










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.36 no.3 Bogotá July/Sept. 2016
https://doi.org/10.7705/biomedica.v36i3.2639
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2639
Contribución de los autores:
Nancy Gélvez, Derly Castro y Greizy López: análisis y procesamiento de las muestras e interpretación de los datos
Johanna Acosta, Martha Bermúdez, Juan Carlos Prieto y Marta L. Tamayo: consecución de las muestras, evaluación clínica de los pacientes, y análisis e interpretación de los datos
Todos los autores participaron en la escritura del manuscrito.
Recibido: 05/02/15; aceptado: 05/04/16
Introducción. La fenilcetonuria es un trastorno metabólico caracterizado por un compromiso neurológico grave y por alteraciones del comportamiento. Su diagnóstico temprano permite establecer un tratamiento efectivo que evita las secuelas y modifica el pronóstico.
Objetivo. Caracterizar a una familia con fenilcetonuria en Colombia, a nivel clínico, bioquímico y molecular.
Materiales y métodos. Se estudió una población de siete individuos de una familia consanguínea en la que cuatro hijos presentaban signos clínicos sugestivos de fenilcetonuria. Una vez firmado el consentimiento informado, se tomaron muestras de sangre y orina para las pruebas colorimétricas, la cromatografía de capa fina y la cromatografía líquida de alta eficacia. Se extrajo el ADN y se secuenciaron los 13 exones del gen PAH de todos los sujetos estudiados. Se diseñaron iniciadores para cada exón con el programa Primer 3; la secuenciación automática se hizo con el equipo Abiprism 3100 Avant y, el análisis de las secuencias, con el programa SeqScape v2.0.
Resultados. Se describieron las características clínicas y moleculares de una familia colombiana con fenilcetonuria en la que se identificó la mutación c.398_401delATCA; se presentó una correlación fenotipo-genotipo con una interesante variabilidad clínica entre los afectados, a pesar de tener la misma mutación.
Conclusiones. Es importante el reconocimiento temprano de esta enfermedad para evitar sus secuelas neurológicas y psicológicas, pues los pacientes llegan a edades avanzadas sin diagnóstico ni tratamiento adecuados.
Palabras clave: fenilcetonurias, fenilalanina hidroxilasa, discapacidad intelectual, mutación, genética, diagnóstico precoz, dieta.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2639
Phenotypic and molecular characterization of a Colombian family with phenylketonuria
Introduction: Phenylketonuria is a metabolic disorder characterized by severe neurological involvement and behavioral disorder, whose early diagnosis enables an effective treatment to avoid disease sequelae, thus changing the prognosis.
Objective: To characterize a family with phenylketonuria in Colombia at clinical, biochemical and molecular levels.
Materials and methods: The population consisted of seven individuals of a consanguineous family with four children with suggestive symptoms of phenylketonuria. After signing an informed consent, blood and urine samples were taken for colorimetric tests and high performance liquid and thin layer chromatographies. DNA extraction and sequencing of the 13 exons of the PAH gene were performed in all subjects. We designed primers for each exon with the Primer 3 software using automatic sequencing equipment Abiprism 3100 Avant. Sequences were analyzed using the SeqScape, v2.0, software.
Results: We described the clinical and molecular characteristics of a Colombian family with phenylketonuria and confirmed the presence of the mutation c.398_401delATCA. We established a genotype-phenotype correlation, highlighting the interesting clinical variability found among the affected patients despite having the same mutation in all of them.
Conclusions: Early recognition of this disease is very important to prevent its neurological and psychological sequelae, given that patients reach old age without diagnosis or proper management.
Key words: Phenylketonurias, phenylalanine hydroxylase, intellectual disability, mutation, genetics, early diagnosis, diet.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2639
La fenilcetonuria (PKU, MIM#261600) es un error innato del metabolismo de los aminoácidos, genéticamente heterogéneo y de herencia autosómica recesiva (1,2). Es la forma más grave del grupo de enfermedades denominadas hiperfenilalaninemias (HPA) (1), y causa discapacidad y otras secuelas neurológicas en los individuos afectados (1).
