohemaglutinina ( phytohemagglutinin , PHA) (Sigma).
Las células se incubaron a 37 °C en atmósfera con 5 % de CO 2 y se pulsaron a las 72 horas (cultivos con PHA) o a las 120 horas (cultivos con candidina o toxoide tetánico) con 1 µCi por pozo de 3 [H]-timidina (MP Biomedicals Inc., Irvine, CA) y se incubaron durante 20 horas adicionales.
Finalmente, las células se recogieron en filtros de papel (Packard-PerkinElmer, Boston, MA) con un recolector de células (Inotech, Rockville, MD), se colocaron en platos de lectura (OmniFilter microplate, PerkinElmer, Boston, MA) y luego se agregaron 25 µl de cóctel de centelleo / vial (Ultima Gold, PerkinElmer), y se determinó la incorporación del isótopo radioactivo en un contador de centelleo (Plate Chameleon, Hidex, Finlandia).
Los resultados se presentan como índices de pro liferación correspondientes a la proliferación de las células estimuladas, comparada con la pro liferación observada en las células sin estímulo (cuentas por minuto [cpm] de células estimuladas/cpm de células sin estímulo).
Evaluación de la hipersensibilidad retardada
En la superficie anterior del antebrazo, se aplicó por vía intradérmica 0,1 ml de un extracto de Candida albicans en dilución de 1:10 (Greer Laboratories) o 5 unidades de derivado proteínico purificado de tuberculina ( Purified Protein Derivative , PPD, Mantoux-Tubersol, Aventis Pasteur, Toronto, Canadá). A las 72 horas se determinó el diámetro de la induración alrededor del sitio de aplicación y la prueba se consideró positiva cuando el área de induración fue de 5 mm o mayor.
Enriquecimiento de los linfocitos B
La sangre venosa periférica, anticoagulada con citrato de sodio, se centrifugó a 375 g durante 10 minutos a 20 °C para obtener la capa rica en leucocitos, la cual se diluyó en PBS 1X ( Dulbecco´s Phosphate Buffered Saline , DPBS, Sigma-Aldrich) con 0,6 % de citrato de sodio (Sigma-Aldrich) y 0,1 % de albúmina sérica humana (Sigma-Aldrich), y se separaron las células mononucleares de sangre periférica con medio de separación de linfocitos a 350 g durante 20 minutos a 20 °C. Las células se lavaron con PBS con 0,1 % de albúmina sérica humana y con 0,6 % de citrato de sodio a 200 g durante ocho minutos a 4 °C, y se estableció su número y viabilidad (>98 %).
Diez millones de células mononucleares de sangre periférica se resuspendieron en PBS con 0,1 % de albúmina sérica humana y con 20 µl de la mezcla de anticuerpos (de ratón) del kit de selección negativa (anti-CD2, CD3, CD7, CD14, CD16, CD56 y CD235a, Dynal Biotech, Brown Deer, WI), se incubaron durante 20 minutos a 4 °C y luego se lavaron con PBS con 0,1 % de albúmina sérica humana a 500 g durante ocho minutos a 4 °C y se mezclaron con 1 ml de una dilución de 1:10 de esferas magnéticas (Depletion Dynalbeads, Dynal Biotech) cubiertas con anticuerpos IgG4 humanos específicos para la porción Fc de la IgG de ratón.
Posteriormente, las células se incubaron durante 15 minutos a 2-8 °C y 25 rpm en un mezclador-rotador (RKDynal, Dynal Biotech), para después exponerlas a una columna magnética concentra dora de partículas (Dynal MPC-L, Dynal Biotech) durante dos minutos. Se extrajo el sobrenadante rico en linfocitos B y se repitió la incubación con una nueva suspensión de esferas magnéticas para incrementar la pureza.
Por último, se determinaron la viabilidad (>97 %) y el número de linfocitos B por exclusión con azul de tripano, y se evaluó la eficiencia de la selección mediante citometría de flujo para linfocitos B CD19+ (pureza >85 %).
Determinación de la subpoblación total de linfocitos en sangre periférica o de linfocitos B enriquecidos
A partir de sangre venosa periférica anticoagulada con ácido etilendiamino-tetraacético (EDTA ), se tomaron 100 µl y se agregaron 20 µl de cada uno de los siguientes anticuerpos monoclonales mar cados con fluorocromos o sus isotipos respectivos: CD3, CD4, CD8, CD16, CD19, CD21, CD24, CD27, CD38, CD56, IgD e IgM (BD Biosciences o eBioscience Inc., San Diego, CA). Las células se incubaron con los anticuerpos durante 20 minutos a temperatura ambiente en la oscuridad y después con solución de lisis de glóbulos rojos ( BD Bioscience) durante 10 minutos a temperatura ambiente. Posteriormente, las células se lavaron dos veces con PBS y se fijaron en solución de paraformaldehído al 2 % (Sigma-Aldrich). Cuando las tinciones se hicieron directamente en linfocitos B enriquecidos, estos se volvieron a suspender en PBS 1X con 0,1 % de azida de sodio (Sigma- Aldrich) y 1 % de SBFI (suero bovino fetal inac tivado) (BioWhittaker®, Walkersville, MD) y pH de 7,4, se incubaron con los anticuerpos de interés durante 20 minutos a temperatura ambiente y, posteriormente, se lavaron y se fijaron en solución de paraformaldehído al 2 %.
La lectura se hizo en un citómetro de flujo equipado con un láser de argón de 488 nm (FACScan, BD Bioscience). Cada ensayo incluyó los con troles de isotipo respectivos (IgG1-FITC, IgG1-PE, IgG1-PE-Cy5, BD Bioscience). El análisis de las subpoblaciones se hizo con el programa FlowJo, versiones 7.8 y 8.0 para Macintosh (Tree Star, Inc., Ashland, OR).
