










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.36 supl.1 Bogotá Dec. 2016
https://doi.org/10.7705/biomedica.v36i2.2797
PRESENTACIÓN DE CASO
doi: http://dx.doi.org/10.7705/biomedica.v36i2.2797
1 Departamento de Nefrología y Trasplante Renal, Hospital Pablo Tobón Uribe, Medellín, Colombia
2 Departamento de Medicina Interna y Nefrología, Universidad de Antioquia, Medellín, Colombia
3 Departamento de Medicina Interna, Universidad Pontificia Bolivariana, Medellín, Colombia
4 Departamento de Pediatría y Puericultura, Universidad de Antioquia, Medellín, Colombia
5 Departamento de Enfermedades Infecciosas, Hospital Pablo Tobón Uribe, Medellín, Colombia
6 Departamento de Enfermedades Infecciosas, Universidad Pontificia Bolivariana, Medellín, Colombia
7 Departamento de Enfermedades Infecciosas, Universidad de Emory, Atlanta, Georgia, Estados Unidos
Contribución de los autores:
John Fredy Nieto, Sandra Milena Gómez, Diana Carolina Moncada y Lina María Serna: concepción y diseño del estudio; análisis e interpretación de los datos; toma de muestras y seguimiento del paciente; análisis estadístico de los datos; redacción y revisión del artículo, y aprobación final de la versión enviada para publicación.
Todos los autores participaron en la redacción y la revisión del artículo.
Recibido: 27/04/15; aceptado: 09/09/15
La linfohistiocitosis hemofagocítica es un síndrome poco frecuente que resulta de una activación incontrolada de los macrófagos y linfocitos, la cual compromete múltiples órganos y es potencialmente fatal sin el tratamiento oportuno. El síndrome puede ser de origen hereditario o secundario a procesos infecciosos, neoplásicos o autoinmunitarios.
Se presenta el caso de un paciente con HIV/sida que desarrolló linfohistiocitosis hemofagocítica y coagulación intravascular diseminada asociadas a histoplasmosis, y que fue exitosamente tratado con anfotericina B, esteroides y tratamiento dialítico transitorio.
Palabras clave: linfohistiocitosis hemofagocítica, coagulación intravascular diseminada, síndrome de inmunodeficiencia adquirida, histoplasmosis.
doi: http://dx.doi.org/10.7705/biomedica.v36i2.2797
Successful treatment of hemophagocytic lymphohistiocytosis and disseminated intravascular coagulation secondary to histoplasmosis in a patient with HIV/AIDS
Haemophagocytic lymphohistiocytosis is an uncommon syndrome that results from an uncontrolled activation of macrophages and lymphocytes resulting in the compromise of multiple organs that is potentially fatal without timely treatment. It can be hereditary or a secondary result of infectious processes, neoplasms or autoimmune conditions.
We present the case of a patient with HIV/AIDS who developed hemophagocytic lymphohistiocytosis as well as disseminated intravascular coagulation associated with histoplasmosis and who was successfully treated with amphotericin B, steroids and transitory dialytic support.
Key words: Lymphohistiocytosis, hemophagocytic, disseminated intravascular coagulation, acquired immunodeficiency syndrome, histoplasmosis.
doi: http://dx.doi.org/10.7705/biomedica.v36i2.2797
La linfohistiocitosis hemofagocítica es un síndrome caracterizado por una activación anormal del sistema inmunitario después de una estimulación excesiva de las células asesinas naturales ( natural killer , NK) y los linfocitos T CD8; puede ser primaria (de origen genético) o secundaria a condiciones neoplásicas, infecciosas o autoinmunes (1). Se caracteriza clínicamente por fiebre, hepato-esplenomegalia, citopenias, hiperferritinemia, hiper- trigliceridemia e hipofibrinogenemia. El tratamiento se basa principalmente en el control de la enfermedad de base, sin embargo, en muchos casos se requieren esteroides y quimioterapia (2,3), y de todas maneras, la mortalidad es alta (4).