La fenilcetonuria se caracteriza por un aumento del aminoácido fenilalanina y sus metabolitos (fenil-lactato, fenil-acetato, fenil-piruvato y fenil-acetilglutamina), en todos los líquidos corporales, debido a una mutación en el gen de la enzima fenilalanina hidroxilasa ( PAH ) (1-5). La principal ruta metabólica de la fenilalanina es su hidroxilación a tirosina por medio de la enzima PAH y el uso de la tetrahidrobiopterina (BH 4 ) como cofactor; esta enzima se reduce después de la reacción y se regenera por medio de la reductasa de dihidropterina (DHPR) (1,2,4,6).
El diagnóstico de la enfermedad se basa en la detección de niveles elevados de fenilalanina en el plasma, aumento que resulta en una acumulación tóxica de fenilalanina en el cerebro, con el consecuente daño neurocognitivo y neuromotor, así como en dificultades psicológicas y trastornos del comportamiento (7).
Aunque la fisiopatología de la enfermedad no es completamente clara, se la ha relacionado con el exceso de fenilalanina en la sangre. En diversos estudios se ha demostrado que existe un desequilibrio en el transporte a través de la barrera hematoencefálica de la fenilalanina y de otros aminoácidos neutros largos, como la tirosina, el triptófano, la valina, la leucina, la isoleucina, la treonina, la metionina y la histidina, lo cual afecta la síntesis de otras aminas biógenas (dopamina, noradrenalina y serotonina) (4,5,8).
La principal causa de la hiperfenilalaninemia es la deficiencia de PAH (98 % de los casos). Otros defectos en la síntesis o la regeneración de la tetrahidrobiopterina representan el 2 % restante de los casos, lo cual debe considerarse en el diagnóstico diferencial, ya que su tratamiento y pronóstico son diferentes (1).
El gen PAH está ubicado en la región 12q22-24.1 y tiene 13 exones (3,4). Hasta el momento, se han reportado más de 500 mutaciones diferentes en este gen (1-3). En la base de datos Phenylalanine Hydroxylase Locus Knowledgebase (PAHdb), se encuentra que los exones en los que más se han descrito mutaciones son: el exón 7 (15,54 %), el 6 (13,57 %), el 11 (8,93 %), el 10 (8,75 %), el 3 (7,68 %), el 5 (5,71 %), el 12 (5 %), el 9 (4,29 %), el 8 (4,11 %) y el 2 (3,93 %) (9). La mutación más frecuente (9,23 %) a nivel mundial es la p.R408W en el exón 12 (9).
En Latinoamérica, las mutaciones varían según el país, pero la p.V388M en el exón 11 es común a todos, aunque con frecuencias variables: en Brasil oscila entre 9 y 21 % (São Paulo y Minas Gerais, respectivamente), en Chile es de 13 % y en México se ha reportado un caso (9-12); en Venezuela se reportó la mutación p.S349L en el exón 10 (9). Entre estas se han descrito mutaciones de cambio de sentido (62 %), mutaciones sin sentido (5 %), mutaciones en el sitio de empalme (11 %), deleciones pequeñas (13 %) e inserciones (2 %), que afectan tanto a los exones como a los intrones (1,3).
El espectro de las mutaciones varía en función de la población y el grupo étnico (1). Generalmente, cada mutación individual tiene una frecuencia muy baja, así que hay pocas mutaciones recurrentes y muchas nuevas en una misma población (1). La fenilcetonuria es una enfermedad con gran heterogeneidad alélica (3). En la literatura científica se ha reportado un estado de heterocigoto compuesto con una variabilidad fenotípica, hasta en 70 % de los pacientes con fenilcetonuria (1). Su prevalencia en la población caucásica fluctúa entre 1:4.000 y 21.000 recién nacidos, con un promedio de 1:10.000 nacidos vivos, dependiendo de la zona geográfica (1,4).