Análisis estadísticos
Los resultados de las variables continuas (clínicas) se presentan en forma de frecuencias absolutas y relativas. Para el análisis de los resultados de proliferación de linfocitos, se compararon los grupos mediante la prueba de Mann-Whitney y, para el análisis de la hipersensibilidad retardada, se aplicó la prueba exacta de Fisher, siendo significativo un valor de p<0,05 en ambos casos. Además, a las subpoblaciones de linfocitos B de los controles se les determinó la media ± 2 DE (desviación estándar). Los análisis estadísticos se hicieron mediante los programas GraphPad Prism, versión 5.00 (GraphPad Software Inc., San Diego, CA), e Epi Info 6.04 (Centers for Disease Control and Prevention, Atlanta, GA).
Resultados
Hallazgos clínicos
En los doce pacientes estudiados (7 mujeres y 5 hombres) con diagnóstico clínico de inmuno-deficiencia común variable, se observaron dos intervalos de inicio de la enfermedad: en la infancia (3 a 14 años) y en la adultez (18 a 71 años). En cuanto a posible segregación familiar, los pacientes 7 y 8 eran primos hermanos, mientras que el paciente 12 era hijo de padres consanguíneos (primos terceros) y tuvo una hermana con antecedentes de hipogammaglobulinemia y enfermedad diarreica que murió a los tres años de edad.
En todos los pacientes se documentaron infecciones recurrentes del tubo digestivo (particularmente enfermedad diarreica) o de las vías respiratorias superior e inferior (cuadro 1). Nueve pacientes tenían historia de infecciones en otros sistemas (ojos, piel, vías urinarias y médula ósea), en dos se documentaron manifestaciones de tipo autoinmunitario (artritis y hepatitis autoinmune), tres tenían diagnóstico de alergia (rinitis y asma) y uno presentó enteropatía asociada al gluten. La paciente 2 murió 12 años después del diagnóstico, debido a un adenocarcinoma metastásico. La tasa de supervivencia para esta cohorte de pacientes desde el momento del diagnóstico, fue de 11/12.
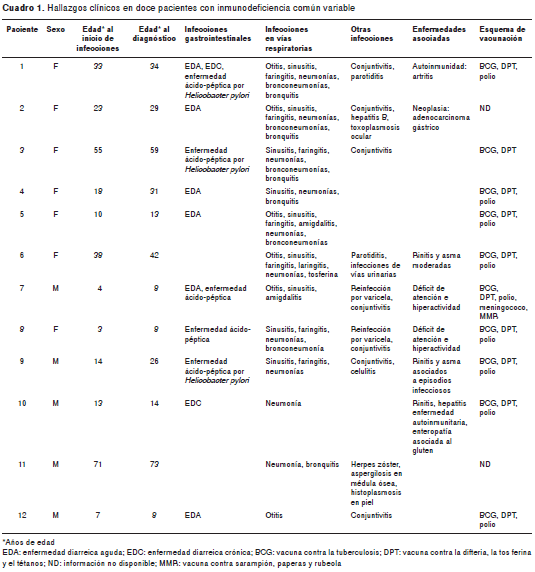
Inmunoglobulinas séricas
Una de las manifestaciones cardinales de la inmunodeficiencia común variable es la hipo-gammaglobulinemia y en esta cohorte todos los pacientes presentaron IgG sérica con <2 DE de los valores normales para la edad, mientras que en nueve pacientes se detectó disminución significativa de la IgA y la IgM. Los pacientes 6 y 10 presentaron valores normales de IgA e IgM, y el paciente 7 presentó valores normales de IgA pero disminución de la IgM (cuadro 2).
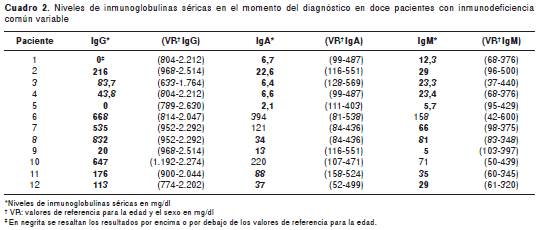
Evaluación de la inmunidad celular
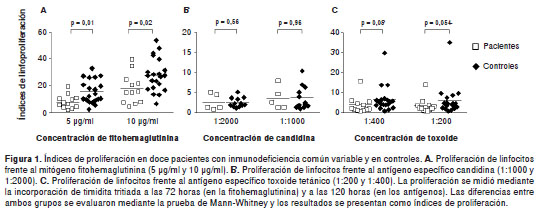
En la inmunodeficiencia común variable se han documentado respuestas de hipersensibilidad retardada que van desde normales a ausentes (1,33). Tales respuestas frente a la candidina y la prueba de PPD, solo se evaluaron en los pacientes 1 a 5. Estos cinco pacientes presentaron una respuesta de hipersensibilidad retardada negativa a la candidina (0/5), mientras que la proporción de reacciones positivas entre los sujetos sanos fue significativamente mayor (8/14, p=0,007). Además, la hipersensibilidad retardada en la prueba de PPD, solo fue positiva en la paciente número 3.
En pacientes con este tipo de inmunodeficiencia se han documentado respuestas variables de proliferación de los linfocitos T frente a mitógenos y antígenos específicos (4). Los mitógenos, como la fitohemaglutinina, pueden inducir la activación y proliferación de ´clonas´ de linfocitos T sin importar la especificad de su receptor de antígeno ( T cell receptor , TCR), por lo que pueden proporcionar información global de la capacidad de prolifera ción de los linfocitos T. En este trabajo se evaluó la proliferación de linfocitos en respuesta a la fitohemaglutinina (figura 1A) y al toxoide tetánico (figura 1C) en los doce pacientes, mientras que la respuesta a la candidina solo se evaluó en los pacientes 1 a 5 (figura 1B). Se encontró que la proliferación de linfocitos en respuesta a la fitohemaglutinina fue significativamente menor en los pacientes que en los controles con las dos concentraciones evaluadas (índices de prolife ración 49 y 37 % más bajos para 5 y 10 µg/ml, respectivamente). Por el contrario, no se observaron diferencias estadísticamente significativas en los índices de proliferación con los antígenos específicos (candidina y toxoide tetánico).
