










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.36 no.2 Bogotá Apr./June 2016
https://doi.org/10.7705/biomedica.v36i3.2998
TOPIC REVIEW
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2998
^rND^1A01^nIrais^sPoblete-Naredo^rND^1A01^nArnulfo^sAlbores^rND^1A01^nIrais^sPoblete-Naredo^rND^1A01^nArnulfo^sAlbores^rND^1A01^nIrais^sPoblete-Naredo^rND^1A01^nArnulfo^sAlbores TOPIC REVIEW doi: http://dx.doi.org/10.7705/biomedica.v36i3.2998
Author´s contributions:
Irais Poblete-Naredo: design and drafting of the manuscript
Arnulfo Albores: design and drafting of the manuscript and critical revision
Both authors contributed equally to this paper.
Received: 23/07/15; accepted: 21/12/15
Biomarkers, or bioindicators, are metric tools that, when compared with reference values, allow specialists to perform risk assessments and provide objective information to decision makers to design effective strategies to solve health or environmental problems by efficiently using the resources assigned.
Health risk assessment is a multidisciplinary exercise, and molecular biology is a discipline that greatly contributes to these evaluations because the genome, transcriptome, proteome and metabolome could be affected by xenobiotics causing measurable changes that might be useful biomarkers. Such changes may greatly depend on individual genetic background; therefore, the polymorphic distribution of exposed populations becomes an essential feature for adequate data interpretation.
The aim of this paper is to offer an up-to-date review of the role of different molecular biomarkers in health risk assessments.
Key words: Biological markers, risk assessment, environmental pollutants, molecular biology, molecular epidemiology.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2998
Biomarcadores moleculares para evaluar el riesgo para la salud debido a la exposición a contaminantes ambientales
Los biomarcadores, o bioindicadores, son herramientas métricas que, al compararse con los valores de referencia, permiten evaluar los riesgos y generar información objetiva que ayude a las autoridades a planificar estrategias efectivas, solucionar problemas de salud o ambientales y utilizar los recursos asignados de manera eficiente.
La evaluación de riesgos en salud es un ejercicio multidisciplinario y la biología molecular contribuye enormemente a estos estudios, dado que el genoma, el transcriptoma, el proteoma y el metaboloma pueden verse afectados por xenobióticos, lo que causa cambios cuya medición resulta útil en la adopción de decisiones. Dichos cambios pueden variar por la carga genética de cada individuo y su distribución polimorfa en las poblaciones expuestas se convierte en un factor esencial para una adecuada interpretación de resultados.
Por lo tanto, el objetivo del presente artículo fue hacer una revisión de los diferentes biomarcadores moleculares aplicables a la evaluación de riesgos para la salud provenientes de los contaminantes ambientales.
Palabras clave: marcadores biológicos, medición del riesgo, contaminantes ambientales, biología molecular, epidemiología molecular.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2998
Indicators or biomarkers
Indicators or markers are widely used metric tools that assess the degree of agreement or accomplishment of qualitative or quantitative data with respect to a given reference value. Specialists and the general public routinely apply them in practically all fields, such as economics, development, politics, academics and health. Indicators provide precise and specialized information to the general population and/or professionals to acquire an accurate perception of specific issues on the status of society or on personal performances. Changes in specific indicators can be used to estimate the status of a system and assess its evolution, progress, improvement or risk; then, indicators provide the basis to implement political changes or for informed decisions towards the efficient achievement of particular goals.
In Biology and Health Sciences, the US National Academy of Sciences defined biomarkers as "cellular or biochemical components or processes altered by a xenobiotic; those changes can be measured in the entire system or in a biological sample" (1). Thus, according to biochemistry or physiology, reference values of certain metabolic activities or physiological functions are used to assess the health status of an individual. Based on biomarker data, the extent of changes caused by internal or external factors can be assessed and used to design strategies to solve such situations.
The environment and health issues
The need to know the incidence of disease and its etiology, particularly due to environmental occupational exposures, started with Bernardino Ramazzini (1633-1714), an Italian physician from Padova, who successfully associated occupational environmental exposures with specific pathologies. In his book "De morbis artificum diatriba" (1700), he associated 54 work activities with illnesses. The book was the first attempt to systematically classify diseases according to the chemicals present in the work environment and the pathologies developed (2). Presently, biomarkers are used as specialized parameters to assess particular conditions in a cell up to a population. Biomarkers are useful to make informed decisions at all levels –they are necessary to implement policies aimed at solving challenges or preventing situations efficiently. However, bio-markers should be reliable and opportunely available to decision makers.
Biomarkers characteristics
Biomarkers are classified as biomarkers of exposure, effect or susceptibility (3) (figure 1). Each one is useful to assess the health status of an individual, a population or even an ecosystem. A biomarker can be used alone; however, sometimes biomarkers of different types need to be analyzed jointly because some of them may not be highly specific and may require correlation with other parameters for adequate interpretations.
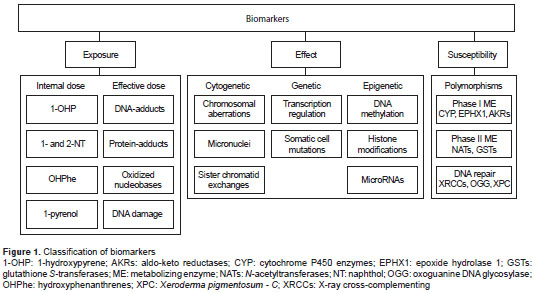
To be considered reliable, a good biomarker should:
a) Respond to a biologically active contaminant,
b) have a dose-effect response correlated with the contaminant levels,
c) have a response that persists even if the exposure to a pollutant ceases because the compound may not remain indefinitely in the environment,
d) preferably be non-invasive in the case of humans and should be easily measurable, and
e) be specific so that an effect can be attributed to a chemical, thus contributing to the certainty of an assessment, and sensitive because the biomarker response should be related to the amount of toxicant capable of eliciting an effect.
These parameters indicate the biological and environmental relevance of pollutant concentrations (4) (figure 2).

Blood is the preferred tissue to analyze biomarkers; its collection is of low invasiveness, and it is a readily available tissue that contains different cell types (erythrocytes and lymphocytes) and fluids (plasma). One of its characteristics is that it distributes nutrients, collects metabolic products that carry information among organs, or serves as vehicle for their excretion. However, blood has its own features, cells belong to different lineages, and metabolites come from different organs. When blood or blood cells are used as surrogates for other tissues, toxicity and risk assessment results are obtained faster than with solid tissues, and the physiological changes in organs or fluids may be reflected by changes in the blood. In other words, changes in blood measurements should be closely correlated with the effects in the parent organ.
Adequate biomarkers are necessary to develop a successful biological monitoring program, and the following steps should be taken:
a) Select a biomarker that fits with almost all criteria of reliability;
b) delimit the size of the biological sample to be studied in terms of the sensitivity and specificity of the biomarker;
c) consider all confounding factors that may alter results, as well as co-exposure to other chemical agents, and
d) select the appropriate tissue for analysis.
The results will reflect the total burden of chemical agents that enter into an individual through all body routes.
Biomarkers of exposure
A biomarker of exposure refers to the quantitation of a parent compound or its metabolites in a tissue or body fluid, or the reaction product(s) of any of those with a biological molecule (4). According to this definition, biomarkers of exposure can be classified into:
1) Markers of internal dose, which indicate the concentration of a given pollutant in an organism, and
2) markers of effective dose, which indicate the extent of the interaction of a pollutant with a target molecule (figure 3).

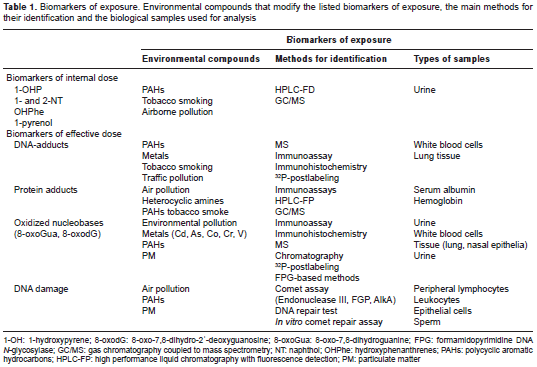
A brief summary of the methods used to study several types of common biomarkers, surrogate tissues for analysis and environmental factors that modify their levels is shown in table 1.
Biomarkers of internal dose
Because direct determinations of environmental pollutants do not necessarily reflect the biologically available concentrations in an organism, the evaluation of internal doses becomes necessary to assess the current exposure status in terms of the quantity of the parent compound or its metabolites present in the body fluids or tissues of exposed individuals or populations (4). Some metabolic products from the biotransformation of a xenobiotic are useful biomarkers of internal dose. The environmental or occupational exposure to polycyclic aromatic hydrocarbons (PAH) can be monitored by the quantitation of metabolic intermediates of frequent and abundant chemicals in the mixture, such as 1-hydroxypyrene (1-OHP), the main pyrene metabolite (90%), with an average half-life of approximately 18-20 hours after pyrene inhalation (5). Urinary 1-OHP is routinely used as a biomarker in workers occupationally exposed to PAHs (6-8), tobacco smokers (9,10) and the general population exposed to airborne pollution (11-13). Urinary 1-OHP is readily measured by high performance liquid chromatography with fluorescence detection (HPLC-FD), and/or gas chromatography coupled to mass spectrometry (GC-MS); its quantitation has shown a good correlation with PAH human exposure at both high and low environmental levels (5).
Other examples of internal dose biomarkers for environmental contaminants are 1- and 2-naphthol (NT), naphthalene metabolites that have been proposed as PAH biomarkers of air or inhalation exposure (14-16); phenanthrene hydroxylated metabolites (OHPhe), which are considered good biomarkers for occupational PAH exposure (10), and 1-pyrenol, another pyrene metabolite, which is less frequently used because its concentration does not necessarily correlate with the levels of exposure to carcinogenic PAHs (17).
Biomarkers of effective dose
Biomarkers of effective dose indicate the interaction of a chemical with a biological target, such as DNA and proteins, or its damaging effect to a biological molecule. Considering that there may be substantial interindividual variations in the metabolism of the compounds, the determination of the effective dose is required for the evaluation of the health effects of pollutants in organisms (4). The classical biomarkers of effect determined by the exposure to chemical pollutants are DNA and protein adducts; the levels of DNA oxidized nucleobases and DNA damage.
DNA adducts. A DNA adduct is the product of covalent binding of a small electrophilic chemical compound to DNA (18). The nature of the electrophilic moiety of the DNA adduct can be a parent compound or a product of its metabolism. Although DNA adducts are targets of multiple DNA repair enzymes, some modifications may persist and produce several undesirable effects, such as nucleotide substitutions, leading to DNA mutations and synthesis of altered protein products (19). Many environmental air pollutants (20), tobacco smoke (20-22) and PAHs from traffic exhaust (23,24) or from occupational exposure (21) have shown a positive correlation with DNA adduct formation in exposed populations. For further information, see reference (25).
DNA adducts have been assessed and quantified in almost all human tissues, but the preferred samples of study are white blood cells due to their availability, and lung tissue because the lung is the point of entry for most carcinogenic environmental contaminants (26). DNA adducts are evaluated by three main methodological approaches, all characterized by their sensitivity, specificity, cost and instrumentation (19). Mass spectrometry (MS) is the most specific methodology and is able to identify DNA adducts according to their molecular weight. This is a high-throughput platform that allows the identification of several adducts in a sample. However, DNA adduct determination by MS requires complex instrumentation and is expensive. Immunoassays and immunohistochemistry are techniques that use antisera to identify specific DNA adducts in DNA samples or directly in the studied tissues. These are low-cost and high-throughput assays with the limitation that the adduct species to identify are restricted by the availability of specific antisera; therefore, the discovery of new adducts using these methodologies is not possible. 32 P-postlabeling includes DNA digestion and radio-active labeling, and then normal and modified DNA molecules are separated by chromatographic techniques. Despite the fact that this methodology is unable to elucidate the structure of DNA adducts, only the DNA adduct profile of the sample, it is the approach of choice in several studies on DNA damage caused by environmental pollution at very low exposure levels (27,28). According to Poirier et al. (19), approximately 40 different DNA adducts have been identified in human samples using the aforementioned techniques.
Protein adducts. Protein adducts are the product of a covalent interaction between a reactive chemical compound and a protein. The most common protein adducts evaluated are those formed with serum albumin [particularly with cysteine at position 34 (Cys 34 )] and hemoglobin; these proteins are found in very large quantities in blood that react with electrophilic compounds via their reactive carboxyl, amino and/or sulfhydryl groups (29). The study of protein adducts as surrogate markers of effective dose offers the advantages that proteins often have longer half-lives than other markers (parent compound or its metabolites), they are much more abundant than DNA adducts, and they are not subjected to repair (29). The methodological approaches employed for protein adducts research are based on the number of samples, time, and available methodologies. For large-scale studies, immunoassays are recommended, but chromatographic-based techniques, such as HPLC-FP or GC-MS, are also suitable.
The protein adducts assessed in environmental, smoking-related or occupational exposure to PAHs are those formed with benzo[ a ]pyrene (BaP) or the carcinogenic metabolite of BaP (r-7,t-8-dihydroxy-t-9,t-10-epoxy-7,8,9,10-tetrahydrobenzo[ a ]pyrene), BPDE. BaP and BPDE form adducts with serum albumin and hemoglobin. BaP protein adducts are well correlated with smoking, but the adduct levels showed only a marginal association or even no association at all with environmental PAH exposure (30). Moreover, BaP adducts have been found at highe associated with cell metabolism (31). Different enzymatic pathways are dedicated to maintenance of the correct balance between pro-oxidative and anti-oxidative status, but some exogenous stimuli may shift this equilibrium and induce a pro-oxidant condition that can damage cell integrity. As is well known, ROS are able to modify the DNA nucleobases and generate mutations when those are not repaired by the corresponding mechanisms (25). Although a wide number of oxidative modifications of DNA have been described, the oxidation of guanine generates 8-oxo-7,8-dihydroguanine (8-oxoGua) and its deoxynucleoside, 8-oxo-7,8-dihydro-2´-deoxyguanosine (8-oxodG), which are the most commonly measured products of oxidative DNA dama ge (31). Modified guanine products originate from direct oxidation of guanine in double-stranded DNA or by the modification of dGTP to 8-oxo-dGTP in the general pool of nucleotides in the cell, which is then incorporated in the DNA during duplication or repair (32). The main repair mechanism of guanine-oxidized lesions is mediated by the enzyme oxoguanine DNA glycosylase 1 (OGG1) via the excision of 8-oxoGua from the DNA (31), with the final product released in the urine. However, other minor repair systems have been described (32). If the corresponding repair system fails in the removal of 8-oxoGTP, it results in mutagenesis due to the production of GC→TA transversions during DNA replication (33).