Gracias al establecimiento de programas de tamización neonatal a nivel mundial, el cuadro típico de la fenilcetonuria clásica es poco común, ya que se inicia la restricción en la dieta y la administración de preparados de aminoácidos libres de fenilalanina desde el nacimiento (6,8). Sin embargo, es importante señalar que en Colombia aún no se cuenta con este tipo de programas.
Los pacientes que no reciben tratamiento presentan alteraciones en el desarrollo cerebral (problemas de mielinización) visibles en el electroencefalograma, en la síntesis de neurotransmisores (dopamina, noradrenalina y serotonina), y en la presencia de microcefalia, epilepsia, temblor, retardo mental (IQ=50), espasticidad en las extremidades y signos extrapiramidales, así como alteraciones del comportamiento (hiperactividad, estereotipias, ansiedad y agresividad) (1,5). La excreción excesiva de fenilalanina y sus metabolitos crea un olor a humedad en el cuerpo y favorece la aparición de eccema en la piel (1,5). La inhibición secundaria de la tirosina es la responsable de la hipopigmentación generalizada (piel, pelo e iris) (1,5).
La fenilcetonuria se puede diagnosticar mediante diferentes pruebas de laboratorio. Las pruebas colorimétricas y la cromatografía en capa fina son formas cualitativas de evaluar la presencia de fenilalanina y sus metabolitos en la orina (4,5). Los resultados positivos en estas pruebas deben ser confirmados con pruebas cuantitativas (4,5). La cuantificación en sangre de la fenilalanina y de otros aminoácidos como la tirosina, y la medición en orina de la tetrahidrobiopterina, ayudan al diagnóstico de la enfermedad (4,5). La cromatografía líquida de alto rendimiento ( High Performance Liquid Chromatography , HPLC) es uno de los métodos cualitativos más utilizados para confirmar el diagnóstico en estos pacientes (5).
El diagnóstico molecular es una alternativa viable, fácil de realizar y con un alto porcentaje de detección de la mutación (hasta 99 % al secuenciar el gen PAH ), aunque no sería costo-efectivo para un programa de tamización (3).
El objetivo principal del tratamiento es disminuir suficientemente los niveles de fenilalanina en la sangre para prevenir los efectos neuropatológicos en el cerebro y, en lo posible, mantener los niveles de este aminoácido entre 2 y 6 mg/dl (120-360 µmol/L) (1,4,13). La ingestión de proteínas de origen animal debe restringirse dependiendo de los niveles de la fenilalanina en la sangre en el momento del diagnóstico (1,4,13). Además de la administración de preparados comerciales de otros aminoácidos esenciales libres de feni-lalanina, algunos estudios han confirmado los efectos benéficos de la administración conjunta de tetrahidrobiopterina (4,5). Cuando existe déficit del cofactor, se administra tetrahidrobiopterina con levadopa y L-5-hidroxitriptofano (1). Deben vigilarse en forma estricta los niveles de fenilalanina en la sangre en quienes reciben tratamiento.
En este trabajo se logró caracterizar a una familia con fenilcetonuria en Colombia, a nivel clínico, bioquímico y molecular.
Materiales y métodos
Población de estudio
La población de estudio incluyó a siete individuos pertenecientes a una familia consanguínea procedente de Tasco, en el departamento de Boyacá. En esta familia, cuatro hijos presentaban manifestaciones clínicas sugestivas de fenilcetonuria.
Análisis de las muestras
Se tomaron muestras de sangre y orina, y se hicieron las correspondientes pruebas bioquími cas (colorimétricas, cromatografía de capa fina y HPLC para la confirmación de la fenilcetonuria). Posteriormente, se extrajo ADN mediante el método de fenol cloroformo; se amplificó mediante reacción en cadena de la polimerasa (PCR) convencional, y se secuenciaron los 13 exones del gen PAH en todos los integrantes de la familia. El árbol genealógico y las fotografías de los afectados se muestran en la figura 1.