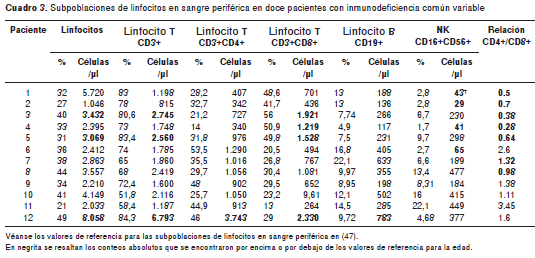
Subpoblaciones de linfocitos totales en sangre periférica
Se han reportado alteraciones variables en la frecuencia y el número absoluto de linfocitos T, linfocitos B y células NK ( natural killer ), en sangre periférica de pacientes con la inmunodeficiencia común variable (43-46), por lo tanto, nosotros evaluamos estas subpoblaciones en todos los pacientes. En el paciente 12, se encontró linfocitosis leve con aumento de los linfocitos T CD4+ y CD8+ y los linfocitos B (CD19+) (cuadro 3). En los pacientes 3 y 5, la linfocitosis fue a expensas de los linfocitos T CD8+, aunque el paciente 4 también presentó un aumento de los linfocitos T CD8+, pero, sin hallazgo de linfocitosis. La alteración más común en todos fue la disminución en la relación CD4+/CD8+ (en siete pacientes). En cuanto a los linfocitos B, con excepción del paciente 12, todos presentaron conteos totales que estaban dentro del rango normal para la edad. Los niveles de los linfocitos NK CD3-CD16+CD56+ se encontraron disminuidos en cuatro pacientes: 1, 2, 4 y 6.
Linfocitos B de memoria
El esquema usado para la identificación de las distintas subpoblaciones de linfocitos B (CD19+) está ilustrado en la figura 2. Los linfocitos B de memoria humanos se identifican mediante la expre sión en superficie del marcador CD27 (30,31), y en combinación con la IgD y la IgM de superficie permiten definir tres subpoblaciones adicionales que incluyen linfocitos vírgenes ( CD27-IgD+) , de memoria que no han hecho cambio de isotipo (CD27+IgD+) y de memoria que han hecho cambio de isotipo (CD27+IgD-) (32) (figura 2C).
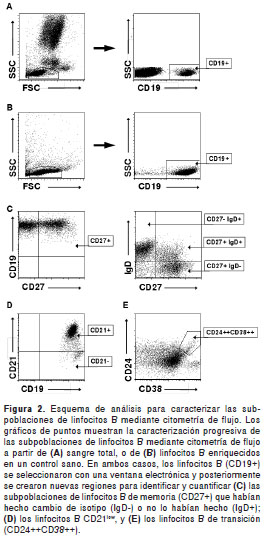
En esta inmunodeficiencia se han descrito alteraciones en los linfocitos B CD27+ , así como anormalidades en el número y porcentaje de linfocitos B con cambio de isotipo o sin él (33,48); por lo tanto, se evaluaron las subpoblaciones de linfocitos B de memoria en los doce pacientes y se compararon los resultados con once controles sanos y con valores de referencia (33,38,40).
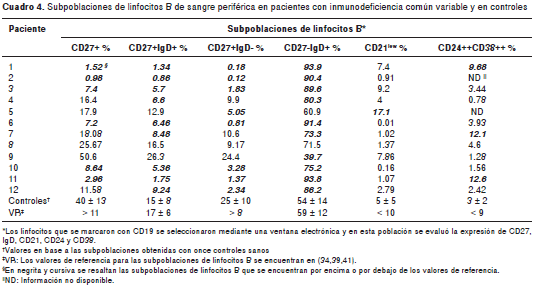
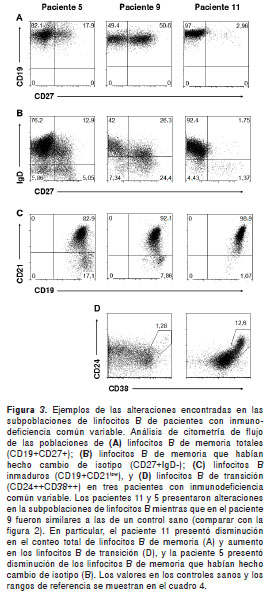
Como se ilustra en el cuadro 4, seis de los pacientes tenían porcentajes bajos de linfocitos B de memoria (CD27+) (véase paciente 11 en la figura 3A); en particular, los niveles de linfocitos B de memoria del paciente 2 estaban casi ausentes (<1 %) y la mayoría de los linfocitos B de memoria de los pacientes 3 y 6 no habían hecho cambio de isotipo ( IgD+). De los que presentaron porcentajes normales de linfocitos B de memoria, los pacientes 4, 7 y 12 tenían disminución en los linfocitos B de memoria sin cambio de isotipo (IgD+), el paciente 5 presentó menor frecuencia de aquellos con cambio de isotipo (IgD-, figura 3B) y el paciente 10 presentó disminución en ambos. Los pacientes 8 y 9 presentaron porcentajes similares a los controles (figura 3B). La expresión de IgM fue similar a la expresión de IgD en los linfocitos B CD27+ y CD27- de todos los controles y los pacientes evaluados (datos no mostrados).
Linfocitos B CD21 low y de transición en sangre periférica
Algunos pacientes con inmunodeficiencia común variable suelen presentar un aumento significativo de linfocitos B CD21 low y linfocitos B de transición (CD24++CD38++) (33,37,40). Se evaluó la expre sión de los marcadores de superficie CD21, CD24 y CD38 en sangre periférica de pacientes y controles sanos mediante citometría de flujo (figuras 2D y 2E).
Como se muestra en el cuadro 4, la mayoría de los pacientes exhibieron porcentajes normales de linfocitos B CD21 low (<10 %), mientras que la paciente 5 (con disminución en los linfocitos B de memoria que hicieron cambio de isotipo), tuvo un aumento significativo de linfocitos B CD21 low (17,1 %) (figura 3C). Además, tres pacientes, el 1, el 7 y el 11, presentaron aumento en los porcentajes de linfocitos B de transición (>9 %) (figura 3D); resultó interesante observar que dos de estos pacientes tenían porcentajes normales de linfocitos de memoria (pacientes 7 y 11), mientras que en uno de ellos estaban casi ausentes (paciente 1).