The level of guanine-oxidized products is mainly determined in leukocyte DNA and in urine, even though it has also been detected in different tissues, such as nasal epithelia. Despite the fact 8-oxoGua is the major product of DNA repair, the determination of 8-oxodG is preferred because there are many standardized and validated methods for 8-oxodG determination by chromatographic methods and by commercial ELISA kits (32). Moreover, the 8-oxoGua product is less stable than its deoxynucleoside counterpart, which makes its extraction, separation and detection difficult (32).
In general terms, the levels of 8-oxodG in a given organ or tissue reflect the oxidative state in that particular sample, whereas the levels of 8-oxodG in urine is a measure of the oxidative damage in the whole organism (32). The detection and analysis of 8-oxodG is accomplished by chromatographic approaches, mainly HPLC with electrochemical (EC) detection (HPLC-EC), GC-MS and liquid chromatography tandem mass spectrometry (LC/MS-MS); immunoassays and immunohistochem-istry; 32 P-postlabeling, and the formation of strand breaks by the removal of 8-oxodG with the use of formamidopyrimidine DNA N -glycosylase (FPG) (31,32).
Many environmental pollutants have the ability to induce ROS production, such as particulate matter (PM), traffic-related contaminants, wood smoke and heavy metals, among others (25,32,33). Overall, there is strong evidence that there is an association between environmental air pollution and the urinary excretion of 8-oxodG or 8-oxoGua, biomarkers that are widely used to assess endogenous oxidative DNA damage (25).
DNA damage: comet assay. The comet assay or the single cell gel electrophoresis assay is an easy, fast and sensitive method to evaluate DNA transient damage at the level of single- and double-stranded breaks, abasic sites, and DNA replication and repair intermediates. DNA is isolated from the rest of the cell components with a high saline solution with detergents in a gelatin-covered slice. The obtained DNA is incubated in an alkaline media to unwind DNA with strand breaks and then it is subjected to electrophoresis. The broken DNA portions will migrate towards the anode resembling a "comet tail" and are visualized with a fluorescent DNA-binding dye (34,35). In general terms, the more DNA damage the longer the DNA tail. Modifications of the basic comet assay include the use of lesion-specific enzymes for the identification of modified nucleobases to increase the specificity and sensitivity of the method (34). Usually, the endonuclease III enzyme is selected to detect oxidized pyrimidines,
FPG for 8-oxoGua identification (oxidized purines) and AlkA for alkylated bases (35). Other variants of the traditional comet assay are the DNA repair test and the in vitro comet repair assay. The former measures the DNA repair kinetics and the degree of repair after a genotoxic cellular stimulus and the latter, the capacity of cell extracts to repair damaged DNA (35).
The comet assay is considered a highly sensitive assay, and its results correlate well with other cytogenetic tests, such as micronucleus (MN), sister chromatid exchange (SCE) and chromosomal aberration (CA) analysis (34). The preferred biological material for the comet assay comes from isolated peripheral lymphocytes or leukocytes, but other type of cells have been used satisfactorily, such as epithelial cells obtained from the mouth, nose and tear ducts, exfoliated bladder cells, and sperm, all of them appropriate surrogate tissues for those directly affected by chemicals (36). Moreover, in the case of air contamination, the tissue of choice is the nasal epithelia because the nose is the point of entry of the air pollutants, and these cells have shown great sensitivity in sensing environmental xenobiotics (34).
The application of the comet assay in human biomonitoring for environmental and occupational exposure to genotoxic agents has been extensively reviewed (34,36). Briefly, the majority of the studies reveal a good correlation between exposure to environmental pollution and DNA damage, as well as associations with the results obtained from other biomarkers of exposure (36). For example, occupational exposure to carcinogenic PAHs reduced the ability of cells to repair damaged DNA (37), and the environmental exposure to PM10 from the emissions of an oil refinery increased DNA damage in lymphocytes obtained from exposed subjects (38). Moreover, in buccal epithelial cells from women chronically exposed to biomass smoke, there was a positive correlation between PM10 and PM2.5 indoor levels and DNA damage (39). Tobacco smoke effects on DNA integrity are, in contrast, a controversial issue due to the increased, synergistic, discrepant or null effects observed in several studies. While in some studies there was a strong association of DNA damage with tobacco smoking, in others there was only an effect in conjunction with other genotoxic agents, and in others, the effect was greater in control groups compared with exposure groups (with co-exposure with other chemicals) or there was no association (34,40).
Biomarkers of effect
The presence of chemical contaminants or adverse agents in the environment will induce physiological and biochemical changes in a living being, and such changes are used as biomarkers of effect. These alterations must be a response to a chemical exposure and are manifested as the regulation, overexpression or inhibition of a specific behavioral, biochemical, anatomical, physiological or molecular function (27). Biomarkers of effect assess the levels of genetic damage in DNA and chromosomes, alteration of enzymatic activity, gene transcription, and/or gene mutation as well as epigenetic modifications, among others. In terms of this review, the molecular biomarkers of effect will be divided into three categories: cytogenetic, genetic and epigenetic biomarkers, which are summarized in figure 4 and table 2.


Cytogenetic biomarkers
The evaluation of cytogenetic biomarkers constitutes the first-choice tool in the assessment of human exposure to different agents, as contact with environmental pollutants is a common area of study (41). The cytogenetic assays that have been widely and extensively used involve the determination of CAs and MN, but other approaches have been used successfully, such as the determination of SCEs. Nevertheless, cytogenetic biomarkers are unspecific to the type of agent of exposure, and there are many confounding factors, such as age, cigarette smoking, gender, and irradiation exposure, that should be considered to reach correct conclusions (42).
Chromosomal aberrations. The alteration of the normal arrangement of chromosomes by deletions, duplications or reorganizations of the genome that can be visualized microscopically is defined as a chromosomal aberration (CA) (25,43). Double-stranded breaks of DNA (DSB) are the main source for CAs and are naturally produced during DNA synthesis by the accumulation of single stranded breaks through excision DNA repair, oxidative DNA modifications, and other processes or by external stimuli, such as UV and ionizing radiation, mutagens, antibiotics or nucleases (43). DSBs are generally repaired by three different mechanisms in eukaryotes: Homologous recombination repair (HRR), single-strand annealing and non-homologous DNA end joining. If DSBs in DNA are left unrepaired, the chromosome remains broken and leads to cell lethality. On the other hand, if the DNA repair mechanisms fail in the correct reestablishment of the genetic information, it may generate rearrangements, mutations and cell transformation (43).
The microscopic evaluation of structural CAs relies on staining chromosomes using different techniques. Human lymphocytes are cultured (metaphase or interphase), and chromosomes are stained mainly by three different techniques: solid staining, chromosome banding and fluorescence in situ hybridization (FISH). Solid staining, which consists of labeling the chromosomes with only one color, is a very limited method that only allows for the identification of dicentric and acentric products of asymmetrical exchanges, underestimating the genetic damage of the cell (43). Moreover, as asymmetric exchanges are lethal to cells, the cytogenetic assessment can only be performed during a short period of time due to the instability of the aberrations (44). The chromosome banding technique consists of different chromosomal banding stains, such as Giemsa staining (G- banding), reverse Giemsa (R-banding) staining, centromeric (heterochromatin) (C-banding) or multi-colored banding stain with hybridization labels for specific regions of the chromosome (mBANDs). Chromosome banding identifies all types of structural arrangements, including intrachromosomal exchanges (that cannot be identified by FISH), but this methodology is time consuming and demands a skillful analyzer (41,44), which is a disadvantage in large-scale studies. In contrast, FISH is the most powerful technique and uses chromosome-specific probes, allowing for the detection of complex genetic configurations involving several breaks in two or more chromosomes in addition to symmetric and asymmetric rearrangements (43). Single and multicolored FISH protocols increase the number of chromosomes "painted," and the number of aberrations identified is increased as well as the precision of the analysis (43). The greater specificity and speed of this approach allows the evaluation of larger quantities of cells, which increases the sensitivity of this method for the analysis of low frequency aberrations due to low-concentration exposures (44). One disadvantage of this method is that, to gain specificity, it requires augmenting the number of labeled chromosomes (with different colors or intensities), making the study costly and the analysis complex (41).
The association of environmental pollution with cytogenetic damage in exposed populations has been discussed in several reports and review papers (25,27), which have noted CAs are good biomarkers of effect and good predictors of cancer risks (41,44) The most suitable cell type for CA assessment in humans is the peripheral lymphocyte. This cell type offers many advantages: 1) It easily grows in vitro; 2) a high number of metaphase cells are attainable; 3) it provides a whole perspective of the systemic exposure to a chemical; 4) it is a cell type with a relatively long life, and 5) it is an abundant tissue that does not require invasive methods to be obtained (44). In occupational or environmental exposure assessments, the most popular technique for CA characterization is FISH (41).
Micronuclei. Micronuclei (MN) are cytoplasmic chromosomes or chromosomal fragments that have been excluded from the nucleus of both daughter cells after cell division. Eventually, the omitted fragments or the whole chromosomes are enveloped by a nuclear membrane that resembles a small nucleus that can be observed by light microscopy (45). Two main mechanisms of MN induction have been defined: Chromosome breakage and disruption of the cell spindle during mitosis (44,45).
Different methodologies are available for MN assessment. Routine staining with May-Grünwald Giemsa or Papanicolaou´s stain is suitable for identification of MNs, but DNA-specific stains such as Feulgen or acridine orange are more specific (45). These staining methods are routine procedures that can be performed and afforded by almost all laboratories but are laborious, and scoring may be tedious. The automation of MN scoring by flow cytometry and image analysis offers to increase the efficiency of the evaluation process (41). Recently, the most widespread technique for MN assays has been the cytokinesis-block MN assay (CBMN). CBMN evaluates chromosome abnormalities in dividing cultured cells (binucleated) after cell division has been blocked with cytochalasin-B, a cytokinesis inhibitor. Using this method, other parameters of DNA misrepair, telomere end fusions, DNA elimination or amplification can be measured along with the evaluation of cytostatic effects and cytotoxicity in the same assay (46).
CBMN can be combined with other tools such as in situ hybridization or immunostaining to provide more information about the origin of the MN and its content. The identification of a centromeric structure with antibodies against kinetochore proteins would indicate either the presence of one or more chromosomes in the MN or its origin by disruption of the spindle or other components during mitosis. Likewise, the detection of consensus repetitive centromeric sequences with pancentromeric probes allows the identification of whole or partial chromosomes in the MN. Moreover, the use of DNA probes directed against specific sequences provides clues about the specific chromosome contained in a MN (44,46).
Micronuclei assays can be performed in almost any cell, but the majority of surrogates come from the hematopoietic linage (lymphocytes and erythrocytes) and bone marrow. From those, lymphocytes are the most widely used because micronucleated erythrocytes are only obtained from splenectomized subjects; on the other hand, the use of bone marrow requires a very invasive method. Recently, some studies evaluated MN in exfoliated epithelial cells obtained from buccal, nasal, urothelial, vaginal or cervical mucosa (41,44,46). These surrogates are easy to collect and, as they are readily in contact with the genotoxic compounds, they are the most vulnerable cells for MN appearance (45).
Many agents and stimuli produce MN, such as pollutants, chemical drugs, radiation, infections, nutritional state and inflammation or even aging (45). The validation of the MN assay as a biomarker for cancer risk assessment remains to be established (44), but some preliminary data has found that MN frequency in lymphocytes is a good predictor for carcinogenesis (47). Despite its lower predictability of cancer, the MN assay was recently substituted for CA determination in studies of exposure to genotoxicants. MN analysis has been widely applied in environmental monitoring, and it is the second most preferred method (only after 32 P-postlabeling) to evaluate exposure to traffic pollution (25), as the assessment of PAHs is a popular issue (41). Moreover, Demetriou, et al. , suggested that there is some evidence of an association between MN and exposure to air pollutants (25).
Sister chromatid exchanges. The process whereby two sister chromatids break and rejoin during DNA replication, interchanging genetic material at homologous regions, is called sister chromatid exchanges (SCE) (44,48). This is a natural process in mammalian cells that is associated with DNA replication when the two chromatids are tightly joined by the centromere, and the mechanism underlying its formation involves a single-stranded break of DNA. During DNA synthesis, a replication fork is formed between the two parental and the nascent strands. When a gap is encountered in one of them, the replication fork is broken by a DNA repair mechanism, and a 3 ´ single-stranded break forms. The replication fork is restored likely by HRR with the exchange of specific DNA regions between both chromatids (48).
SCE is microscopically analyzed using the thymidine analog 5-bromodeoxyuridine (BrdUrd). BrdUrd is added to the cell culture and incorporated during the first two rounds of DNA replication. During the first replication, each original DNA strand is duplicated with a daughter strand containing BrdUrd. During the second round, the original
DNA strand will be again duplicated with a BrdUrd-containing strand, but the other strand will be double labeled. Therefore, both sister chromatids can be visualized and distinguished with a staining procedure, such as fluorescence plus Giemsa staining, acridine orange followed by Giemsa staining, or with the use of anti-BrdUrd-labeling counter-stained with DAPI or propidium iodide (48-50). Exchanges are recognized by the interchange of physical regions from one chromatid to the other. The ease of analysis has made this assay popular for the assessment of the genotoxic potential of chemicals with the consequent generation of large SCE databases (44).
SCEs are evaluated in any cell that can be maintained in culture for two duplication cycles in the presence of BrdUrd (44), and by far, human peripheral blood lymphocytes are the cell type most widely used for this purpose. Although SCE assays have been extensively used for the determination of the genotoxicity of multiple compounds, some evidence suggests that it is not a reliable biomarker of cancer risk; nevertheless, it can be used as a valuable tool for the assessment of cytogenetic damage in exposed individuals, as well as a biomarker of exposure (44). The effect of ambient pollutants, such as exposure to traffic exhaust, has been assessed and reviewed previously (51).