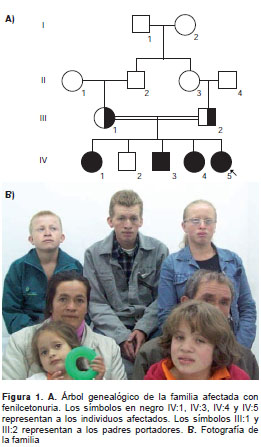
Se diseñaron los iniciadores para cada uno de los exones con el programa Primer 3. La secuenciación automática se hizo con el equipo Abiprism 3100 Avant y las secuencias obtenidas se analizaron utilizando el programa SeqScape, v2.0.
Consideraciones éticas
Este estudio fue aprobado por el Comité de Investigaciones y Ética de la Pontificia Universidad Javeriana, como consta en el Acta N o 02-2011. Cada uno de los participantes del estudio firmó el debido consentimiento informado, aprobado por este mismo Comité.
Resultados
Descripción clínica
Caso 1. Era una niña de cuatro años de edad en el momento de la evaluación, producto del quinto embarazo. Se hicieron controles prenatales desde los dos meses de gestación; no se registró daño perinatal y se trató de un parto normal, a término y sin complicaciones. Desde los primeros meses de vida, se evidenció el retardo en el desarrollo psicomotor y del lenguaje. En una primera evaluación genética clínica, se reportó bajo peso (<P3), baja talla (<P3), pliegue epicántico bilateral, braquidactilia, hipotonía periférica, ausencia de lenguaje, piel hipopigmentada, (fototipo 2: blanca beige ), ojos muy claros y cabello castaño claro.
Caso 2. Era una niña de 13 años de edad producto del cuarto embarazo; se hicieron los controles prenatales y no se reportó daño perinatal. Desde los seis meses de vida, se evidenció retardo del desarrollo psicomotor y del lenguaje, además de retardo pondoestatural significativo. A los cuatro años de edad, la niña comenzó a tener episodios convulsivos con pérdida del tono y, posteriormente, crisis tonicoclónicas generalizadas, controladas solo parcialmente con la medicación. En el examen físico se encontró baja talla (<P3), peso y perímetro cefálico adecuados, heterocromía parcial del iris, braquidactilia en manos y pies, ausencia de lenguaje verbal, hiperreflexia en miembros inferiores, piel blanca hipopigmentada, ojos muy claros y pelo castaño claro.
Caso 3. Era un niño de 14 años de edad producto del tercer embarazo; hubo controles prenatales, el parto fue normal, a término y en el domicilio, al parecer, sin complicaciones. Presentó problemas de aprendizaje desde el inicio de la etapa escolar y repitió dos veces cada año académico debido al mal desempeño. En el momento del estudio, presentaba agresividad hacia otros y ansiedad. En el examen físico se encontró frente amplia, pelo, cejas y pestañas rojizas, ojos azules y raíz nasal alta; establecía contacto con el examinador, obedecía órdenes sencillas y su lenguaje era poco expresivo.
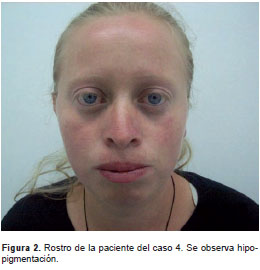
Caso 4. Era una mujer de 21 años de edad producto del primer embarazo; no hubo controles prenatales, el parto fue eutócico, podálico a término, y la niña fue internada en la unidad de cuidados del recién nacido durante dos días (no se especificó la causa). Desde el inicio de la etapa escolar, presentó dificultades en el aprendizaje asociadas a déficit de atención y ansiedad. En el examen físico se encontró que el peso, la talla y el perímetro cefálico estaban dentro de los límites normales; en cuanto a la apariencia, tenía frente amplia, pelo, cejas y pestañas rojizas, ojos azules, raíz nasal alta, hipoplasia de alas nasales, dientes hiperpigmentados, y presentaba abstracción normal, bradipsiquia y bradilalia (figura 2).