Clasificación de pacientes en grupos homogéneos según las subpoblaciones de linfocitos B
Según la clasificación de EUROclass de pacientes con inmunodeficiencia común variable (41), los pacientes 1 y 11 pertenecían al grupo smB- 21 norm Tr hi (CD27+IgD- <2 % y linfocitos B de transición >9 %); los pacientes 3 y 6, al grupo smB- 21 norm Tr norm (linfocitos B CD27+IgD- <2 % y linfocitos B de transición <9 %), y la paciente 2 podría haberse clasificado en cualquiera de estos dos grupos, ya que se desconocía el estado de sus linfocitos B de transición.
Seis pacientes se clasificaron en el grupo smB + (linfocitos B CD27+IgD+ >2 %): la paciente 5 en el subgrupo smB+ 21 lo (linfocitos B CD21 low >10 %) y los pacientes 4, 8, 9, 10 y 12 , al subgrupo smB+ 21 norm (linfocitos B CD21 low <10 %).
No fue posible asignar el paciente 7 a ninguno de los grupos propuestos, ya que mostró un porcentaje elevado de linfocitos B de transición, pero no presentó alteraciones en los porcentajes de linfocitos B que habían hecho cambio de isotipo.
Discusión
La inmunodeficiencia común variable es una inmunodeficiencia primaria con manifestaciones clínicas e inmunológicas heterogéneas. De forma similar a lo reportado previamente, todos los pacientes en este estudio presentaron infecciones recurrentes como las descritas en otras cohortes (1). Además, se documentaron otras manifestaciones, como autoinmunidad, alergia, enteropatía y adenocarcinoma, que también son de alta incidencia en este tipo de inmunodeficiencia.
Por otro lado, en esta cohorte, la capacidad de proliferación de los linfocitos T in vitro en respuesta a la fitohemaglutinina, fue significativamente menor que en los controles sanos, y esta disminución podría explicarse por el aumento en la apoptosis de los linfocitos T y la menor producción de inter-leucina 2 que se han reportado previamente en la inmunodeficiencia común variable (43).
Por el contrario, la reacción a los antígenos específicos parece conservarse. Esta diferencia en la proliferación de linfocitos T como reacción a la fitohemaglutinina y a los antígenos específicos, puede ser el resultado del aumento preferencial de linfocitos T de memoria central y la disminución de linfocitos T efectores causada por la fitohema glutinina (49). Por lo tanto, es posible que la fitohemaglutinina ponga en evidencia un defecto en la generación de estas células de memoria central en los pacientes con inmunodeficiencia común variable, en tanto que en la respuesta a antígenos específicos se espera una mayor contribución de las células con memoria efectora, las cuales parecen responder normalmente en estos pacientes.
Aunque la proliferación de los linfocitos T en respuesta a antígenos específicos in vitro fue normal, la hipersensibilidad retardada a la candi dina y al PPD fueron negativas en la mayoría de los pacientes. Sin embargo, se debe tener en cuenta que, debido a que la hipersensibilidad retardada se evaluó únicamente en cinco de los pacientes, es posible que esta tendencia en los resultados cambie si la prueba se realiza en un número mayor de pacientes. No obstante, se ha documentado que la respuesta de proliferación de linfocitos frente a antígenos en la inmunodeficiencia común variable puede situarse en rangos entre normales y disminuidos, sin que exista una correlación directa con el grado de la respuesta de hipersensibilidad retardada (4,33,50), por lo que el significado biológico o clínico de estos hallazgos no es claro.
Es interesante anotar que los resultados concernientes a la hipersensibilidad retardada en este estudio, sugieren que algunas anormalidades en la actividad de los linfocitos T podrían ser evidentes bajo las condiciones que modulan las respuestas de proliferación in vivo (microambiente, presencia de ciertas células, citocinas y accesibilidad de los linfocitos T de memoria al antígeno, entre otras). Una posible explicación de esta respuesta dife rencial podría encontrarse en un estudio llevado a cabo en un modelo de ratón, en el cual se demostró que la ausencia de anticuerpos circulantes supri me la fase efectora inicial de las respuestas de hipersensibilidad retardada a antígenos específicos (51). Esto sugiere que este tipo de reacción dependería, además, de mecanismos efectores que posiblemente involucran anticuerpos (o el complemento).
Se han descrito diversas alteraciones en las sub poblaciones de linfocitos en sangre periférica de pacientes con inmunodeficiencia común variable. En algunos se ha documentado específicamente un aumento significativo del porcentaje de linfocitos T CD8+CD28- (52,53), los cuales se asocian a funciones reguladoras negativas de la respuesta inmunitaria (54); también, se ha demostrado una disminución en el número total de los linfocitos T CD4+ (44) que puede resultar de defectos intrín secos de los linfocitos T, tales como defectos en la señalización (55-58) o aumento de la apoptosis (59).
En siete de los pacientes de este estudio, se esta bleció una disminución de la relación CD4+/CD8+ que, en algunos casos, se debió al aumento en el número total de linfocitos T CD8+ . Aunque actualmente no existe evidencia experimental que explique satisfactoriamente la aparente expansión de los linfocitos T CD8+ en sangre periférica, ni la alteración de la relación CD4+/CD8+ en la inmunodeficiencia común variable, es posible que la evaluación detallada de la función de las sub-poblaciones de linfocitos T y B en el contexto de la cooperación T-B in vitro , permita identificar diferencias en la habilidad que tienen para activarse y expresar diferentes moléculas coestimuladoras, lo que podría explicar el defecto inmunológico de estos pacientes. Además, en cuatro pacientes observamos una reducción en el número total de los linfocitos NK, un hallazgo común en la inmu nodeficiencia común variable, la cual, según se postula, resultaría de una permanencia prolongada de estas células en el intestino de algunos pacientes, lo que , a su vez, reduciría su frecuencia en la circulación periférica (45).