Genetic biomarkers
Transcriptional regulation. The gene expression induction by diverse stimuli reflects the biological response of the cell to the stimulus. In the case of exposure to environmental pollutants, the transcriptional profile of a given cell or organism indicates the defense mechanisms activated, the minimal dose at which the effect is observed, and the time window of gene induction or repression, as well as the signaling pathways involved in the process. Therefore, the knowledge of the repertoire of genes transcribed under a given circumstance aid in the dissection of the mode of action of toxicants.
The available methodologies to assess gene expression profiles include high-throughput platforms, such as cDNA microarrays, and gene-specific methods, i.e., RT-PCR or Northern blots. Microarray analysis is, by far, the preferred methodology, and this procedure includes total RNA extraction and cDNA synthesis for microarray hybridization. Subsequently, the validation of the results is performed by real-time RT-PCR of specific genes of interest.
The transcript profile can be evaluated in multiple types of samples, with availability depending on the model of study. In the case of animal models, the transcriptome can be determined from any type of tissue or fluid, while in human studies, the use of subrogate tissues, such as peripheral blood lymphocytes (PBL) (52) or leukocytes (53), is preferred. Accordingly, Siravastava, et al., noted that PBLs (54) and peripheral blood mononuclear cells (PBMC) (55) are appropriate models to study the effect of diesel exhaust particles (DEP) in animal airways due to the similarities in gene expression patterns in comparison to lung tissue. However, human B lymphocytes (56), alveolar macrophages (57), and airway epithelium (58) have also been used in several reports. Another alternative is the use of cell lines, which serve as models of lung, liver, endothelium, epithelium, lymphocytes, etc.
The evaluation of the transcript levels of some xenobiotic-metabolizing enzymes is a valuable tool for the assessment of exposure to specific environmental pollutants. These biomarkers comprise phase I and II enzymes, such as members of the cytochrome P450 (CYP450) superfamily and conjugating enzymes. Phase I metabolism consists of a series of oxidative reactions that biotransform the parent compound into bioactive or detoxified metabolites. The transcriptional regulation of CYP1A1 is a typical biomarker of exposure to PAHs that has been widely used in environmental biomonitoring (15). The induction of CYP1A1 is initiated by the ligand binding to the aryl hydrocarbon receptor (AhR) and its translocation into the nucleus. There, AhR forms a heterodimer with the aryl hydrocarbon receptor nuclear translocator (Arnt) and binds to xenobiotic response elements (XRE) located in the promoter region of target genes, including CYP1A1 , CYP1A2 and CYP1B1 , promoting gene transcription (59).
CYP1A1 induction by exposure to PAHs, 3-methyl-cholanthrene and 2,3,7,8-tetrachlorodibenzo- p -dioxin (TCDD) has been used for the assessment of exposure to these compounds in several vertebrate models (60-62) and cell lines (61,63,64). Moreover, CYP1A1 transcript upregulation has been described in various experimental models exposed to airborne PM (65), DEPs (54,55,66) and cigarette smoking (67-69). Alternatively, the CYP1B1 transcription regulation is also postulated as a sensitive biomarker of ambient pollution (15) because increased mRNA levels have been documented in the presence of PAH mixtures (63,64), BaP (61,63,70), TCDD (61,71), PM (72), DEPs (54,55,73,74) and tobacco smoking (58,68,69) as well.
Other phase I enzymes mostly reported to be upregulated at the transcriptional level by environmental toxicants are CYP1A2 (55,75) and various aldoketo reductases (AKR). AKRs are a family of soluble NAD(P)H oxidoreductases that catalyze the metabolisms of a wide range of compounds, such as PAHs, aldehydes and drugs. AKRs have been associated with exposure to tobacco smoke (52,58,68), quinones derived from diesel exhaust (DE) (76), BaP and TCDD (61,70).
On the other hand, phase II metabolism involves conjugation reactions to increase chemical hydrophilicity for ease of excretion and elimination of possible reactive metabolites. The phase II metabolizing enzymes comprise mainly transferases, including UDP-glucuronosyltransferases (UGT), sulfotransferases (SULT), N -acetyltransferases (NAT), and glutathione S -transferases (GST), among others (77). At the transcriptional level, the most inducible phase II genes by environmental pollutants are the UGTs (72,78) and GSTs (54,55,72,74). Other phase II enzymes, such as the NAD(P)H: Quinone oxidoreductase-I ( NQO1 ) and heme oxygenase-1 ( HO-1 ) genes, are also upregulated by a wide range of airborne contaminants (55,56,58,61,69,72,74,79).
A master transcriptional regulator for the expression of antioxidant and phase II detoxifying genes is nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 is a transcription factor that recognizes specific DNA elements known as antioxidant response elements (ARE) located in the enhancer region of target genes, such as GST subunits A 1 and A 2 , NQO1 , HO-1 , UGTs and others (80). As expected and associated with the upregulation of phase II enzyme transcription by atmospheric pollutants; also, Nrf2 is also robustly induced by these contaminants. Nrf2 transcriptional activity and expression is increased in the presence of airborne particulate matter (79), BaP and TCDD (61), and cigarette smoke (67). Moreover, it has been suggested that the mechanisms of Nrf2 activation by BaP in HepG2 cells depends on the metabolic transformation of the parent compound and subsequent ROS generation (unpublished data).
Additionally, the exposure to DE or ultrafine carbon particles (UFP) in experimental chambers has provided useful information about transcriptional activation in human volunteers. DE is a model to study the effects of airborne particles, and the UFP are suggested as main causative agent of the adverse effects of airborne pollutants. Peripheral blood leukocytes are the human tissue of study due to their availability, their inflammatory response activation following PM exposure and their role as cell messengers from the site of entry of the pollutants to the rest of the organs (81,82). Human exposure to DE induced the activation of oxidative stress (82,83), inflammation (83), and the proteasome and coagulation cell pathways (82), as well as UFP-activated pathways associated with inflammation, growth factors, NrF2-mediated oxidative stress and xenobiotic metabolism (81).
Somatic cell mutations. A somatic cell mutation is any change in the genomic DNA caused by internal factors, such as natural errors produced during replication or endogenous agents, or by the action of environmental mutagens (84). The index of mutations can be estimated via the study of reporter genes, and the most frequently used is the HPRT gene.
HPRT is an X-linked gene that encodes hypoxanthine-guanine phosphoribosyltransferase (HPRT), an enzyme involved in the phospho-ribosylation of guanine and hypoxanthine as well as purine analogues, such as 6-thioguanine (TG), 6-mercaptopurine and 8-azaguanine, which require this metabolic step to exert their cytotoxic effects (85). Two different methods have been developed for the assessment of in vivo HPRT mutations, an auto-radiographic and a clonal assay.
The first method has time and cost advantages, but it is unable to detect the origin of the mutational phenotype of the cells. The cell model of study is T lymphocytes, which are cryopreserved, followed by stimulation for cell division with phytohemagglutinin (PHA) for 3 H-thymidine incorporation in selection media containing TG. Only mutant cells ( HPRT - ) are able to incorporate 3 H-thymidine and TG, while normal cells ( HPRT + ) are sensitive to TG and do not synthesize DNA. After the isolation of cellular nuclei, they are fixed and autoradiographed, and variant frequencies are scored (86).
On the other hand, the clonal assay allows the isolation, clonal expansion and characterization of the mutant cells. Isolated T lymphocytes are activated with PHA and seeded in microplates containing HPRT -deficient feeder cells (B-lymphoblastoid cells) in media supplemented with T cell growth factor in the presence or absence of TG. The cell density to be seeded in each plate depends on TG supplementation; TG-containing wells receive a high cell density for the growth of rare HPRT - cells (10 5 cells/well), but wells without TG will receive approximately one cell/well. Cells are incubated for 10-14 days and the colonies are characterized by either microscopy, 3 H-thymidine incorporation or cell expansion (86). Furthermore, TG-resistant cells may be assayed for HPRT enzymatic activity, expression of surface markers and for genomic DNA changes.
Accordingly, variant frequencies (VF, autoradio-graphic method) and mutant frequencies (MF, cloning assay) have been determined in different populations, and it has been determined that both parameters increase with the age of the individuals (86,87). Several exposure situations also increase the parameters of VF and MF in the HPRT gene, such as accidental irradiation, radiotherapy and chemotherapy (84). Smoking significantly increases VF and MF in active smokers compared with former or never smokers, and an association was found between cotinine levels and the number of smoked cigarettes in this VF (87,88); however, this association has not always been found (85). For instance, in utero exposure to cigarette smoke was not associated with an increased MF in newborns (89). However, the study of the in utero exposure to environmental pollutants in a highly polluted Polish city showed an increased level of DNA adducts in the fetus in correlation with an augmented MF, an association that was not found in the mothers (90). Similarly, the occupational exposure to DE did not show an increased MF in the HPRT gene in bus mechanic maintenance workers, but there was a correlation between MF and adduct levels in the most heavily exposed individuals (91). The exposure to moderate and low environmental pollution, as well as the occupational exposure to PAHs, however, did not reflect a statistical association with HPRT MF (92). Consequently, HPRT mutation frequency is not considered a sensitive biomarker of exposure to genotoxicants, but it can be valuable in chemoprevention programs for individuals with a known exposure risk (85).
Epigenetic biomarkers. Epigenetics is the study of hereditary gene expression modifications that do not involve a DNA sequence change (93). Three main mechanisms of epigenetic control in mammalian cells have been described: 1) DNA methylation, 2) histone modifications, and 3) microRNA (miRNAs). It is well established that many environmental factors are able to modify the epigenome of an organism as an adaptive response. These epigenomic modifications are persistent and, in some cases, impact future generations. Therefore, the epigenomic "imprinting" of an organism reflects its previous exposure to a given contaminant and its response capacity to environmental stressors (94).
There are some challenges for epigenetic investigations that studies need to overcome to draw confident conclusions. The main limitation in the study of environmental epigenetics is that modifications in the epigenome may be negligible but cumulative, and their manifestation could be evident only after a considerable period of time (95,96). However, the number of publications in environmental epigenetics is constantly growing but lags far behind in comparison with cancer epigenetics, for instance (97). The second challenge is the accessibility of the appropriate target tissue or even the cell type. It is well known that genomic DNA is practically identical in any cell type of an organism, but the epigenomic profile of each cell may be unique and constantly changing (97,98). Therefore, the precise selection of a target or a surrogate tissue, its availability, and even the separation and classification of specific cell types may be crucial for the success of the study.
DNA methylation. DNA methylation consists of the addition of a methyl group to the fifth carbon of a cytosine residue present in regions enriched with the dinucleotide CpG, known as "CpG islands". This process is catalyzed by DNA methyltransferases (DNMT), enzymes that are either constitutively active (DNMT1) or inducible (DNMT3A and 3B). Some CpG islands are located in the promoter region of protein-coding genes, and their hypermethylation is generally associated with gene transcription suppression. Nevertheless, most of the CpG dinucleotides (90%) are located in repetitive transposable elements that are generally heavily methylated (99). DNA methylation is associated with natural processes, such as DNA imprinting, chromosome stability and X-chromosome inactivation, but in aberrant situations, it is associated with cancer and the silencing of tumor suppressor genes (100).
The evaluation of DNA methylation is possible from many different biological samples, such as urine, plasma, sputum, lymphocytes, buccal cells, saliva, and tissues (96), and some technological advances have been developed for the analysis of DNA methylation from very small amounts of a sample. Nevertheless, there is some debate about the usefulness of surrogate tissues, such as peripheral blood, in the study of epigenetic modifications in target tissues (98). Several approaches are available for the study of gene-specific or genome-wide analysis of DNA methylation status. In the first type of procedures, the most popular method consists of the conversion of unmethylated cytosines to uracils using sodium bisulfite, leaving the methylated residues without any change. This technique is followed by different approaches to identify the specific methylated DNA region. PCR amplification with specific primers directed to the methylated DNA sequence, called methylation-specific PCR (MSP); combined bisulfite conversion restriction analysis (COBRA) that identifies restriction enzyme sites that have been preserved or lost after bisulfite conversion, and DNA sequencing by the Sanger method or pyrosequencing are some methodologies used for the identification of specific DNA-methylated regions (96,101).
Some methods for the identification of global DNA methylation content are high performance capillary electrophoresis (HPCE) and HPLC-MS after DNA digestion to single oligonucleotides (96,101). The evaluation of methylation levels of transposable highly repetitive sequences in DNA is another method to estimate genome-wide methylation levels. The multiple repetition elements Alu and LINE-1 (long interspersed nuclear element-1) are two well-characterized elements that normally are heavily methylated, and any decrease in their methylation degree indicates a change in the whole-genome methylation status (99). Likewise, different genome-wide methylation platforms are based on microarrays and combine different technologies, such as immunoprecipitation of 5-methylcytosine with specific antibodies (methylated DNA immunoprecipitation or MeDIP), methylation-sensitive restriction enzymes and PCR amplification, bisulfite treatment coupled with hybridization to microarrays, and sequencing, among others.
The modification of the DNA methylation profile in individuals exposed to environmental contaminants is the most studied epigenetic modification due to the relative stability of methylation, to the multiple technical approaches that are available for its study (98) and to the feasibility to preserve DNA samples in comparison to chromatin and RNA (97). Metals such as nickel, cadmium, lead and arsenic are recognized for their capacity to alter the methylation profile of DNA (102). Ambient air pollution is associated with the hypermethylation of the forkhead box transcription factor 3 ( FOXP3 ) and is involved in asthma pathogenesis (103,104); specifically, the exposure to particulate matter (PM 2.5), black carbon and sulfates produces global DNA hypomethylation (assessed by methylation levels of ALU and/or LINE1) (105,106) as well as aberrant methylation levels in the promoter of the inducible nitric oxide synthase ( iNOS ) (107-109). In mouse models acutely exposed to inhaled DEP or particulate air pollution, hypermethylation of different CpGs in the interferon gamma (IFN- ? ) promoter and hypomethylation at one CpG island in the interleukin ( IL ) -4 promoter (110), as well as persistent hypermethylation of sperm DNA (111), were found, respectively.
The exposure to PAHs is also related to an increase in global methylation levels of DNA, to the hypermethylation of cytokine IL-6 and to p53 hypomethylation in coke-oven workers (112). In neonates, PAH exposure induces the hypermethylation of 5 ´ -CpG islands of acyl-CoA synthetase long-chain family member 3 ( ACSL3 ) (113) and IFN- ? (114) in white blood cells extracted from the umbilical cord. In firefighters, the level of dual specificity phosphatase 22 ( DUSP22 ) promoter methylation decreases in relation to the years of service, an effect that is reproduced with BaP at low doses (115).