Exámenes paraclínicos
Las pruebas colorimétricas con cloruro férrico, dinitro-fenilhidrazina (DNFH), nitrosonaftol y nitro-prusiato se hicieron en los cinco hijos de la familia, y el resultado fue positivo con el cloruro férrico y la DNFH en los cuatro individuos afectados. Además, se hizo la cromatografía de aminoácidos en plasma y orina en todos ellos, y se evidenció una banda que migra a la altura de fenilalanina en los cuatro individuos con pruebas colorimétricas positivas.
Se cuantificaron los aminoácidos mediante HPLC y se encontraron niveles de fenilalanina superiores a 1.200 µmol/L en los cuatro individuos afectados, con lo cual se determinó la presencia de la variante clásica de la enfermedad.
Mediante secuenciación automática, se identificó una deleción de 4 pb, la c.398_401delATCA, en estado homocigoto en el exón 4 en todos los afectados, y en estado heterocigoto en los padres, cambio que ya ha sido reportado previamente (14). El hermano asintomático no es portador de la mutación. En la figura 3 se muestra el electroferograma con la mutación descrita.
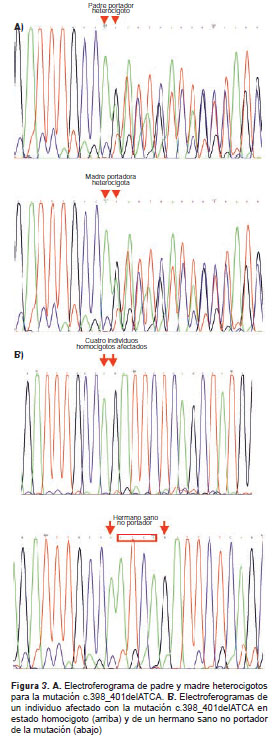
Además, en la región que flanquea el intrón 4, se detectó una variante, un cambio de C>T, que no se había reportado previamente. La madre y el hijo sano fueron heterocigotos para este cambio, y el padre y los hijos afectados, homocigotos.
Manejo médico
Después de la confirmación diagnóstica, se inició la restricción en la ingestión de proteínas y la administración de otros preparados comerciales de aminoácidos libres de fenilalanina; asimismo, se implementó un programa vigilado de pedagogía familiar sobre el tratamiento médico y nutricional de la enfermedad, con lo que se evidenció una importante mejoría en los casos 1 y 2 con respecto al retardo en su desarrollo psicomotor y lingüístico, así como de las convulsiones. En los casos 3 y 4, hubo mejoría significativa de los síntomas de ansiedad y agresividad hacia otros.
Discusión
Se describe una familia colombiana con fenil-cetonuria clásica confirmada mediante métodos bioquímicos y moleculares, y se da cuenta del hallazgo de la mutación c.398_401delATCA.
En los individuos afectados con la misma mutación, se evidenció una interesante variabilidad fenotípica familiar, tanto en la gravedad del compromiso neurológico como en la forma y el grado de la hipopigmentación. La variabilidad fenotípica se ha reportado previamente en varios estudios. El genotipo puede correlacionarse con la tolerancia a la fenilalanina y la reacción a tratamientos con tetrabiopterinas, pero no es un predictor totalmente confiable del fenotipo (15).