Varias investigaciones sobre la inmunodeficiencia común variable se han orientado a clasificar pacientes en grupos homogéneos para tratar de determinar defectos moleculares que expliquen la hipogammaglobulinemia y, en consecuencia, la gran frecuencia de infecciones recurrentes. Warnatz, et al. (34), y Piqueras, et al. (38), hicieron las primeras clasificaciones, y, posteriormente, un consorcio de varios centros europeos publicó una nueva clasificación ( EUROclass trial ) (40).
En esta cohorte de pacientes fue posible identificar anormalidades en las subpoblaciones de linfocitos B en sangre periférica (cuadro 4) y agrupar a diez de ellos en grupos homogéneos según la clasificación propuesta por el consorcio europeo (cuadro 5). Warnatz y EUROclass clasifican los grupos según los defectos en el porcentaje de linfocitos B de memoria que han hecho cambio de isotipo, y es difícil establecer si el defecto está en todos los linfocitos B de memoria o solo en el proceso de cambio de isotipo.

Según la clasificación de EUROclass, los pacientes con bajo porcentaje de linfocitos B, p robablemente poseen defectos graves en la diferenciación temprana de dichas células; los defectos del grupo con linfocitos B que han hecho cambio de isotipo, ocurren durante la diferenciación en los centros germinales, en tanto que, en el grupo con subpoblaciones normales, están aquellos con problemas en la diferenciación terminal de los linfocitos B hacia células plasmáticas. Por el contrario, Piqueras agrupa a los pacientes según el porcentaje de linfocitos B de memoria y diferencia el porcentaje que ha hecho cambio de isotipo, lo cual facilita distinguir en qué fase de la diferenciación de los linfocitos B está el defecto celular; sin embargo, no agrupa a aquellos pacientes con ausencia de linfocitos B CD19+.
Por su parte, Warnatz y EUROclass incluyen el porcentaje de linfocitos B CD21 low , subpoblación que comparte características con los linfocitos B senescentes (defectos en la proliferación y bajas respuestas de calcio al estimular el BCR, pero también, con linfocitos B de memoria periférica (expresión de moléculas coestimuladoras y mayor producción de IgM en los linfocitos B estimulados) (60). Se ha encontrado que los pacientes con expansión de los linfocitos B CD21 low presentan menor porcentaje de linfocitos T reguladores CD4+CD25 hi Foxp3+ en sangre periférica, lo que podría explicar la alta incidencia de autoinmu nidad y esplenomegalia en este grupo (61). La de EUROclass es la primera clasificación que considera el porcentaje de linfocitos B de transición. Sin embargo, todavía se desconocen las alteraciones que podrían explicar la expansión de los linfocitos B CD21 low o de transición.
En todas estas clasificaciones se establecieron correlaciones clínicas con algunos subgrupos de pacientes. En este estudio, solo dos pacientes pre sentaron autoinmunidad o neoplasia, ambos con niveles bajos de linfocitos B de memoria que habían hecho cambio de isotipo y con linfocitos B CD21 low normales (smB- 21 norm ), y uno de ellos con aumento de los linfocitos B de transición (Tr hi ), lo cual es similar a las correlaciones reportadas en el estudio EUROclass. Sin embargo, debido al tamaño de esta muestra y a la baja incidencia de manifesta ciones tales como la enfermedad granulomatosa y la hepatoesplenomegalia, no fue posible establecer una correlación estadística entre los defectos en las subpoblaciones de linfocitos B y el riesgo de desarrollar complicaciones como las descritas.
A diferencia de la clasificación de EUROclass, que identificó los linfocitos B de transición según la expresión de CD38 e IgM ( CD38++IgM++), en el presente estudio se evaluó la expresión de los marcadores CD24 y CD38, alternativa que también se ha utilizado para la identificación de esta subpoblación (62,63). En esta cohorte no fue posible clasificar al paciente 7 en ninguno de los grupos establecidos por EUROclass. Sin embargo, en el momento del estudio, el paciente tenía 10 años, y se ha reportado que los porcentajes de linfocitos B de transición son elevados en la infan cia (representan entre el 30 y el 50 % en sangre periférica durante el primer año), y disminuyen con la edad hasta alcanzar niveles similares a los del adulto a partir de los 14 años (63). EUROclass no establece cortes variables según la edad, a pesar de haber incluido pacientes desde los 10 años de edad. Por ello se propuso un sistema de clasificación diferente para pacientes entre los 2 y los 19 años, en el cual solo se evalúa el porcentaje de linfocitos B de memoria CD19+CD27+IgM- y que encontró mayor incidencia de sepsis, enfermedad granulomatosa y autoinmunidad en pacientes con menos de 5 células/ml (64). Según esta clasificación, el paciente 7 haría parte del grupo con menor riesgo de desarrollar este tipo de complicaciones al tener valores normales de linfocitos B de memoria CD19+CD27+IgM- (>5 células/ml).
Aunque se acepta que para evaluar los linfocitos B de memoria que han hecho cambio de isotipo no se requiere el uso concomitante de IgM e IgD, y que la expresión de IgD es suficiente, en este estudio se sugiere incluir la evaluación de ambos marcadores. En este contexto, se demostró previamente que pacientes con síndrome de hiperinmunoglobulinemia IgM de tipo II causado por mutaciones en el gen AID (HIGM-II, MIM#605258), presentan un aumento significativo de linfocitos B CD27+IgM+IgD- (65), por lo cual es necesario descartar mutaciones en el gen AID en pacientes con inmunodeficiencia común variable que presentan aumento en esta subpoblación. Se subraya la importancia de la valo ración tanto de IgM como de IgD en el estudio de las subpoblaciones de linfocitos B, ya que orientaría mejor el estudio del defecto molecular .