Histone modifications. The post-translational modification of the amino-terminal tail domains of histones is another important mechanism of epigenetic regulation. Histones are globular proteins that fold, organize and compact DNA in structural units called nucleosomes that form chromatin. The nucleosome is a histone octamer consisting of one H3-H4 tetramer and two H2A-H2B dimers, and the histone H1 acts as a linker (116).
Several mechanisms regulate histone function, and these proteins are highly dynamic actors in gene regulation. Histone modifications comprise methylation, acetylation, phosphorylation, ubiquitination, sumoylation, citrullination and ADP-ribosylation (95); enzymes such as histone acetyltranferases (HAT), deacetylases (HDAC), methyltransferases, and demethylases are involved in these processes. The methylation, acetylation and phosphorylation status of specific amino-terminal tail domains of histones defines chromatin structure and gene expression. In general terms, histone hyperacetylation is associated with loosely packed chromatin or euchromatin and gene transcription. On the other hand, hypoacetylated and hypermethylated histones generate condensed chromatin or heterochromatin and are associated with methylated DNA and transcriptional silencing (101,116). Nevertheless, the "histone code hypothesis" indicates that specific histone modification patterns, present at specific residues, determine the overall activation or repression of gene transcription (116).
Several histone modifications have been analyzed in cell lines (117-119) and peripheral mononuclear cells (120), blood leukocytes (121) and respiratory epithelia (122) from different human populations. The methodologies applied for these studies include genome-wide and gene-specific approaches. The most accurate method for the identification of histone modifications is MS, but the combination of other methods such as HPLC, HPCE or Western blotting can also be applied to identify histone modifications globally (101). Recently, ELISA kits have been developed and are being used for the analysis of specific histone modifications (120,121). The most popular current method for the study of single gene-associated histone modifications is chromatin immunoprecipitation (ChIP) (117-119,122) that uses specific antibodies against histone chemical markers and a primer-specific PCR for a candidate gene. For the genome-wide study of histone modifications, ChIP-on-chip is a platform that allows the detection of patterns of histone modifications and combines the use of antibodies against histone modifications with a DNA array (101).
There is strong evidence that environmental factors lead to histone modification changes and, therefore, to gene transcription modulation. The most studied environmental stressors that modify histone patterns in different models are metals such as nickel, cobalt, arsenic and chromium. For instance, nickel is associated with many histone modifications in H2, H3 and H4 by methylation, acetylation, phosphorylation and ubiquitination, whereas arsenic is related to methylation modifications in H3 and acetylation in H4, and, finally, chromium and cobalt are associated with H3 methylation at different lysine residues. In a Chinese population occupationally exposed to nickel, elevated levels of H3K4me3 (histone 3, lysine 4 trimethylation) and reduced H3K9me2 (histone 3, lysine 9 dimethylation) were found in blood mononuclear cells (120). Moreover, in peripheral blood leukocytes from steel workers exposed to PM enriched in metal particles, two histone modifications associated with open actively transcribed chromatin, H3K4me2 (histone 3, lysine 4 dimethylation) and H3K9ac (histone 3, lysine 9 acetylation), were increased and correlated with the years of service (121). With respect to some environmental pollutants, DE particulate matter caused an overregulation of the pro-inflammatory mediator cyclooxygenase-2 (COX-2) via the hyper-acetylation of histone H4 and recruitment of p300 HAT to the COX2 promoter and degradation of HDAC1 in a human bronchial epithelial cell line (BEAS-2B) (117). In addition, in A549 cells, PM10 exposure increased the levels of the pro-inflammatory cytokine IL-8 via an increase in H4 acetylation of the IL-8 promoter region (118). Additionally, BaP and cigarette smoke condensate (CSC) exposures were associated with an increase in the transcriptionally active H3K4me3 and H3K9ac chromatin indicators in HeLa cells and normal human respiratory epithelia, respectively (119,122).
MicroRNAs. MicroRNAs (miRNAs) are short non-protein coding single-stranded RNAs that interfere with the translation of complementary mRNAs or induce their degradation (95). miRNAs are transcribed by RNA polymerase II as primary miRNAs (pri-miRNAs) containing a 5 ´ -capping and a 3 ´ -polyadenylation tail. In the nucleus, the pri-miRNA is processed by RNase III Drosha/DGCR8 into a precursor miRNA (pre-miRNA), which is then exported to the cytoplasm by exporting 5/RAN-GTP. The RNase III Dicer complex processes the pre-miRNA into a mature single-stranded miRNA of approximately 20 nucleotides in length, which is incorporated into the RISC (RNA-induced silencing complex). Mature miRNAs regulate mRNAs expression by two different mechanisms according to its complete or incomplete complementarity to the target mRNA. The complete complementarity leads to the degradation of the target mRNA; on the other hand, incomplete complementarity leads to translation inhibition (100).
The evaluation of miRNA levels as a molecular biomarker has many advantages:
1) miRNAs are considered less complex compared to proteins;
2) they do not accumulate modifications after their processing;
3) different molecular tools are available for their isolation, and
4) they can be easily and sensitively detected by PCR-based technologies.
Nevertheless, there are still some challenges to overcome before miRNAs can be used as good biomarkers of exposure. For instance, the technical procedure should be standardized in terms of sample storage, collection and transportation, as well as miRNA extraction, detection, normalization, reproducibility, validation and analysis (123,124). The identification of miRNA targets is another limitation that has to be overcome to elucidate the biological function of specific miRNAs (124). According to their mechanisms of action, each miRNA may present incomplete complementarity with several target mRNAs and therefore regulate the expression of numerous transcripts and vice versa , as one mRNA can be modulated by many miRNAs. The available tools for the identification of potential miRNA targets comprise computational and experimental approaches (124).
The most widely used technique for miRNA detection is the miRNA microarray for genome-wide screening and real-time PCR for gene-specific approaches, but sequencing is also being used, albeit less frequently. miRNAs have been assessed in cell lines (125), human primary cell cultures (126-128), human peripheral blood leukocytes (129), tissues (122,130,131) and different body fluids (123,132,133). The latter surrogate samples have been widely used in oncology studies due to miRNA localization in microparticles and exosomes that confer high stability (123). Accordingly, serum, plasma, urine, amniotic fluid, and sputum, among others, can be used for miRNA analysis.
Some studies have highlighted the sensitivity of miRNAs to respond against environmental insults and their usefulness as powerful biomarkers of exposure and for biomonitoring and prevention. miRNA expression has been linked to exposure to environmental stressors, and altered miRNA levels have been associated with exposure to ambient particulate matter (127), DE and DEP (126,127), metals (129,133), cigarette smoke (122,125,134), asbestos (128), carbon black nanoparticles (131), ozone (132) and others.
Metals such as arsenic, cadmium, aluminum and lead alter miRNA expression (124,129). For instance, exposure to residual oil flay ash (ROFA), a fine particle product from oil combustion containing transition metals such as Ni, V and Fe, elicited cardiac dysfunction and altered miRNA expression levels in hypertensive rats (130). The study performed by Bollati, et al. (2010), in peripheral blood leukocytes from a population of steel workers exposed to metal-rich particulate matter also identified overexpression of two miRNAs related to oxidative stress and inflammation regulation (miR-222 and miR-21) in exposed individuals. Furthermore, the expression of miR-222 was directly correlated with exposure to lead, and miR-146a expression was inversely correlated with lead and cadmium (129). Other upregulated miRNAs in workers exposed to metal-rich PM were miR-421, miR-146a, miR-29a and let-7g (133).
DE particles, likewise, were associated with the modification in expression levels of 197 out of 313 miRNAs identified in human airway epithelial cells, some of which are associated with the inflammatory response and tumorigenic processes (126). Human microRNA-375 is one of the miRNAs upregulated following exposure to DEP and ambient PM and possibly regulates the expression of the AhR receptor (127). Furthermore, BaP exposure upregulated the expression of miR-320 and miR-494 and was associated with cell cycle progression and tumorigenesis in murine bronchial epithelial cells (135); however, the in vivo BaP exposure does not significantly modify hepatic miRNA expression regardless of marked changes in mRNA transcription in mice (136). Other environmental pollutants, such as hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX), caused modulation of several miRNAs associated with cancer and tumor suppression in mouse brain and liver (137).
Susceptibility biomarkers
It is well documented that the development of adverse health effects by exposure to different environmental factors is determined by the susceptibility of each individual. These susceptibility factors are cataloged as genetic predisposition, age, gender and ethnicity (138). The balance between exposure time and dose with susceptibility factors determines individual biological response and the risk for disease development.
In terms of the influence of chemical pollutants on human health, the participation of genes encoding metabolic or detoxification enzymes plays a key role in the genetic susceptibility to xenobiotics; however, genes associated with DNA damage repair likewise predispose individuals to a susceptibility phenotype against toxicological insults. The biomarkers of susceptibility most frequently used are the genetic polymorphisms located in metabolic genes associated with phase I and II metabolizing enzymes.
Genetic polymorphisms
Genetic polymorphisms are natural occurring variations in DNA sequence with a frequency equal to or higher than 1% in the general population (139). A polymorphism may be located in coding or non-coding sequence, and they might be "silent" or "functional" if the polymorphism impacts the activity, stability and/or expression levels of a protein. Some of the effects of a polymorphism in the expression of a gene are: 1) Amino acid substitution, 2) duplication, 3) deletion, 4) protein variants, 5) modulation of the expression levels, and 6) modification of RNA splicing and stability (139).
Single nucleotide polymorphisms (SNP) are a case in point and consist of the substitution of a single base pair, resulting in variants of the same gene or alternative alleles (140). SNPs are the most frequent type of polymorphisms in humans, accounting for approximately 90% of them (140). SNP identification is possible from different human samples, and venous blood is the tissue sample of choice; however, specific blood cell populations are used, for example leukocytes (141,142), lymphocytes (143-145) and the mononuclear fraction (146), and tissues such as placenta (20), or even buccal cells (147,148) are also appropriate for genotyping. Several methods have been developed for their identification, with DNA sequencing being the gold standard (149). The most common method for SNP analysis uses restriction endonucleases in cases where the polymorphism creates or destroys a specific nucleotide recognition sequence for a restriction enzyme (restriction fragment length polymorphism or RFLP). Other useful met ods are real-time PCR using TaqMan probes (13,148,150), and primer extension-based methods (145,151,152) (table 3).


Phase I enzymes. CYP1A1 polymorphisms are the most studied variants in terms of human genetic susceptibility (15). The variant CYP1A1*2A (T3801C) in the 3 ´ non-coding region, which generates an Msp1 restriction site, has been linked to a variety of outcomes related to exposure to different types of environmental pollutants. The presence of this polymorphism is associated with an increased urinary 1-OHP concentration related to air pollution (12), PAH exposure (10) and light tobacco consumption (154). Individuals carrying the CYP1A1*2A allele are predisposed to higher DNA adduct levels and an increased percent of aberrant cells when exposed to environmental tobacco smoke (ETS) (144). Likewise, in an Asian population, the presence of the MspI polymorphism correlated with reduced birth weight following high PM10 exposure (155), oral cancer risk associated with tobacco smoking and chronic obstructive pulmonary disease (156).
Additionally, the variant CYP1A1*2B or 2C (A2455G), which has an amino acid substitution of Ile462 to Val in exon 7, has been associated with higher 1-OHP (9,12,13), 2-NT (9), DNA damage (13) and PAH-DNA adducts (157) in high PAH or tobacco smoking populations, as well as reduced birth weight in pregnant women exposed to high PM10 levels (155). The interaction of smoking or ETS with the CYP1A1 (Ile462Val) polymorphisms and their association with adverse health effects is controversial. Because, while cervical cancer development has shown a significant association (142), others like lung cancer (150) and asthma development in children (158) did not. Both variants, CYP1A1*2A and CYP1A1*2B, have been associated with a higher enzymatic induction and/or enhanced catalytic activity and are correlated with an increased risk of several types of cancer; therefore, these polymorphisms are useful susceptibility biomarkers for PAH exposure and carcinogenesis (15).
In addition, other family members of the CYP450s have been reported as polymorphic and have shown interactions with environmental pollutants. Among them, CYP2E1, a cytochrome associated with the metabolism of alcohol, acetone, benzene, toluene, styrene and nitrosamines, has at least 34 variants (159). The CYP2E1 RsaI polymorphism is associated with increased urinary levels of 1- and 2-NTs (14,160), and 1-OHP (14), as well as with higher DNA adduct levels in human lung tissue (161) from PAH-exposed individuals. Nevertheless, some studies did not find a correlation of this polymorphism with increased PAH metabolic products (9,16,162).
Other phase I polymorphic enzymes relevant to environmental toxicant metabolism are epoxide hydrolase 1 ( EPHX1 ) and AKRs . EPHX1 , which encodes a protein that catalyzes the addition of water to epoxides for their detoxification, presents two polymorphic variants, T8668C (Y113H) and A15543G (H139R). The former polymorphism has been associated with high OHPhe urinary levels, whereas the latter was associated with low risk in Caucasians (165) and no relation with mRNA levels of different AKRs ( AKR1A1, AKR1C1-AKR1C3 ) in smokers (52).
Phase II enzymes. NATs (NAT1 and NAT2), the key enzymes in the conjugation of arylamine compounds, display a high inter-individual variability in their capability to acetylate certain drugs, leading to the classification of individuals as fast, slow and intermediate "metabolizers" (77,166). Thirty-six NAT2 polymorphic variants have been described in humans (166). NAT2*4 , the wild-type allele, is the fast acetylation variant, although variants such as NAT2*12 and NAT2*13 are also associated with the rapid acetylator phenotype in some populations. Several alleles are associated with the slow acetylator phenotype, and the most common ones are NAT2*5 , NAT2*6 , NAT2*7 and NAT2*14 , which contain one or more of the G191A, T341C, A434C and G590A polymorphisms (166). The slow acetylator genotype has been associated with a higher risk of bladder cancer by exposure to arylamine pollutants from tobacco smoke, occupational exposure to benzidine-based dyes, or by alcohol intake; higher risk of urothelial cancer of the renal pelvis in dinitrotoluene-exposed workers; and an increased risk of breast cancer in postmenopausal smoking women and in non-smoker women exposed to ETS for long periods (148,153,166). Likewise, the NAT2 slow genotype has been associated with higher DNA adduct levels (21) and higher urinary PAH metabolites (167) in occupational and non-occupational PAH-exposed populations. On the other hand, the NAT2 fast acetylator genotype was associated with higher 8-OHdG levels by tobacco smoking and PAH exposure (16) and predisposed carriers to lung cancer (168) and breast cancer in the case of long-time heavy smoker women (148). NAT1 allelic variants have also been described (26 alleles in humans), but their functional effects have been less studied; nevertheless, the variant NAT1*10 (associated with a fast acetylation phenotype) was related to an increased risk of bladder cancer in smokers (166).