Por un lado, la variabilidad del fenotipo metabólico está determinada por el genotipo, es decir, por la mutación encontrada en el gen PAH ; sin embargo, no predice la gravedad del compromiso neurológico (16). Incluso los hermanos con un mismo genotipo, como en este caso, presentan un fenotipo clínico, e incluso bioquímico, diferente. Los mecanismos causantes de estas diferencias en la patogenia de la enfermedad aún no son del todo claros, y varios autores han propuesto que podrían estar influenciados por factores ambientales y epigenéticos e, incluso, mecanismos reguladores como la distribución de la fenilalanina en la barrera hematoencefálica (17), el transporte de otros aminoácidos en dicha barrera, y polimorfismos en otros genes reguladores, como el gran trasportador de aminoácidos neutros (LAT1) (18), entre otras hipótesis.
Según la literatura científica, el fenotipo correspondiente a la mutación encontrada (c.398_ 401delATCA) es el de una fenilcetonuria clásica, en la cual se reportan niveles de fenilalanina sérica de más de 1.800 µmol/L, lo cual concuerda con los hallazgos bioquímicos en los individuos afectados en este estudio. La mutación se encontró en estado homocigoto, aunque se ha reportado el estado de heterocigoto compuesto con una frecuencia de hasta 70 % (2,19,20).
La mutación detectada en el presente estudio fue reportada en 1996 por Guldberg, et al. , en el estudio colaborativo de fenilcetonuria materna (MPKUCS), en una mujer de origen francés, holandés y alemán procedente de Estados Unidos, quien presentaba la forma clásica (14).
En este estudio, después de iniciar el tratamiento con restricción proteica de alto valor biológico y administración de una fórmula de aminoácidos libre de fenilalanina, se observaron resultados contradictorios, pues los pacientes menos afectados (casos 3 y 4) mejoraron su rendimiento académico, su atención e hiperactividad, en tanto que en el caso 1 se observó mejoría en el tono muscular y el control de las convulsiones, pero, en el caso 2, no se observaron cambios significativos en su autismo, ni en el déficit cognitivo y, aunque se logró disminuir la frecuencia de las crisis convulsivas, no se pudieron controlar totalmente.
Lamentablemente, a pesar de las intervenciones nutricionales, el asesoramiento y el acompañamiento, no todos los pacientes mantuvieron el tratamiento, básicamente por problemas socio-culturales, y dos de ellos lo abandonaron. Esto resalta aún más la importancia de un diagnóstico temprano mediante la tamización neonatal de esta y otras condiciones relacionadas con los aminoácidos, ya que, como se pudo comprobar con esta familia y como se reporta en otros estudios de pacientes diagnosticados tardíamente, una vez instauradas, las secuelas neurológicas son irreversibles y es difícil conseguir que el tratamiento se mantenga (7).
La importancia de los programas de tamización neonatal es evidente, pues permiten la detección temprana de la enfermedad y logran un cometido crucial en la medicina actual: evitar que se instauren secuelas irreversibles de enfermedades que pudieron haber sido tratadas y controladas si se hubieran diagnosticado en el momento del nacimiento (21,22).
Es muy importante que los médicos generales, los pediatras y otros especialistas aprendan a reconocer esta enfermedad y a diagnosticarla tempranamente, para evitar el daño neurológico irreversible. Este caso lamentable de detección tardía en una zona rural apartada, lo cual seguramente se ha observado también en otros países en desarrollo, nos lleva a proponer que en las zonas rurales del país y en sectores económicamente deprimidos, se implementen adecuadamente los programas de tamización neonatal para la detección temprana, y que el sistema de salud refuerce las medidas pedagógicas de prevención y promoción de la salud, así como la capacitación del personal de atención primaria y secundaria (médicos generales, pediatras e internistas, entre otros) en torno a enfermedades raras o huérfanas. Debe tenerse presente siempre el cuadro clínico de la fenilcetonuria y la forma de diagnosticarla, pues solo así es posible su detección.
La búsqueda de mutaciones ya reportadas en la literatura científica mundial, o la secuenciación completa del gen, cuando sea posible, permitirá ampliar el conocimiento del genotipo de familias colombianas con diagnóstico clínico de fenilcetonuria y, consecuentemente, establecer una adecuada correlación del genotipo y el fenotipo.