Los valores obtenidos en las subpoblaciones de linfocitos B en el presente estudio, confirman las observaciones previas que demuestran la existencia de grupos diferentes de pacientes cuyas alteraciones en este compartimiento celular pare cen sugerir diferentes defectos en la maduración, la diferenciación y el cambio de isotipo. Por ello, la clasificación de EUROclass se ha adoptado como herramienta de rutina para pacientes con diagnóstico de inmunodeficiencia común variable en el Grupo de Inmunodeficiencias Primarias de la Universidad de Antioquia. Es posible que sea necesario incluir un grupo adicional en las clasificaciones actuales para aquellos que presentan elevación en los linfocitos B de transición, con niveles normales de linfocitos B de memoria (como el paciente 7), pero sería necesario evaluar cohortes de mayor tamaño para determinar la prevalencia de este grupo en adolescentes y adultos con inmunodeficiencia común variable. Además, sería importante evaluar la presencia de linfocitos B de memoria que no expresan CD27 en pacientes con esta inmunodeficiencia, ya que dicha subpoblación está prácticamente ausente en individuos sanos pero aumenta en pacientes con enfermedades autoinmunitarias (66).
La importancia de esta clasificación radica, no solo en que permite establecer grupos homogéneos de pacientes, sino también en que permite determinar las complicaciones asociadas a defectos inmunitarios específicos. Con respecto a esto, se han identificado pacientes con inmunodeficiencia común variable afectados por neumonías recurrentes, bronquiectasias y daño pulmonar (evidenciado en tomografía computadorizada), y reducción significativa en los linfocitos B CD27+ IgM+, lo que se correlaciona significativamente con una producción baja de los anticuerpos antipolisacáridos del neumococo de tipo IgM (67).
En resumen, los resultados de este estudio respaldan hallazgos previos que proponen la tipificación de subpoblaciones de linfocitos B en sangre periférica como una herramienta importante para la identificación de grupos homogéneos de pacientes con inmunodeficiencia común variable que tiene valor pronóstico en cuanto al desarrollo de complicaciones y facilita la caracterización del defecto genético y molecular que subyace en estos pacientes. Además, la caracterización de otras anormalidades inmunitarias (relativas, por ejemplo, a subpoblaciones de linfocitos T), tiene el potencial de mejorar los esquemas de clasificación actuales con el objetivo de lograr un esquema ideal.
A Carlos J. Montoya, Margarita Olivares, Javier E. Toro, Jorge Botero, Juan C. López, Mauricio Arias, Sara Robledo, Wildeman Zapata, Sebastián Muñoz, Luisa Fernanda Cardona y Diego E. Góngora, por su colaboración, disponibilidad y asesoría técnica y científica, así como a Klaus Warnatz y Bodo Grimbacher, por las discusiones e ideas para el desarrollo de este trabajo.
Los autores del presente artículo manifestamos que no tenemos ningún conflicto de intereses con respecto a la información aquí suministrada.
Este trabajo fue financiado por el Instituto Colom biano para el Desarrollo de la Ciencia y la Tecnología (Colciencias, Proyectos 1115-04-11924 y 1115- 05-16874), por el Comité para el Desarrollo de la Investigación (CODI) de la Universidad de Antioquia, y por la Fundación Diana García de Olarte para las Inmunodeficiencias Primarias (FIP).
Correspondencia: José Luis Franco, Sede de Investigación Universitaria, Universidad de Antioquia, Calle 62 N° 52-59, laboratorio 530, Medellín, Colombia
Teléfono: (574) 219 6474; fax: (574) 219 1065 jlfrancor@une.net.co
1. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood. 2008;112:277-86. http://dx.doi.org/10.1182/blood-2007-11-124545 [ Links ]
2. Gathmann B, Binder N, Ehl S, Kindle G, ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: Update 2011. Clin Exp Immunol. 2012;167:479-91. http://dx.doi. org/10.1111/j.1365-2249.2011.04542.x [ Links ]
3. Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709-27. http://dx.doi.org/10.1111/j.1365-2141.2009.07669.x [ Links ]
4. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34-48. http://dx.doi. org/10.1006/clim.1999.4725 [ Links ]
5. Sorensen RU, Moore C. Antibody deficiency syndromes. Pediatr Clin North Am. 2000;47:1225-52. http://dx.doi.org/10.1016/S0031-3955(05)70269-8 [ Links ]
6. Ballow M. Primary immunodeficiency disorders: Antibody deficiency. J Allergy Clin Immunol. 2002;109:581-91. http://dx.doi.org/10.1067/mai.2002.122466 [ Links ]
7. Pandolfi F, Paganelli R, Cafaro A, Oliva A, Giovannetti A, Scala E, et al. Abnormalities of lymphocyte subpopulations in CVI do not correlate with increased production of IL-6. Immunodeficiency. 1993;4:19-23. [ Links ]
8. Farrant J, Spickett G, Matamoros N, Copas D, Hernández M, North M, et al . Study of B and T cell phenotypes in blood from patients with common variable immunodeficiency (CVID). Immunodeficiency. 1994;5:159-69. [ Links ]
9. Yong PF, Thaventhiran JE, Grimbacher B. "A rose is a rose is a rose," but CVID is Not CVID: Common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47-107. http://dx.doi.org/10.1016/B978-0-12-385991-4.00002-7 [ Links ]
10. Park JH, Resnick ES, Cunningham-Rundles C. Pers pectives on common variable immune deficiency. Ann N Y Acad Sci. 2011;1246:41-9. http://dx.doi.org/10.1111/j.1749-6632.2011.06338.x [ Links ]
11. Salzer U, Unger S, Warnatz K. Common variable immuno deficiency (CVID): Exploring the multiple dimensions of a heterogeneous disease. Ann N Y Acad Sci. 2012;1250:41-9. http://dx.doi.org/10.1111/j.1749-6632.2011.06377.x [ Links ]
12. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al . Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261-8. http://dx.doi.org/10.1038/ni902 [ Links ]
13. Salzer U, Maul-Pavicic A, Cunningham-Rundles C, Urschel S, Belohradsky BH, Litzman J, et al . ICOS deficiency in patients with common variable immuno-deficiency. Clin Immunol. 2004;113:234-40. http://dx.doi.org/10.1016/j.clim.2004.07.002 [ Links ]