Different classes of GST polymorphisms are associated with increased levels of health risks according to the modification of several biomarkers of exposure and effect to environmental pollutants. From them, the GSTM polymorphism GSTM1*0, which results in a deleted allele of high prevalence between populations, is associated with higher levels of urinary 1-OHP (8,13), 1- and 2-NT (14,160), 8-OHdG (169-171) and DNA adduct levels (17,20,167,172,173). Moreover, it is considered a risk factor for preterm delivery in association with exposure to PM10 (174). Conversely, the GSTT null mutant is neither associated with increased urinary 1-OHP levels (12,13) nor higher urinary 8-OHdG (169) or DNA adduct concentrations (163), but it has been linked to a higher predisposition for acute leukemia in Chinese children living in proximity to industrial plants (151) and coronary artery disease in smokers (141). As for the GSTP polymorphisms, the A2627G genotype (I105V) is related to an increase (162) or decrease in the urinary 1-OHP levels (12) in occupational exposed workers. Notwithstanding, the genotype GSTP1 C5317T (A114V) was related to higher urinary 1-OHP and OHPhe in German workers with high PAH exposures (143).
DNA repair gene polymorphisms. Polymorphisms present in DNA repair genes are also considered variability factors that predispose individuals to health outcomes in high-risk situations of toxicological exposure. High asbestos pollution, for instance, is associated with malignant mesothelioma, and the individuals who carry the variants 399Q of XRCC1 , N118N of ERCC1 and 241T of XRCC3 are at increased risk for disease development (145,152). Other DNA repair gene polymorphic variants reported as susceptibility factors are: 8-oxo-guanine-DNA glycosylase/AP lyase ( hOGG1) 326C, that correlates with higher oxidative DNA damage in bus drivers exposed to PAHs and volatile compounds (146) and with increased risk for lung cancer by indoor PAH exposure (164); exon 23 variant of the Xeroderma pigmentosum-D ( XPD) A35931C Lys751Gln, that induces higher levels of DNA strand breaks in the aforementioned population (146), and Xeroderma pigmentosum-C ( XPC) polyAT insertion of 83 bp in intron 9 ( PAT +), that associates with higher anti-BPDE-DNA adducts under low (172) and high levels of PAH exposure (173).
Gene-gene interactions. Finally, multiple gene-gene interactions with environmental pollutants have been observed in individuals carrying more than one polymorphism, leading to an extremely complex scenario of genetic susceptibility. Genetic interactions are observed with the CYP1A1*2A ( MspI ) polymorphism and a variety of phase II enzymes. The interaction of the CYP1A1*2 polymorphism with the GSTM1 null, GSTP1 (Ile/Val) and the EPHX "slow" variants results in higher DNA adduct levels (24,163). The combination of the GSTM1 null with the NAT2 slow acetylator genotype correlates with higher urinary PAH metabolites (167) as well as increased CAs (175) in individuals exposed to rich PAH environments. Likewise, an increased risk for lung and breast cancer has been found in individuals carrying the GSTM1 null, GSTT1 null and the NAT2 rapid acetylator genotypes (176,177).
Conclusion
In the present review, we summarized the main and extensively used molecular biomarkers applied to environmental monitoring for human risk assessment. Some of them are highly associated with the exposure condition, while others are not statistically significant or display contradictory results. Despite the presence of some discrepancies, there is cumulative evidence that environmental pollution indeed affects several molecular markers, including all three biomarker types: exposure, effect and susceptibility (table 1).
The usefulness of biomarkers is of great importance for studies on risk evaluation and impact to health because they provide a platform to offer solutions leading to the amelioration of contamination and the adverse effects that pollutants produce. Equally important is to bear in mind that the response will depend on an individual´s genetic background, thus the study of polymorphisms in human populations will also become useful for accurate interpretations. However, some of the issues remain to be discussed in depth, such as the selection of the most appropriate surrogate tissues for the study to produce reliable results with the least invasive methods for sample collection.
As peripheral blood is the surrogate of choice for most analyses, the acquisition of precise and correct results is still debatable, as blood is neither the first contact organ with xenobiotics nor the location of pollutant metabolism. Therefore, the exploration of new surrogate types of cells or tissues should consider the collection methods. The study design should also take into account the size of the sample required, its susceptibility to cancer and the interaction of the surrogate with the xenobiotic or its metabolites.
Therefore, with the development of new technologies and the discovery of novel biomarkers, risk assessment evaluation of pollutant exposure should comprise a set of biomarkers that includes diverse aspects of the individual´s toxicological response to integrate the data to dissect the mechanisms of action of xenobiotics and target organs and to predict adverse health effects.
The authors declare that they have no conflicts of interest.
This work was supported by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACyT), México, reference number 162391.
Corresponding author: Arnulfo Albores, Avenida Instituto Politécnico Nacional 2508, Colonia San Pedro Zacatenco, Delegación Gustavo A. Madero, CP 07369, Ciudad de México, México Teléfono: (01555) 747 3800, extensión 5476 aalbores@cinvestav.mx
1. Gil F, Pla A. Biomarkers as biological indicators of xenobiotic exposure . J Appl Toxicol. 2001;21:245-55. http://dx.doi.org/10.1002/jat.769 [ Links ]
2. Ramazzini B. Las enfermedades de los trabajadores: "De morbis artificum diatriba". Obra completa, Bernardino Ramazzini. Tomo II. México: Miguel Ángel Porrúa; 2012. p. 373. [ Links ]
3. Owen R, Galloway TS, Hagger JA, Jones MB, Depledge MH. Biomarkers and environmental risk assessment: Guiding principles from the human health field . Mar Pollut Bull. 2008;56:613-9. http://dx.doi.org/10.1016/j.marpolbul.2008.01.022 [ Links ]
4. Timbrell JA. Biomarkers in toxicology . Toxicology. 1998;129: 1-12. http://dx.doi.org/10.1016/S0300-483X(98)00058-4 [ Links ]
5. Castano-Vinyals G, D´Errico A, Malats N, Kogevinas M. Biomarkers of exposure to polycyclic aromatic hydrocarbons from environmental air pollution . Occup Environ Med. 2004;61:e12. http://dx.doi.org/10.1136/oem.2003.008375 [ Links ]
6. Castorena-Torres F, Mendoza-Cantu A, de León MB, Cisneros B, Zapata-Pérez O, López-Carrillo L, et al. CYP1A2 phenotype and genotype in a population from the Carboniferous Region of Coahuila, México . Toxicol Lett. 2005;156:331-9. http://dx.doi.org/10.1016/j.toxlet.2004.12.005 [ Links ]
7. do Vale Bosso RM, Amorim LM, Andrade SJ, Rossini A, de Marchi MR, de León AP , et al. Effects of genetic polymorphisms CYP1A1, GSTM1, GSTT1 and GSTP1 on urinary 1-hydroxypyrene levels in sugarcane workers . Sci Total Environ. 2006;370:382-90. http://dx.doi.org/10.1016/j.scitotenv.2006.07.025 [ Links ]
8. Zare M, Shahtaheri SJ, Mehdipur P, Abedinejad M, Zare S. The influence of CYP1A1 and GSTM1 polymorphism on the concentration of urinary 1-hydroxypyrene in cPAHs exposed Iranian anode plant workers . Mol Cell Toxicol. 2013;9:303-9. http://dx.doi.org/10.1007/S13273-013-0038-8 [ Links ]
9. Lee CY, Lee JY, Kang JW, Kim H. Effects of genetic polymorphisms of CYP1A1, CYP2E1, GSTM1, and GSTT1 on the urinary levels of 1-hydroxypyrene and 2-naphthol in aircraft maintenance workers . Toxicol Lett. 2001;123:115-24. http://dx.doi.org/10.1016/S0378-4274(01)00374-5 [ Links ]
10. Chuang CY, Chang CC. Urinary 1-hydroxypyrene level relative to vehicle exhaust exposure mediated by metabolic enzyme polymorphisms . J Occup Health. 2007;49:140-51. http://dx.doi.org/10.1539/joh.49.140 [ Links ]
11. Ruchirawat M, Navasumrit P, Settachan D, Tuntaviroon J, Buthbumrung N, Sharma S. Measurement of genotoxic air pollutant exposures in street vendors and school children in and near Bangkok . Toxicol Appl Pharmacol. 2005;206:207-14. http://dx.doi.org/10.1016/j.taap.2004.11.025 [ Links ]
12. Petchpoung K, Kaojarern S, Yoovathaworn K, Sura T, Sirivarasai J. The influence of metabolic gene polymorphisms on urinary 1-hydroxypyrene concentration in Thai bus drivers . Environ Toxicol Pharmacol. 2011;31:160-4. http://dx.doi.org/10.1016/j.etap.2010.10.006 [ Links ]
13. Sánchez-Guerra M, Pelallo-Martínez N, Díaz-Barriga F, Rothenberg SJ, Hernández-Cadena L, Faugeron S, et al. Environmental polycyclic aromatic hydrocarbon (PAH) exposure and DNA damage in Mexican children . Mutat Res. 2012;742:66-71. http://dx.doi.org/10.1016/j.mrgentox.2011.12.006 [ Links ]
14. Nan HM, Kim H, Lim HS, Choi JK, Kawamoto T, Kang JW, et al. Effects of occupation, lifestyle and genetic polymorphisms of CYP1A1, CYP2E1, GSTM1 and GSTT1 on urinary 1-hydroxypyrene and 2-naphthol concentrations . Carcinogenesis. 2001;22:787-93. http://dx.doi.org/10.1093/carcin/22.5.787 [ Links ]
15. Yi B, Yang JY, Yang M. Past and future applications of CYP450-genetic polymorphisms for biomonitoring of environmental toxicants . J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:353-77. http://dx.doi.org/10.1080/10590500701704037 [ Links ]
16. Kim YD, Lee CH, Nan HM, Kang JW, Kim H. Effects of genetic polymorphisms in metabolic enzymes on the relationships between 8-hydroxydeoxyguanosine levels in human leukocytes and urinary 1-hydroxypyrene and 2-naphthol concentrations . J Occup Health. 2003;45:160-7. http://dx.doi.org/10.1539/joh.45.160 [ Links ]
17. Viezzer C, Norppa H, Clonfero E, Gabbani G, Mastrangelo G, Hirvonen A, et al. Influence of GSTM1, GSTT1, GSTP1, and EPHX gene polymorphisms on DNA adduct level and HPRT mutant frequency in coke-oven workers . Mutat Res. 1999;431:259-69. http://dx.doi.org/10.1016/S0027-5107(99)00169-4 [ Links ]
18. Sturla SJ. DNA adduct profiles: Chemical approaches to addressing the biological impact of DNA damage from small molecules . Curr Opin Chem Biol. 2007;11:293-9. http://dx.doi.org/10.1016/j.cbpa.2007.05.021 [ Links ]
19. Poirier MC. Chemical-induced DNA damage and human cancer risk . Nat Rev Cancer. 2004;4:630-7. http://dx.doi.org/10.1038/nrc1410 [ Links ]
20. Topinka J, Binkova B, Mrackova G, Stavkova Z, Peterka V, Benes I, et al. Influence of GSTM1 and NAT2 genotypes on placental DNA adducts in an environmentally exposed population . Environ Mol Mutagen. 1997;30:184-95. http://dx.doi.org/10.1002/(SICI)1098-2280(1997)30:2<184::AID-EM11>3.0.CO;2-9 [ Links ]
21. Binkova B, Topinka J, Mrackova G, Gajdosova D, Vidova P, Stavkova Z, et al. Coke oven workers study: The effect of exposure and GSTM1 and NAT2 genotypes on DNA adduct levels in white blood cells and lymphocytes as determined by 32P-postlabelling . Mutat Res. 1998;416:67-84. http://dx.doi.org/10.1016/S1383-5718(98)00061-8 [ Links ]
22. Piipari R, Savela K, Nurminen T, Hukkanen J, Raunio H, Hakkola J, et al. Expression of CYP1A1, CYP1B1 and CYP3A, and polycyclic aromatic hydrocarbon-DNA adduct formation in bronchoalveolar macrophages of smokers and non-smokers . Int J Cancer. 2000;86:610-6. http://dx.doi.org/10.1002/(Sici)1097-0215(20000601)86:5<610::Aid-Ijc2>3.0.Co;2-M [ Links ]
23. Binkova B, Chvatalova I, Lnenickova Z, Milcova A, Tulupova E, Farmer PB, et al. PAH-DNA adducts in environmentally exposed population in relation to metabolic and DNA repair gene polymorphisms . Mutat Res. 2007;620:49-61. http://dx.doi.org/10.1016/j.mrfmmm.2007.02.022 [ Links ]
24. Topinka J, Sevastyanova O, Binkova B, Chvatalova I, Milcova A, Lnenickova Z, et al. Biomarkers of air pollution exposure - a study of policemen in Prague . Mutat Res. 2007;624:9-17. http://dx.doi.org/10.1016/j.mrfmmm.2007.02.032 [ Links ]
25. Demetriou CA, Raaschou-Nielsen O, Loft S, Moller P, Vermeulen R, Palli D, et al. Biomarkers of ambient air pollution and lung cancer: A systematic review . Occup Environ Med. 2012;69:619-27. http://dx.doi.org/10.1136/oemed-2011-100566 [ Links ]