Asesoría genética
Gracias a la detección de la mutación en esta familia, fue posible brindarle una completa asesoría genética sobre el mecanismo de herencia de la enfermedad y las probabilidades de descendencia afectada en cada una de las generaciones.
A la Fundación Derecho a la Desventaja (Fundalde), por la colaboración global en el proyecto; a Metabólica SAS, por el tratamiento que le donó a los pacientes durante un año; al Instituto de Genética Humana de la Pontificia Universidad Javeriana, por la financiación recibida, y al Laboratorio de Genética del Hospital de La Victoria, por la colaboración en algunas de las pruebas de laboratorio.
Los autores declaran no tener conflictos de intereses con entidades privadas o públicas en lo concerniente a la presente publicación. Ninguno de los autores recibió honorarios, participó en eventos, o recibió financiación alguna de casas farmacéuticas o laboratorios que procesen o distribuyan medicamentos relacionados con el tratamiento de los pacientes incluidos en el estudio.
El proyecto "Caracterización clínica, bioquímica y genética de una familia con fenilcetonuria (PKU) procedente del departamento de Boyacá" (ID # 00004472) fue financiado por el Instituto de Genética Humana de la Pontificia Universidad Javeriana, Bogotá, D.C., Colombia.
Correspondencia:
Marta L. Tamayo, Instituto de Genética Humana, Pontificia Universidad Javeriana, Carrera 7 N° 40-62, edificio 32, Bogotá, D.C., Colombia
Teléfono: (571) 320 8320, extensiones 2788 y 2787; fax: (571) 320 8320, extensión 2793
1. Vilarinho L, Queirós A, Leandro P, Tavares de Almeida I, Rivera I. Fenilcetonuria revisitada. Arq Med. 2006;20: 161-72. [ Links ]
2. Santos LL, Fonseca CG, Starling AL, Januario JN, Aguiar MJ, Peixoto MG, et al . Variations in genotype-phenotype correlations in phenylketonuria patients. Genet Mol Res. 2010;9:1-8. http://dx.doi.org/10.4238/vol9-1gmr670 [ Links ]
3. Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831-45. http://dx.doi.org/10.1002/humu.20526 [ Links ]
4. Scriver CR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited disease. Eighth edition. New York: McGraw Hill; 2001. p. 1667-724. [ Links ]
5. Fernandes J, Saudubray JM, van den Berghe G, Walter JH. Inborn metabolic diseases, diagnosis and treatment. Fourth edition. Würzbur, Germany: Springer; 2006. p. 226-31. [ Links ]
6. Berry SA, Brown C, Grant M, Greene CL, Jurecki E, Koch J, et al . Newborn screening 50 years later: Access issues faced by adults with PKU. Genet Med. 2013;15:591-9. http://dx.doi.org/10.1038/gim.2013.10 [ Links ]
7. Hagedorn TS, van Berkel P, Hammerschmidt G, Lhotakova M, Saludes RP. Requirements for a minimum standard of care for phenylketonuria: The patients' perspective. Orphanet J Rare Dis. 2013;8:191. http://dx.doi.org/10.1186/1750-1172-8-191 [ Links ]
8. de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses. Mol Genet Metab. 2010;99(Suppl.1):S86-9. http://dx.doi.org/10.1016/j.ymgme.2009.10.016 [ Links ]
9. Scriver CR, Prevost L, Hurtubise M, Konecki D, Dobrowolski SF. Phenylalanine Hydroxylase Locus Knowl-edgebase. 2002. Fecha de consulta: 3 de diciembre de 2014. Disponible en: http://www.pahdb.mcgill.ca/cgi-bin/pahdb/mutation_statistics-1.cgi. [ Links ]