14. Salzer U, Chapel HM, Webster AD, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al . Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005; 37:820-8. http://dx.doi.org/10.1038/ng1600 [ Links ]
15. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al . TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005; 37:829-34. http://dx.doi.org/10.1038/ng1601 [ Links ]
16. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarstrom Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967-76. http://dx.doi.org/10.1182/blood-2008-02-141937 [ Links ]
17. Pan-Hammarström Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429-30. http://dx.doi.org/10.1038/ng0407-429 [ Links ]
18. Almejún MB, Sajaroff E, Galicchio M, Oleastro M, Bernasconi A, Zelazko M, et al . Immunological characteristics and two novel mutations in TACI in a cohort of 28 pediatric patients with common variable immuno- deficiency. J Clin Immunol. 2012;32:89-97. http://dx.doi.org/10.1007/s10875-011-9613-8 [ Links ]
19. van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, et al. An antibody- deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901-12. http://dx.doi.org/10.1056/NEJMoa051568 [ Links ]
20. Kanegane H, Agematsu K, Futatani T, Sira MM, Suga K, Sekiguchi T, et al . Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun. 2007;8:663-70. http://dx.doi.org/10.1038/sj.gene.6364431 [ Links ]
21. van Zelm MC, Smet J, van der Burg M, Ferster A, Le PQ, Schandené L, et al. Antibody deficiency due to a missense mutation in CD19 demonstrates the importance of the conserved tryptophan 41 in immunoglobulin superfamily domain formation. Hum Mol Genet. 2011;20:1854-63. http://dx.doi.org/10.1093/hmg/ddr068 [ Links ]
22. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA. 2009;106:13945-50. http://dx.doi.org/10.1073/pnas.0903543106 [ Links ]
23. van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, et al . CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265-74. http://dx.doi.org/10.1172/JCI39748 [ Links ]
24. Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, et al . Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801-910. http://dx.doi.org/10.1016/ j.jaci.2011. 09.027 [ Links ]
25. Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214-22. http://dx.doi.org/10.1172/JCI40231 [ Links ]
26. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al . CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129:787-93. http://dx.doi.org/10.1016/j.jaci. 2011.11.013 [ Links ]
27. López-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al . Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986-1001. http://dx.doi.org/10.1016/j.ajhg.2012.04.015 [ Links ]
28. Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. 2005;105:4390-8. http://dx.doi.org/10.1182/blood-2004-11-4284 [ Links ]
29. Rolink AG, Andersson J, Melchers F. Molecular mechanisms guiding late stages of B-cell development. Immunol Rev. 2004;197:41-50. http://dx.doi.org/10.1111/j.0105-2896.2004.0101.x [ Links ]
30. Maurer D, Holter W, Majdic O, Fischer GF, Knapp W. CD27 expression by a distinct subpopulation of human B lymphocytes. Eur J Immunol. 1990;20:2679-84. http://dx. doi.org/10.1002/eji.1830201223 [ Links ]
31. Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. 1998;188:1691- 703. http://dx.doi.org/10.1084/jem.188.9.1691 [ Links ]
32. Agematsu K, Nagumo H, Yang FC, Nakazawa T, Fukushima K, Ito S, et al. B cell subpopulations separated by CD27 and crucial collaboration of CD27+ B cells and helper T cells in immunoglobulin production. Eur J Immunol. 1997;27:2073-9. http://dx.doi.org/10.1002/eji.1830270835 [ Links ]
33. Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: A new approach to classify a heterogeneous disease. Blood. 2002;99:1544-51. http://dx.doi.org/10.1182/blood.V99.5.1544 [ Links ]
34. Bonhomme D, Hammarström L, Webster D, Chapel H, Hermine O, Le Deist F, et al. Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol. 2000;165:4725-30. http://dx.doi.org/10.4049/jimmunol.165. 8.4725 [ Links ]
35. Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30:2516-20. http://dx.doi.org/10.1002/1521-4141(200009)30:9<2516::AID-IMMU2516>3.0.CO;2-Z [ Links ]
36. Jacquot S, Maçon-Lemaître L, Paris E, Kobata T, Tanaka Y, Morimoto C, et al . B cell co-receptors regulating T cell-dependent antibody production in common variable immunodeficiency: CD27 pathway defects identify subsets of severely immuno-compromised patients. Int Immunol. 2001;13:871-6. http://dx.doi.org/10.1093/intimm/13.7.871 [ Links ]
37. Cuss AK, Avery DT, Cannons JL, Yu LJ, Nichols KE, Shaw PJ, et al . Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. 2006;176:1506-16. http://dx.doi.org/10.4049/jimmunol.176.3.1506 [ Links ]
38. Piqueras B, Lavenu-Bombled C, Galicier L, Bergeron-van der Cruyssen F, Mouthon L, Chevret S, et al . Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385-400. http://dx.doi.org/10.1023/A:1025373601374 [ Links ]
39. Ferry BL, Jones J, Bateman EA, Woodham N, Warnatz K, Schlesier M, et al . Measurement of peripheral B cell subpopulations in common variable immunodeficiency (CVID) using a whole blood method. Clin Exp Immunol. 2005;140:532-9. http://dx.doi.org/10.1111/j.1365-2249.2005.02793.x [ Links ]
40. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: Defining subgroups in common variable immunodeficiency. Blood. 2008;111:77-85. http://dx.doi.org/10.1182/blood-2007-06-091744 [ Links ]
41. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-7. http://dx.doi.org/10.1006/clim.1999.4799 [ Links ]
42. Montoya CJ, Salgado H, Olivares MM, Patiño PJ. Evaluación de la respuesta inmune en el paciente con infección recurrente sospechoso de padecer inmuno- deficiencia. Salud UIS. 2000;32:105-11. [ Links ]
43. Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, et al . Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932-43. http://dx.doi.org/10.4049/jimmunol.178.6.3932 [ Links ]