26. Povey AC. DNA adducts: Endogenous and induced . Toxicol Pathol. 2000;28:405-14. http://dx.doi.org/10.1177/019262330002800308 [ Links ]
27. Mussali-Galante P, Tovar-Sánchez E, Valverde M, Rojas-Del Castillo E. Biomarkers of exposure for assessing environmental metal pollution: From molecules to ecosystems . Revista Internacional de Contaminación Ambiental. 2013;29:117-40. [ Links ]
28. Nielsen PS, Okkels H, Sigsgaard T, Kyrtopoulos S, Autrup H. Exposure to urban and rural air pollution: DNA and protein adducts and effect of glutathione-S-transferase genotype on adduct levels . Int Arch Occup Environ Health. 1996;68:170-6. http://dx.doi.org/10.1007/BF00381627 [ Links ]
29. Meyer MJ, Bechtold WE. Protein adduct biomarkers: State of the art . Environ Health Perspect. 1996;104(Suppl.5): 879-82. [ Links ]
30. Scherer G, Frank S, Riedel K, Meger-Kossien I, Renner T. Biomonitoring of exposure to polycyclic aromatic hydrocarbons of nonoccupationally exposed persons . Cancer Epidemiol Biomarkers Prev. 2000;9:373-80. [ Links ]
31. Pilger A, Rudiger HW. 8-Hydroxy-2´-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures . Int Arch Occup Environ Health. 2006;80:1-15. http://dx.doi.org/10.1007/s00420-006-0106-7 [ Links ]
32. Loft S, Danielsen P, Lohr M, Jantzen K, Hemmingsen JG, Roursgaard M, et al. Urinary excretion of 8-oxo-7,8-dihydroguanine as biomarker of oxidative damage to DNA . Arch Biochem Biophys. 2012;518:142-50. http://dx.doi.org/10.1016/j.abb.2011.12.026 [ Links ]
33. Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2´ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis . J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120-39. http://dx.doi.org/10.1080/10590500902885684 [ Links ]
34. Faust F, Kassie F, Knasmuller S, Boedecker RH, Mann M, Mersch-Sundermann V. The use of the alkaline comet assay with lymphocytes in human biomonitoring studies . Mutat Res. 2004;566:209-29. http://dx.doi.org/10.1016/j.mrrev.2003.09.007 [ Links ]
35. Dusinska M, Collins AR. The comet assay in human biomonitoring: Gene-environment interactions . Mutagenesis. 2008;23:191-205. http://dx.doi.org/10.1093/mutage/gen007 [ Links ]
36. Valverde M, Rojas E. Environmental and occupational biomonitoring using the Comet assay . Mutat Res. 2009;681: 93-109. http://dx.doi.org/10.1016/j.mrrev.2008.11.001 [ Links ]
37. Cebulska-Wasilewska A, Wiechec A, Panek A, Binkova B, Sram RJ, Farmer PB. Influence of environmental exposure to PAHs on the susceptibility of lymphocytes to DNA-damage induction and on their repair capacity . Mutat Res. 2005;588:73-81. http://dx.doi.org/10.1016/j.mrgentox.2005.08.013 [ Links ]
38. Coronas MV, Pereira TS, Rocha JA, Lemos AT, Fachel JM, Salvadori DM, et al. Genetic biomonitoring of an urban population exposed to mutagenic airborne pollutants . Environ Int. 2009;35:1023-9. http://dx.doi.org/10.1016/j.envint.2009.05.001 [ Links ]
39. Mondal NK, Bhattacharya P, Ray MR. Assessment of DNA damage by comet assay and fast halo assay in buccal epithelial cells of Indian women chronically exposed to biomass smoke . Int J Hyg Environ Health. 2011;214:311-8. http://dx.doi.org/10.1016/j.ijheh.2011.04.003 [ Links ]
40. Piperakis SM, Petrakou E, Tsilimigaki S. Effects of air pollution and smoking on DNA damage of human lymphocytes . Environ Mol Mutagen. 2000;36:243-9. http://dx.doi.org/10.1002/1098-2280(2000)36:3<243::AID-EM8>3.0.CO;2-9 [ Links ]
41. Bonassi S, Ugolini D, Kirsch-Volders M, Stromberg U, Vermeulen R, Tucker JD. Human population studies with cytogenetic biomarkers: Review of the literature and future prospectives . Environ Mol Mutagen. 2005;45:258-70. http://dx.doi.org/10.1002/Em.20115 [ Links ]
42. Motykiewicz G. Application of biomarkers in heavily polluted industrialized areas of countries of central and Eastern Europe . Toxicology. 1995;101:117-23. http://dx.doi.org/10.1016/0300-483X(95)03025-B [ Links ]
43. Obe G, Pfeiffer P, Savage JR, Johannes C, Goedecke W, Jeppesen P, et al. Chromosomal aberrations: Formation, identification and distribution . Mutat Res. 2002;504:17-36. http://dx.doi.org/10.1016/S0027-5107(02)00076-3 [ Links ]
44. Tucker JD, Preston RJ. Chromosome aberrations, micronuclei, aneuploidy, sister chromatid exchanges, and cancer risk assessment . Mutat Res. 1996;365:147-59. http://dx.doi.org/10.1016/S0165-1110(96)90018-4 [ Links ]
45. Samanta S, Dey P. Micronucleus and its applications . Diagn Cytopathol. 2012;40:84-90. http://dx.doi.org/10.1002/dc.21592 [ Links ]
46. Fenech M. Cytokinesis-block micronucleus cytome assay . Nature Protocols. 2007;2:1084-104. http://dx.doi.org/10.1038/nprot.2007.77 [ Links ]
47. Bonassi S, El-Zein R, Bolognesi C, Fenech M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: Evidence from human studies . Mutagenesis. 2011;26:93-100. http://dx.doi.org/10.1093/mutage/geq075 [ Links ]
48. Wilson DM 3rd, Thompson LH. Molecular mechanisms of sister-chromatid exchange . Mutat Res. 2007;616:11-23. http://dx.doi.org/10.1016/j.mrfmmm.2006.11.017 [ Links ]
49. Simpson LJ, Sale JE. Sister chromatid exchange assay . Subcell Biochem. 2006;40:399-403. http://dx.doi.org/10.1007/978-1-4020-4896-8_34 [ Links ]
50. Stults DM, Killen MW, Pierce AJ. The sister chromatid exchange (SCE) assay . Methods Mol Biol. 2014;1105:439-55. http://dx.doi.org/10.1007/978-1-62703-739-6_32 [ Links ]
51. DeMarini DM. Genotoxicity biomarkers associated with exposure to traffic and near-road atmospheres: A review . Mutagenesis. 2013;28:485-505. http://dx.doi.org/10.1093/mutage/get042 [ Links ]
52. Barrón-Vivanco BS, Rothenberg SJ, Medina-Díaz IM, Robledo-Marenco L, Rojas-García AE, Hernández-Cadena L, et al. AKRs expression in peripheral blood lymphocytes from smokers: The role of body mass index . Hum Exp Toxicol. 2013;32:418-26. http://dx.doi.org/10.1177/0960327112455071 [ Links ]
53. Lampe JW, Stepaniants SB, Mao M, Radich JP, Dai HY, Linsley PS, et al. Signatures of environmental exposures using peripheral leukocyte gene expression: Tobacco smoke . Cancer Epidemiol Biomarkers Prev. 2004;13:445-53. [ Links ]
54. Srivastava A, Yadav S, Sharma A, Dwivedi UN, Flora SJ, Parmar D. Similarities in diesel exhaust particles induced alterations in expression of cytochrome P-450 and glutathione S-transferases in rat lymphocytes and lungs . Xenobiotica. 2012;42:624-32. http://dx.doi.org/10.3109/00498254.2011.650732 [ Links ]
55. Srivastava A, Sharma A, Yadav S, Flora SJ, Dwivedi UN, Parmar D. Gene expression profiling of candidate genes in peripheral blood mononuclear cells for predicting toxicity of diesel exhaust particles . Free Radic Biol Med. 2014;67:188-94. http://dx.doi.org/10.1016/j.freeradbiomed.2013.10.820 [ Links ]
56. Wan J, Díaz-Sánchez D. Phase II enzymes induction blocks the enhanced IgE production in B cells by diesel exhaust particles . J Immunol. 2006;177:3477-83. http://dx.doi.org/10.4049/jimmunol.177.5.3477 [ Links ]
57. Woodruff PG, Koth LL, Yang YH, Rodríguez MW, Favoreto S, Dolganov GM, et al. A distinctive alveolar macrophage activation state induced by cigarette smoking . Am J Respir Crit Care Med. 2005;172:1383-92. http://dx.doi.org/10.1164/rccm.200505-686OC [ Links ]
58. Harvey BG, Heguy A, Leopold PL, Carolan BJ, Ferris B, Crystal RG. Modification of gene expression of the small airway epithelium in response to cigarette smoking . J Mol Med (Berl). 2007;85:39-53. http://dx.doi.org/10.1007/s00109-006-0103-z [ Links ]
59. Kawajiri K, Fujii-Kuriyama Y. Cytochrome P450 gene regulation and physiological functions mediated by the aryl hydrocarbon receptor . Arch Biochem Biophys. 2007;464: 207-12. http://dx.doi.org/10.1016/j.abb.2007.03.038 [ Links ]
60. van Ede KI, Gaisch KP, van den Berg M, van Duursen MB. Differential relative effect potencies of some dioxin-like compounds in human peripheral blood lymphocytes and murine splenic cells . Toxicol Lett. 2014;226:43-52. http://dx. doi.org/10.1016/j.toxlet.2014.01.026 [ Links ]
61. Vondracek J, Krcmar P, Prochazkova J, Trilecova L, Gavelova M, Skalova L, et al. The role of aryl hydrocarbon receptor in regulation of enzymes involved in metabolic activation of polycyclic aromatic hydrocarbons in a model of rat liver progenitor cells . Chem Biol Interact. 2009;180:226-37. http://dx.doi.org/10.1016/j.cbi.2009.03.011 [ Links ]
62. García-Tavera JL, Valdés-Lozano D, Poblete-Naredo I, Albores-Medina A, Zapata-Pérez O. Bile benzo[a]pyrene concentration and hepatic CYP1A induction in hypoxic adult tilapia ( Oreochromis niloticus ) . Chemosphere. 2013;92:16-23. http://dx.doi.org/10.1016/j.chemosphere.2013.03.034 [ Links ]
63. Castorena-Torres F, Bermúdez-de León M, Cisneros B, Zapata-Pérez O, Salinas JE, Albores A. Changes in gene expression induced by polycyclic aromatic hydrocarbons in the human cell lines HepG2 and A549 . Toxicol In Vitro. 2008;22:411-21. http://dx.doi.org/10.1016/j.tiv.2007.10.009 [ Links ]
64. Topinka J, Marvanova S, Vondracek J, Sevastyanova O, Novakova Z, Krcmar P, et al. DNA adducts formation and induction of apoptosis in rat liver epithelial ´stem-like´ cells exposed to carcinogenic polycyclic aromatic hydrocarbons . Mutat Res. 2008;638:122-32. http://dx.doi.org/10.1016/j.mrfmmm.2007.09.004 [ Links ]
65. Aung HH, Lame MW, Gohil K, He G, Denison MS, Rutledge JC, et al. Comparative gene responses to collected ambient particles in vitro : Endothelial responses . Physiol Genomics. 2011;43:917-29. http://dx.doi.org/10.1152/physiolgenomics.00051.2011 [ Links ]
66. Totlandsdal AI, Cassee FR, Schwarze P, Refsnes M, Lag M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells . Part Fibre Toxicol. 2010;7:41. http://dx.doi.org/10.1186/1743-8977-7-41 [ Links ]
67. Gebel S, Gerstmayer B, Bosio A, Haussmann HJ, van Miert E, Muller T. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke . Carcinogenesis. 2004;25:169-78. http://dx.doi.org/10.1093/carcin/bgg193 [ Links ]
68. Nagaraj NS, Beckers S, Mensah JK, Waigel S, Vigneswaran N, Zacharias W. Cigarette smoke condensate induces cytochromes P450 and aldo-keto reductases in oral cancer cells . Toxicol Lett. 2006;165:182-94. http://dx.doi.org/10.1016/j.toxlet.2006.03.008 [ Links ]
69. Maunders H, Patwardhan S, Phillips J, Clack A, Richter A. Human bronchial epithelial cell transcriptome: Gene expression changes following acute exposure to whole cigarette smoke in vitro . Am J Physiol Lung Cell Mol Physiol. 2007;292:L1248-56. http://dx.doi.org/10.1152/ajplung.00290.2006 [ Links ]
70. Mahadevan B, Keshava C, Musafia-Jeknic T, Pecaj A, Weston A, Baird WM. Altered gene expression patterns in MCF-7 cells induced by the urban dust particulate complex mixture standard reference material 1649a . Cancer Res. 2005;65:1251-8. http://dx.doi.org/10.1158/0008-5472.CAN-04-2357 [ Links ]
71. Jacob A, Hartz AM, Potin S, Coumoul X, Yousif S, Scherrmann JM, et al. Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1 by TCDD and diesel exhaust particles in rat brain microvessels . Fluids Barriers CNS. 2011;8:23. http://dx.doi.org/10.1186/2045-8118-8-23 [ Links ]
72. Riechelmann H, Deutschle T, Grabow A, Heinzow B, Butte W, Reiter R. Differential response of Mono Mac 6, BEAS-2B, and Jurkat cells to indoor dust . Environ Health Perspect. 2007;115:1325-32. http://dx.doi.org/10.1289/ehp.9874 [ Links ]
73. Verheyen GR, Nuijten JM, van Hummelen P, Schoeters GR. Microarray analysis of the effect of diesel exhaust particles on in vitro cultured macrophages . Toxicol In Vitro. 2004;18: 377-91. http://dx.doi.org/10.1016/j.tiv.2003.10.007 [ Links ]
74. Koike E, Hirano S, Furuyama A, Kobayashi T. cDNA microarray analysis of rat alveolar epithelial cells following exposure to organic extract of diesel exhaust particles . Toxicol Appl Pharmacol. 2004;201:178-85. http://dx.doi.org/10.1016/j.taap.2004.05.006 [ Links ]
75. Nordskog BK, Blixt AD, Morgan WT, Fields WR, Hellmann GM. Matrix-degrading and pro-inflammatory changes in human vascular endothelial cells exposed to cigarette smoke condensate . Cardiovasc Toxicol. 2003;3:101-17. http://dx.doi.org/10.1385/CT:3:2:101 [ Links ]