10. Desviat LR, Pérez B, De Lucca M, Cornejo V, Schmidt B, Ugarte M. Evidence in Latin America of recurrence of V388M, a phenylketonuria mutation with high in vitro residual activity. Am J Hum Genet. 1995;57:337-42. [ Links ]
11. Santos LL, Magalhaes M de C, Reis Ade O, Starling AL, Januario JN, Fonseca CG, et al . Frequencies of phenylalanine hydroxylase mutations I65T, R252W, R261Q, R261X, IVS10nt11, V388M, R408W, Y414C, and IVS12nt1 in Minas Gerais, Brazil. Genet Mol Res. 2006;5:16-23. [ Links ]
12. Acosta AX, Silva WA Jr, Carvalho TM, Zago MA. Ten novel mutations in the phenylalanine hydroxylase gene ( PAH ) observed in Brazilian patients with phenylketonuria. Hum Mutat. 2001;17:77. http://dx.doi.org/10.1002/1098-1004(2001)17:1<77::AID-HUMU19>3.0.CO;2-S [ Links ]
13. Cazzorla C, Cegolon L, Burlina AP, Celato A, Massa P, Giordano L, et al . Quality of Life (QoL) assessment in a cohort of patients with phenylketonuria. BMC Public Health. 2014;14:1243. http://dx.doi.org/10.1186/1471-2458-14-1243 [ Links ]
14. Guldberg P, Levy HL, Hanley WB, Koch R, Matalon R, Rouse BM, et al . Phenylalanine hydroxylase gene mutations in the United States: Report from the Maternal PKU Collaborative Study. Am J Hum Genet. 1996;59:84-94. [ Links ]
15. Scriver CR. Why mutation analysis does not always predict clinical consequences: Explanations in the era of genomics. J Pediatr. 2002;140:502-6. http://dx.doi.org/10.1067/mpd.2002.124316 [ Links ]
16. Scriver CR, Waters PJ. Monogenic traits are not simple: Lessons from phenylketonuria. Trends Genet. 1999;15:267-72. http://dx.doi.org/10.1016/S0168-9525(99)01761-8 [ Links ]
17. Möller HE, Weglage J, Wiedermann D, Ullrich K. Blood-brain barrier phenylalanine transport and individual vulnerability in phenylketonuria. J Cereb Blood Flow Metab. 1998;18:1184-91. http://dx.doi.org/10.1097/00004647-199811000-00004 [ Links ]
18. Møller LB, Paulsen M, Koch R, Moats R, Guldberg P, Güttler F. Inter-individual variation in brain phenylalanine concentration in patients with PKU is not caused by genetic variation in the 4F2hc/LAT1 complex. Mol Genet Metab. 2005;86(Suppl.1):S119-23. http://dx.doi.org/10.1016/j.ymgme.2005.07.031 [ Links ]
19. Langenbeck U, Burgard P, Wendel U, Lindner M, Zschocke J. Metabolic phenotypes of phenylketonuria. Kinetic and molecular evaluation of the Blaskovics protein loading test. J Inherit Metab Dis. 2009;32:506-13. http://dx.doi.org/10.1007/s10545-009-1152-6 [ Links ]
20. Viau KS, Wengreen HJ, Ernst SL, Cantor NL, Furtado LV, Longo N. Correlation of age-specific phenylalanine levels with intellectual outcome in patients with phenylketonuria. J Inherit Metab Dis. 2011;34:963-71. http://dx.doi.org/10.1007/s10545-011-9329-1 [ Links ]
21. Sarkissian CN, Gámez A, Scriver CR. What we know that could influence future treatment of phenylketonuria. J Inherit Metab Dis. 2009;32:3-9. http://dx.doi.org/10.1007/s10545-008-0917-7 [ Links ]
22. van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: Is phenylalanine toxicity the only possible cause? J Inherit Metab Dis. 2009;32:46-51. http://dx.doi.org/10.1007/s10545-008-0946-2 [ Links ]