44. Aukrust P, Müller F, Frøland SS. Elevated serum levels of interleukin-4 and interleukin-6 in patients with common variable immunodeficiency (CVI) are associated with chronic immune activation and low numbers of CD4+ lymphocytes. Clin Immunol Immunopathol. 1994;70:217-24. http://dx.doi.org/10.1006/clin.1994.1032 [ Links ]
45. Aspalter RM, Sewell WA, Dolman K, Farrant J, Webster AD. Deficiency in circulating natural killer (NK) cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia. Clin Exp Immunol. 2000;121:506-14. http://dx.doi.org/10.1046/j.1365-2249.2000.01317.x [ Links ]
46. Jaffe JS, Strober W, Sneller MC. Functional abnormalities of CD8+ T cells define a unique subset of patients with common variable immunodeficiency. Blood. 1993;82:192-201 . [ Links ]
47. García de O D, Patiño PJ, Salgado H, López JA, Montoya CJ, Pérez JE. Evaluación del paciente con inmunodeficiencia. Síndrome de infección recurrente pato-lógica. Med Lab. 1997;7:545-75. [ Links ]
48. Agematsu K, Futatani T, Hokibara S, Kobayashi N, Takamoto M, Tsukada S, et al. Absence of memory B cells in patients with common variable immunodeficiency. Clin Immunol. 2002;103:34-42. http://dx.doi.org/10.1006/clim.2001.5197 [ Links ]
49. Duarte RF, Chen FE, Lowdell MW, Potter MN, Lamana ML, Prentice HG, et al. Functional impairment of human T-lymphocytes following PHA-induced expansion and retro- viral transduction: Implications for gene therapy. Gene Ther. 2002;9:1359-68. http://dx.doi.org/10.1038/sj.gt.3301807 [ Links ]
50. International Union of Immunological Societies Scientific Committee. Primary immunodeficiency diseases. Clin Exp Immunol. 1999;118:1-28. http://dx.doi.org/10.1046/j.1365-2249.1999.00109.x [ Links ]
51. Szczepanik M, Akahira-Azuma M, Bryniarski K, Tsuji RF, Kawikova I, Ptak W, et al . B-1 B cells mediate required early T cell recruitment to elicit protein-induced delayed-type hypersensitivity. J Immunol. 2003;171:6225-35. http://dx.doi.org/10.4049/jimmunol.171.11.6225 [ Links ]
52. North ME, Akbar AN, Borthwick N, Sagawa K, Funauchi M, Webster AD, et al . Co-stimulation with anti-CD28 (Kolt-2) enhances DNA synthesis by defective T cells in common variable immunodeficiency. Clin Exp Immunol. 1994;95:204-8. http://dx.doi.org/10.1111/j.1365-2249.1994.tb06511.x [ Links ]
53. Serrano D, Becker K, Cunningham-Rundles C, Mayer L. Characterization of the T cell receptor repertoire in patients with common variable immunodeficiency: Oligoclonal expansion of CD8(+) T cells. Clin Immunol. 2000;97:248-58. http://dx.doi.org/10.1006/clim.2000.4941 [ Links ]
54. North ME, Webster AD, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): Abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (´cytotoxic´) and CD28- (´suppressor´) CD8+ subsets. Clin Exp Immunol. 1998;111:70-5. http://dx.doi.org/10.1046/j.1365-2249.1998.00479.x [ Links ]
55. Fischer MB, Hauber I, Eggenbauer H, Thon V, Vogel E, Schaffer E, et al . A defect in the early phase of T-cell receptor-mediated T-cell activation in patients with common variable immunodeficiency. Blood. 1994;84:4234-41. [ Links ]
56. Fischer MB, Hauber I, Wolf HM, Vogel E, Mannhalter JW, Eibl MM. Impaired TCR signal transduction, but normal antigen presentation, in a patient with common variable immunodeficiency. Br J Haematol. 1994;88:520-6. http://dx.doi.org/10.1111/j.1365-2141.1994.tb05068.x [ Links ]
57. Fischer MB, Wolf HM, Hauber I, Eggenbauer H, Thon V, Sasgary M, et al . Activation via the antigen receptor is impaired in T cells, but not in B cells from patients with common variable immunodeficiency. Eur J Immunol. 1996;26:231-7. http://dx.doi.org/10.1002/eji.1830260136 [ Links ]
58. Paccani SR, Boncristiano M, Patrussi L, Ulivieri C, Wack A, Valensin S, et al . Defective Vav expression and impaired F-actin reorganization in a subset of patients with common variable immunodeficiency characterized by T-cell defects. Blood. 2005;106:626-34. http://dx.doi.org/10.1182/blood-2004-05-2051 [ Links ]
59. Di Renzo M, Zhou Z, George I, Becker K, Cunningham-Rundles C. Enhanced apoptosis of T cells in common variable immunodeficiency (CVID): Role of defective CD28 co-stimulation. Clin Exp Immunol. 2000;120:503-11. http://dx.doi.org/10.1046/j.1365-2249.2000.01239.x [ Links ]
60. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al . Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci USA. 2009;106:13451-6. http://dx.doi.org/10.1073/pnas.0901984106 [ Links ]
61. Arumugakani G, Wood PM, Carter CR. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol. 2010;30:292-300. http://dx.doi.org/10.1007/s10875-009-9351-3 [ Links ]
62. Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179-91 . http://dx.doi.org/10.1111/j.0105-2896.2004.0109.x [ Links ]
63. Marie-Cardine A, Divay F, Dutot I, Green A, Perdrix A, Boyer O, et al . Transitional B cells in humans: Characterization and insight from B lymphocyte reconstitution after hematopoietic stem cell transplantation. Clin Immunol. 2008;127:14-25. http://dx.doi.org/10.1016/j.clim.2007.11.013 [ Links ]
64. Yong PL, Orange JS, Sullivan KE. Pediatric common variable immunodeficiency: Immunologic and phenotypic associations with switched memory B cells. Pediatr Allergy Immunol. 2010;21:852-8. http://dx.doi.org/10.1111/j.1399-3038.2010.01004.x [ Links ]
65. Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, et al . Human blood IgM "memory" B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104: 3647-54. http://dx.doi.org/10.1182/blood-2004-01-0346 [ Links ]
66. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007;178:6624-33. http://dx.doi.org/10.4049/jimmunol.178.10.6624 [ Links ]
67. Carsetti R, Rosado MM, Donnanno S, Guazzi V, Soresina A, Meini A, et al . The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency. J Allergy Clin Immunol. 2005;115:412-7. http://dx.doi.org/10.1016/j.jaci.2004.10.048 [ Links ]