76. Matsunaga T, Morikawa Y, Haga M, Endo S, Soda M, Yamamura K, et al. Exposure to 9,10-phenanthrenequinone accelerates malignant progression of lung cancer cells through up-regulation of aldo-keto reductase 1B10 . Toxicol Appl Pharmacol. 2014;278:180-9. http://dx.doi.org/10.1016/j.taap.2014.04.024 [ Links ]
77. Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes . Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:103-16. [ Links ]
78. Ueng TH, Hung CC, Kuo ML, Chan PK, Hu SH, Yang PC, et al. Induction of fibroblast growth factor-9 and interleukin-1alpha gene expression by motorcycle exhaust particulate extracts and benzo(a)pyrene in human lung adenocarcinoma cells . Toxicol Sci. 2005;87:483-96. http://dx.doi.org/10.1093/toxsci/kfi251 [ Links ]
79. Zhang H, Liu H, Davies KJ, Sioutas C, Finch CE, Morgan TE, et al. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments . Free Radic Biol Med. 2012;52:2038-46. http://dx.doi.org/10.1016/j.freeradbiomed.2012.02.042 [ Links ]
80. Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: A control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases . Free Radic Res. 2010;44:1267-88. http://dx.doi.org/10.3109/10715762.2010.507670 [ Links ]
81. Huang YC, Schmitt M, Yang Z, Que LG, Stewart JC, Frampton MW, et al. Gene expression profile in circulating mononuclear cells after exposure to ultrafine carbon particles . Inhal Toxicol. 2010;22:835-46. http://dx.doi.org/10.3109/08958378.2010.486419 [ Links ]
82. Pettit AP, Brooks A, Laumbach R, Fiedler N, Wang Q, Strickland PO, et al. Alteration of peripheral blood monocyte gene expression in humans following diesel exhaust inhalation . Inhal Toxicol. 2012;24:172-81. http://dx.doi.org/10.3109/08958378.2012.654856 [ Links ]
83. Peretz A, Peck EC, Bammler TK, Beyer RP, Sullivan JH, Trenga CA, et al. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: An exploratory study of healthy human volunteers . Inhal Toxicol. 2007;19:1107-19. http://dx.doi.org/10.1080/08958370701665384 [ Links ]
84. Albertini RJ, Hayes RB. Somatic cell mutations in cancer epidemiology . IARC Sci Publ. 1997;142:159-84. [ Links ]
85. Albertini RJ. HPRT mutations in humans: Biomarkers for mechanistic studies . Mutat Res. 2001;489:1-16. http://dx.doi.org/10.1016/S1383-5742(01)00064-3 [ Links ]
86. Albertini RJ. Somatic gene mutations in vivo as indicated by the 6-thioguanine-resistant T-lymphocytes in human blood . Mutat Res. 1985;150:411-22. http://dx.doi.org/10.1016/0027-5107(85)90138-1 [ Links ]
87. Curry J, Karnaoukhova L, Guenette GC, Glickman BW. Influence of sex, smoking and age on human hprt mutation frequencies and spectra . Genetics. 1999;152:1065-77. [ Links ]
88. Ammenheuser MM, Hastings DA, Whorton EB Jr., Ward JB Jr. Frequencies of hprt mutant lymphocytes in smokers, non-smokers, and former smokers . Environ Mol Mutagen. 1997;30:131-8. http://dx.doi.org/10.1002/(SICI)1098-2280(1997)30:2<131::AID-EM5>3.0.CO;2-Q [ Links ]
89. Finette BA, Poseno T, Vacek PM, Albertini RJ. The effects of maternal cigarette smoke exposure on somatic mutant frequencies at the HPRT locus in healthy newborns . Mutat Res. 1997;377:115-23. http://dx.doi.org/10.1016/S0027-5107(97)00069-9 [ Links ]
90. Perera F, Hemminki K, Jedrychowski W, Whyatt R, Campbell U, Hsu Y, et al. In utero DNA damage from environmental pollution is associated with somatic gene mutation in newborns . Cancer Epidemiol Biomarkers Prev. 2002;11:1134-7. [ Links ]
91. Hou SM, Lambert B, Hemminki K. Relationship between HPRT mutant frequency, aromatic DNA adducts and genotypes for GSTM1 and NAT2 in bus maintenance workers . Carcinogenesis. 1995;16:1913-7. http://dx.doi.org/10.1093/carcin/16.8.1913 [ Links ]
92. Zanesi N, Mognato M, Pizzato M, Viezzer C, Ferri G, Celotti L. Determination of HPRT mutant frequency and molecular analysis of T-lymphocyte mutants derived from coke-oven workers . Mutat Res. 1998;412:177-86. http://dx. doi.org/10.1016/S1383-5718(97)00190-3 [ Links ]
93. Wolffe AP, Matzke MA. Epigenetics: Regulation through repression . Science. 1999;286:481-6. http://dx.doi.org/10.1126/science.286.5439.481 [ Links ]
94. Mirbahai L, Chipman JK. Epigenetic memory of environmental organisms: A reflection of lifetime stressor exposures . Mutat Res Genet Toxicol Environ Mutagen. 2014;764-765:10-7. http://dx.doi.org/10.1016/j.mrgentox.2013.10.003 [ Links ]
95. Baccarelli A, Bollati V. Epigenetics and environmental chemicals . Curr Opin Pediatr. 2009;21:243-51. http://dx.doi.org/10.1097/Mop.0b013e32832925cc [ Links ]
96. Nogueira da Costa A, Herceg Z. Detection of cancer-specific epigenomic changes in biofluids: Powerful tools in biomarker discovery and application . Mol Oncol. 2012;6:704-15. http://dx.doi.org/10.1016/j.molonc.2012.07.005 [ Links ]
97. Burris HH, Baccarelli AA. Environmental epigenetics: From novelty to scientific discipline . J Appl Toxicol. 2014;34:113-6. http://dx.doi.org/10.1002/jat.2904 [ Links ]
98. Bakulski KM, Fallin MD. Epigenetic epidemiology: Promises for public health research . Environ Mol Mutagen. 2014;55:171-83. http://dx.doi.org/10.1002/Em.21850 [ Links ]
99. Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements . Nucleic Acids Res. 2004;32:e38. http://dx.doi.org/10.1093/nar/gnh032 [ Links ]
100. Chuang JC, Jones PA. Epigenetics and microRNAs . Pediatr Res. 2007;61:24R-29R. http://dx.doi.org/10.1203/pdr.0b013e3180457684 [ Links ]
101. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps . Nat Rev Genet. 2007;8:286-98. http://dx.doi.org/10.1038/nrg2005 [ Links ]
102. Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics . Int J Epidemiol. 2012;41:79-105. http://dx.doi.org/10.1093/ije/dyr154 [ Links ]
103. Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient air pollution impairs regulatory T-cell function in asthma . J Allergy Clin Immunol. 2010;126:845-52. http://dx.doi.org/10.1016/j.jaci.2010.08.008 [ Links ]
104. Brunst KJ, Leung YK, Ryan PH, Khurana Hershey GK, Levin L, Ji H, et al. Forkhead box protein 3 (FOXP3) hypermethylation is associated with diesel exhaust exposure and risk for childhood asthma . J Allergy Clin Immunol. 2013;131:592-4. http://dx.doi.org/10.1016/j.jaci.2012.10.042 [ Links ]
105. Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles . Am J Respir Crit Care Med. 2009;179:572-8. http://dx.doi.org/10.1164/rccm.200807-1097OC [ Links ]
106. Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men . Environ Health Perspect. 2011;119:977-82. http://dx.doi.org/10.1289/ehp.1002773 [ Links ]
107. Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation . Environ Health Perspect. 2009;117:217-22. http://dx.doi.org/10.1289/ehp.11898 [ Links ]
108. Breton CV, Salam MT, Wang X, Byun HM, Siegmund KD, Gilliland FD. Particulate matter, DNA methylation in nitric oxide synthase, and childhood respiratory disease . Environ Health Perspect. 2012;120:1320-6. http://dx.doi.org/10.1289/ehp.1104439 [ Links ]
109. Salam MT, Byun HM, Lurmann F, Breton CV, Wang X, Eckel SP, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children . J Allergy Clin Immunol. 2012;129:232-9. http://dx.doi.org/10.1016/j.jaci.2011.09.037 [ Links ]
110. Liu J, Ballaney M, Al-Alem U, Quan C, Jin X, Perera F, et al. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci. 2008;102:76-81. http://dx.doi.org/10.1093/Toxsci/Kfm290 [ Links ]
111. Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location . Proc Natl Acad Sci USA. 2008;105:605-10. http://dx.doi.org/10.1073/Pnas.0705896105 [ Links ]
112. Pavanello S, Bollati V, Pesatori AC, Kapka L, Bolognesi C, Bertazzi PA, et al. Global and gene-specific promoter methylation changes are related to anti-B[a]PDE-DNA adduct levels and influence micronuclei levels in polycyclic aromatic hydrocarbon-exposed individuals . Int J Cancer. 2009;125:1692-7. http://dx.doi.org/10.1002/ijc.24492 [ Links ]
113. Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. Relation of DNA methylation of 5´-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma . PLoS One. 2009;4:e4488. http://dx.doi.org/10.1371/journal.pone.0004488 [ Links ]
114. Tang WY, Levin L, Talaska G, Cheung YY, Herbstman J, Tang D, et al. Maternal exposure to polycyclic aromatic hydrocarbons and 5´-CpG methylation of interferon-gamma in cord white blood cells . Environ Health Perspect. 2012;120:1195-200. http://dx.doi.org/10.1289/ehp.1103744 [ Links ]
115. Ouyang B, Baxter CS, Lam HM, Yeramaneni S, Levin L, Haynes E, et al. Hypomethylation of dual specificity phosphatase 22 promoter correlates with duration of service in firefighters and is inducible by low-dose benzo[a]pyrene . J Occup Environ Med. 2012;54:774-80. http://dx.doi.org/10.1097/JOM.0b013e31825296bc [ Links ]
116. Jenuwein T, Allis CD. Translating the histone code . Science. 2001;293:1074-80. http://dx.doi.org/10.1126/science.1063127 [ Links ]
117. Cao D, Bromberg PA, Samet JM. COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1 . Am J Respir Cell Mol Biol. 2007;37:232-9. http://dx.doi.org/10.1165/rcmb.2006-0449OC [ Links ]
118. Gilmour PS, Rahman I, Donaldson K, MacNee W. Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particles . Am J Physiol Lung Cell Mol Physiol. 2003;284:L533-40. http://dx.doi.org/10.1152/ajplung.00277.2002 [ Links ]
119. Teneng I, Montoya-Durango DE, Quertermous JL, Lacy ME, Ramos KS. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation . Epigenetics. 2011;6:355-67. http://dx.doi.org/10.4161/epi.6.3.14282 [ Links ]
120. Arita A, Niu J, Qu Q, Zhao N, Ruan Y, Nadas A, et al. Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel . Environ Health Perspect. 2012;120:198-203. http://dx.doi.org/10.1289/ehp.1104140 [ Links ]
121. Cantone L, Nordio F, Hou L, Apostoli P, Bonzini M, Tarantini L, et al. Inhalable metal-rich air particles and histone H3K4 dimethylation and H3K9 acetylation in a cross-sectional study of steel workers . Environ Health Perspect. 2011;119:964-9. http://dx.doi.org/10.1289/ehp.1002955 [ Links ]
122. Xi S, Yang M, Tao Y, Xu H, Shan J, Inchauste S, et al. Cigarette smoke induces C/EBP-beta-mediated activation of miR-31 in normal human respiratory epithelia and lung cancer cells . PLoS One. 2010;5:e13764. http://dx.doi.org/10.1371/journal.pone.0013764 [ Links ]
123. Wittmann J, Jack HM. Serum microRNAs as powerful cancer biomarkers . Biochim Biophys Acta. 2010;1806: 200-7. http://dx.doi.org/10.1016/j.bbcan.2010.07.002 [ Links ]
124. Hou L, Wang D, Baccarelli A . Environmental chemicals and microRNAs . Mutat Res. 2011;714:105-12. http://dx.doi.org/10.1016/j.mrfmmm.2011.05.004 [ Links ]
125. Xi S, Xu H, Shan J, Tao Y, Hong JA, Inchauste S, et al. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis . J Clin Invest. 2013;123:1241-61. http://dx.doi.org/10.1172/JCI61271 [ Links ]
126. Jardim MJ, Fry RC, Jaspers I, Dailey L, Díaz-Sánchez D. Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways . Environ Health Perspect. 2009;117: 1745-51. http://dx.doi.org/10.1289/ehp.0900756 [ Links ]
127. Bleck B, Grunig G, Chiu A, Liu M, Gordon T, Kazeros A, et al. MicroRNA-375 regulation of thymic stromal lymphopoietin by diesel exhaust particles and ambient particulate matter in human bronchial epithelial cells . J Immunol. 2013;190:3757-63. http://dx.doi.org/10.4049/jimmunol.1201165 [ Links ]
128. Nymark P, Guled M, Borze I, Faisal A, Lahti L, Salmenkivi K, et al. Integrative analysis of microRNA, mRNA and aCGH data reveals asbestos- and histology-related changes in lung cancer . Genes Chromosomes Cancer. 2011;50:585-97. http://dx.doi.org/10.1002/gcc.20880 [ Links ]
129. Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes . Environ Health Perspect. 2010;118:763-8. http://dx.doi.org/10.1289/ehp.0901300 [ Links ]
130. Farraj AK, Hazari MS, Haykal-Coates N, Lamb C, Winsett DW, Ge Y, et al. ST depression, arrhythmia, vagal dominance, and reduced cardiac micro-RNA in particulate-exposed rats . Am J Respir Cell Mol Biol. 2011;44:185-96. http://dx.doi.org/10.1165/rcmb.2009-0456OC [ Links ]
131. Bourdon JA, Saber AT, Halappanavar S, Jackson PA, Wu D, Hougaard KS, et al. Carbon black nanoparticle intratracheal installation results in large and sustained changes in the expression of miR-135b in mouse lung . Environ Mol Mutagen. 2012;53:462-8. http://dx.doi.org/10.1002/em.21706 [ Links ]
132. Fry RC, Rager JE, Bauer R, Sebastian E, Peden DB, Jaspers I, et al. Air toxics and epigenetic effects: Ozone altered microRNAs in the sputum of human subjects . Am J Physiol Lung Cell Mol Physiol. 2014;306:L1129-37. http://dx.doi.org/10.1152/ajplung.00348.2013 [ Links ]
133. Motta V, Angelici L, Nordio F, Bollati V, Fossati S, Frascati F, et al. Integrative analysis of miRNA and inflammatory gene expression after acute particulate matter exposure . Toxicol Sci. 2013;132:307-16. http://dx.doi.org/10.1093/toxsci/kft013 [ Links ]
134. Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B, et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci. 2013;135:265-76. http://dx.doi.org/10.1093/toxsci/kft150 [ Links ]
135. Duan H, Jiang Y, Zhang H, Wu Y. MiR-320 and miR-494 affect cell cycles of primary murine bronchial epithelial cells exposed to benzo[a]pyrene . Toxicol In Vitro. 2010;24:928-35. http://dx.doi.org/10.1016/j.tiv.2009.11.013 [ Links ]
136. Yauk CL, Jackson K, Malowany M, Williams A. Lack of change in microRNA expression in adult mouse liver following treatment with benzo(a)pyrene despite robust mRNA transcriptional response . Mutat Res. 2011;722:131-9. http://dx.doi.org/10.1016/j.mrgentox.2010.02.012 [ Links ]
137. Zhang B, Pan X. RDX induces aberrant expression of microRNAs in mouse brain and liver . Environ Health Perspect. 2009;117:231-40. http://dx.doi.org/10.1289/ehp.11841 [ Links ]
138. Perera FP. Environment and cancer: Who are susceptible? Science. 1997;278:1068-73. http://dx.doi.org/10.1126/science.278.5340.1068 [ Links ]
139. Norppa H. Cytogenetic biomarkers and genetic poly-morphisms . Toxicol Lett. 2004;149:309-34. http://dx.doi.org/10.1016/j.toxlet.2003.12.042 [ Links ]
140. Brookes AJ. The essence of SNPs . Gene. 1999;234:177-86. http://dx.doi.org/10.1016/S0378-1119(99)00219-X [ Links ]
134. Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B, et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract . Toxicol Sci. 2013;135:265-76. http://dx.doi.org/10.1093/toxsci/kft150
141. Manfredi S, Federici C, Picano E, Botto N, Rizza A, Andreassi MG. GSTM1, GSTT1 and CYP1A1 detoxification gene polymorphisms and susceptibility to smoking-related coronary artery disease: A case-only study . Mutat Res. 2007;621:106-12. http://dx.doi.org/10.1016/j.mrfmmm.2007.02.014 [ Links ]
142. Roszak A, Lianeri M, Sowinska A, Jagodzinski PP . CYP1A1 Ile462Val polymorphism as a risk factor in cervical cancer development in the Polish population . Mol Diagn Ther. 2014;18 445-50. http://dx.doi.org/10.1007/s40291-014-0095-2 [ Links ]
143. Rihs HP, Pesch B, Kappler M, Rabstein S, Rossbach B, Angerer J, et al. Occupational exposure to polycyclic aromatic hydrocarbons in German industries: Association between exogenous exposure and urinary metabolites and its modulation by enzyme polymorphisms . Toxicol Lett. 2005;157:241-55. http://dx.doi.org/10.1016/j.toxlet.2005.02.012 [ Links ]
144. Georgiadis P, Topinka J, Vlachodimitropoulos D, Stoikidou M, Gioka M, Stephanou G, et al. Interactions between CYP1A1 polymorphisms and exposure to environmental tobacco smoke in the modulation of lymphocyte bulky DNA adducts and chromosomal aberra-tions . Carcinogenesis. 2005;26:93-101. http://dx.doi.org/10. 1093/carcin/bgh294 [ Links ]
145. Betti M, Ferrante D, Padoan M, Guarrera S, Giordano M, Aspesi A, et al. XRCC1 and ERCC1 variants modify malignant mesothelioma risk: A case-control study . Mutat Res. 2011;708:11-20. http://dx.doi.org/10.1016/j.mrfmmm.2011.01.001 [ Links ]
146. Bagryantseva Y, Novotna B, Rossner P Jr, Chvatalova I, Milcova A, Svecova V, et al. Oxidative damage to biological macromolecules in Prague bus drivers and garagemen: Impact of air pollution and genetic polymorphisms . Toxicol Lett. 2010;199:60-8. http://dx.doi.org/10.1016/j.toxlet.2010.08.007 [ Links ]
147. Lan Q, He X. Molecular epidemiological studies on the relationship between indoor coal burning and lung cancer in Xuan Wei, China . Toxicology. 2004;198:301-5. http://dx.doi.org/10.1016/j.tox.2004.02.006 [ Links ]
148. Conlon MS, Johnson KC, Bewick MA, Lafrenie RM, Donner A. Smoking (active and passive), N-acetyltransferase 2, and risk of breast cancer . Cancer Epidemiol. 2010;34:142-9. http://dx.doi.org/10.1016/j.canep.2010.02.001 [ Links ]
149. Kelada SN, Eaton DL, Wang SS, Rothman NR, Khoury MJ. The role of genetic polymorphisms in environmental health . Environ Health Perspect. 2003;111:1055-64. http://dx.doi.org/sc271_5_1835 [ Links ]
150. Yang XR, Wacholder S, Xu Z, Dean M, Clark V, Gold B, et al. CYP1A1 and GSTM1 polymorphisms in relation to lung cancer risk in Chinese women . Cancer Lett. 2004;214:197-204. http://dx.doi.org/10.1016/j.canlet.2004.06.040 [ Links ]
151. Yang Y, Tian Y, Jin X, Yan C, Jiang F, Zhang Y, et al. A case-only study of interactions between metabolic enzyme polymorphisms and industrial pollution in childhood acute leukemia . Environ Toxicol Pharmacol. 2009;28:161-6. http://dx.doi.org/10.1016/j.etap.2009.03.004 [ Links ]
152. Dianzani I, Gibello L, Biava A, Giordano M, Bertolotti M, Betti M, et al. Polymorphisms in DNA repair genes as risk factors for asbestos-related malignant mesothelioma in a general population study . Mutat Res. 2006;599:124-34. http://dx.doi.org/10.1016/j.mrfmmm.2006.02.005 [ Links ]
153. Thier R, Bruning T, Roos PH, Rihs HP, Golka K, Ko Y, et al. Markers of genetic susceptibility in human environmental hygiene and toxicology: The role of selected CYP, NAT and GST genes . Int J Hyg Environ Health. 2003;206:149-71. http://dx.doi.org/10.1078/1438-4639-00209 [ Links ]
154. Merlo F, Andreassen A, Weston A, Pan CF, Haugen A, Valerio F, et al. Urinary excretion of 1-hydroxypyrene as a marker for exposure to urban air levels of polycyclic aromatic hydrocarbons . Cancer Epidemiol Biomarkers Prev. 1998;7:147-55. [ Links ]
155. Suh YJ, Kim BM, Park BH, Park H, Kim YJ, Kim H, et al. Cytochrome P450IA1 polymorphisms along with PM(10) exposure contribute to the risk of birth weight reduction . Reprod Toxicol. 2007;24:281-8. http://dx.doi.org/10.1016/j.reprotox.2007.07.001 [ Links ]
156. Yu KT, Ge C, Xu XF, Zou JC, Zou X, Zhen S. CYP1A1 polymorphism interactions with smoking status in oral cancer risk: Evidence from epidemiological studies . Tumour Biol. 2014;35:11183-91. http://dx.doi.org/10.1007/s13277-014-2422-y [ Links ]
157. Mooney LA, Bell DA, Santella RM, Van Bennekum AM, Ottman R, Paik M, et al. Contribution of genetic and nutritional factors to DNA damage in heavy smokers . Carcinogenesis. 1997;18:503-9. http://dx.doi.org/10.1093/carcin/18.3.503 [ Links ]
158. Muñoz B, Magana JJ, Romero-Toledo I, Juárez-Pérez E, López-Moya A, Leyva-García N, et al. The relationship among IL-13, GSTP1, and CYP1A1 polymorphisms and environmental tobacco smoke in a population of children with asthma in Northern México . Environ Toxicol Pharmacol. 2012;33:226-32. http://dx.doi.org/10.1016/j.etap.2011.12.007 [ Links ]
159. Cao L, Lin J, He B, Wang H, Rao J, Liu Y, et al. A regulatory variant in CYP2E1 affects the risk of lung squamous cell carcinoma . Tumour Biol. 2014;35:455-62. http://dx.doi.org/10.1007/s13277-013-1063-x [ Links ]
160. Yang M, Koga M, Katoh T, Kawamoto T. A study for the proper application of urinary naphthols, new biomarkers for airborne polycyclic aromatic hydrocarbons . Arch Environ Contam Toxicol. 1999;36:99-108. http://dx.doi.org/10.1007/s002449900447 [ Links ]
160. Yang M, Koga M, Katoh T, Kawamoto T. A study for the proper application of urinary naphthols, new biomarkers for airborne polycyclic aromatic hydrocarbons . Arch Environ Contam Toxicol. 1999;36:99-108. http://dx.doi.org/10.1007/s002449900447
161. Kato S, Bowman ED, Harrington AM, Blomeke B, Shields PG. Human lung carcinogen-DNA adduct levels mediated by genetic polymorphisms in vivo. J Natl Cancer Inst. 1995;87:902-7. http://dx.doi.org/10.1093/carcin/bgh303 [ Links ]
162. Chen B, Hu Y, Jin T, Lu D, Shao M, Zheng L, et al. The influence of metabolic gene polymorphisms on urinary 1-hydroxypyrene concentrations in Chinese coke oven workers . Sci Total Environ. 2007;381:38-46. http://dx.doi.org/10.1016/j.scitotenv.2007.02.021 [ Links ]
163. Georgiadis P, Demopoulos NA, Topinka J, Stephanou G, Stoikidou M, Bekyrou M, et al. Impact of phase I or phase II enzyme polymorphisms on lymphocyte DNA adducts in subjects exposed to urban air pollution and environmental tobacco smoke . Toxicol Lett. 2004;149:269-80. http://dx.doi.org/10.1016/j.toxlet.2003.12.038 [ Links ]
164. Lan Q, Mumford JL, Shen M, Demarini DM, Bonner MR, He X, et al. Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions . Carcinogenesis. 2004;25:2177-81. http://dx.doi.org/10.1093/carcin/bgh240 [ Links ]
165. Figueroa JD, Malats N, García-Closas M, Real FX, Silverman D, Kogevinas M, et al. Bladder cancer risk and genetic variation in AKR1C3 and other metabolizing genes . Carcinogenesis. 2008;29:1955-62. http://dx.doi.org/10.1093/carcin/bgn163 [ Links ]
166. Walker K, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. Genetic polymorphism in N-acetyltransferase (NAT): Population distribution of NAT1 and NAT2 activity . J Toxicol Environ Health B Crit Rev. 2009;12:440-72. http://dx.doi.org/10.1080/10937400903158383 [ Links ]
167. Costa DJ. Influence of GSTM1 and NAT2 genotypes on the relationship between personal exposure to PAH and biomarkers of internal dose . Biomarkers. 1998;3:411-24. http://dx.doi.org/10.1080/135475098231057 [ Links ]
168. Cascorbi I, Brockmoller J, Mrozikiewicz PM, Bauer S, Loddenkemper R, Roots I. Homozygous rapid arylamine N-acetyltransferase (NAT2) genotype as a susceptibility factor for lung cancer . Cancer Res. 1996;56:3961-6. [ Links ]
169. Prasad SB, Vidyullatha P, Vani GT, Devi RP, Rani UP, Reddy PP, et al. Association of gene polymorphism in detoxification enzymes and urinary 8-OHdG levels in traffic policemen exposed to vehicular exhaust . Inhal Toxicol. 2013;25:1-8. http://dx.doi.org/10.3109/08958378.2012.745634 [ Links ]
170. Hong YC, Lee KH, Yi CH, Ha EH, Christiani DC . Genetic susceptibility of term pregnant women to oxidative damage . Toxicol Lett. 2002;129:255-62. http://dx.doi.org/10.1016/S0378-4274(02)00014-0 [ Links ]
171. Park EY, Hong YC, Lee KH, Im MW, Ha E, Kim YJ, et al. Maternal exposure to environmental tobacco smoke, GSTM1/T1 polymorphisms and oxidative stress . Reprod Toxicol. 2008;26:197-202. http://dx.doi.org/10.1016/j.reprotox.2008.08.010 [ Links ]
172. Pavanello S, Pulliero A, Clonfero E . Influence of GSTM1 null and low repair XPC PAT+ on anti-B[a]PDE-DNA adduct in mononuclear white blood cells of subjects low exposed to PAHs through smoking and diet . Mutat Res. 2008;638:195-204. http://dx.doi.org/10.1016/j.mrfmmm.2007.10.004 [ Links ]
173. Pavanello S, Pulliero A, Siwinska E, Mielzynska D, Clonfero E. Reduced nucleotide excision repair and GSTM1-null genotypes influence anti-B[a]PDE-DNA adduct levels in mononuclear white blood cells of highly PAH-exposed coke oven workers . Carcinogenesis. 2005;26:169-75. http://dx.doi.org/10.1093/carcin/bgh303 [ Links ]
174. Suh YJ, Ha EH, Park H, Kim YJ, Kim H, Hong YC. GSTM1 polymorphism along with PM10 exposure contributes to the risk of preterm delivery . Mutat Res. 2008;656:62-7. http://dx.doi.org/10.1016/j.mrgentox.2008.07.006 [ Links ]
175. Knudsen LE, Norppa H, Gamborg MO, Nielsen PS, Okkels H, Soll-Johanning H, et al. Chromosomal aberrations in humans induced by urban air pollution: Influence of DNA repair and polymorphisms of glutathione S-transferase M1 and N-acetyltransferase 2 . Cancer Epidemiol Biomarkers Prev. 1999;8:303-10. [ Links ]
176. Lee KM, Park SK, Kim SU, Doll MA, Yoo KY, Ahn SH, et al. N-acetyltransferase (NAT1, NAT2) and glutathione S-transferase (GSTM1, GSTT1) polymorphisms in breast cancer . Cancer Lett. 2003;196:179-86. http://dx.doi.org/10.1016/S0304-3835(03)00311-2 [ Links ]
177. Chen HC, Cao YF, Hu WX, Liu XF, Liu QX , Zhang J, et al. Genetic polymorphisms of phase II metabolic enzymes and lung cancer susceptibility in a population of Central South China . Dis Markers. 2006;22:141-52. http://dx.doi.org/10.1155/2006/436497 [ Links ]