Caso clínico
Se trata de un hombre de 33 años con infección por el virus de la inmunodeficiencia humana (HIV) diagnosticada tres meses antes de la consulta y a quien no se le había administrado tratamiento antirretroviral. El paciente presentaba un cuadro clínico de un mes de evolución de fiebre, astenia, adinamia, pérdida de peso, diarrea abundante, dolor abdominal, sangrado por mucosas, alteración del sensorio y disnea progresiva. Estaba deshidra-tado, hipotenso, taquicárdico y taquipneico, y sufría síndrome encefalopático.
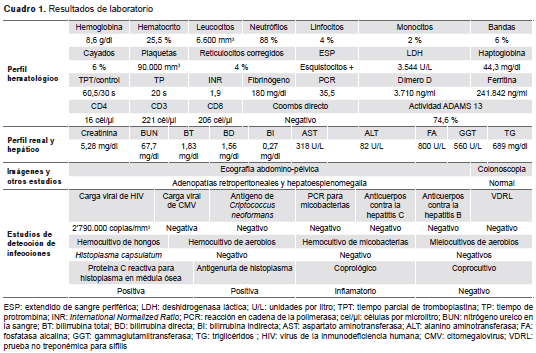
En los exámenes iniciales de laboratorio se detectó anemia, trombocitopenia, escasos esquistocitos en sangre periférica, elevación de los niveles de deshidrogenasa láctica, prolongación de los tiempos de coagulación, aumento de los niveles de reactantes de fase aguda, acidosis metabólica y lesión renal aguda de estadio 3 según la Acute Kidney Injury Network (AKIN) (cuadro 1) (5).
Ante la sospecha inicial de sepsis de origen gastrointestinal con falla orgánica múltiple, se inició tratamiento con reanimación hídrica, soporte hemodinámico y, una vez hechos los cultivos, piperacilina más tazobactam, antibióticos que se suspendieron al día siguiente ante la ausencia de signos clínicos y paraclínicos de infección piógena y por sospecha de infección oportunista del tipo de histoplasmosis o de tuberculosis diseminada. Por este motivo, se solicitaron exá-menes paraclínicos complementarios y se inició el tratamiento empírico con anfotericina B y el tratamiento antituberculoso.
Ante la rápida progresión del cuadro clínico, se consideraron otros diagnósticos, como linfohistio-citosis hemofagocítica, microangiopatía trombótica o coagulación intravascular diseminada (CID) secundaria a infección, y se decidió iniciar el recambio plasmático (alcanzó a recibir dos) y el tratamiento dialítico.
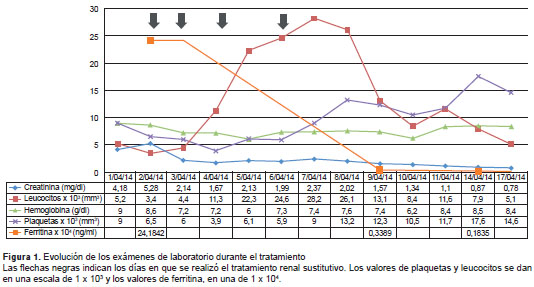
En los estudios adicionales se encontró hiper-ferritinemia, hipertrigliceridemia, consumo de fibri-nógeno, hepatoesplenomegalia, adenomegalias retroperitoneales y actividad normal de la enzima ADAMTS13 (cuadro 1 y figura 1).
En la biopsia renal se encontró necrosis tubular aguda y no se detectaron toxinas Shiga. En el estudio de médula ósea, se observaron escasos hemofa-gocitos y abundantes levaduras intracitoplásmicas indicativas de Histoplasma capsulatum (figura 2a); las levaduras se visualizaron, además, en el extendido de sangre periférica (figura 2b).

Con base en estos hallazgos, se diagnosticó linfohis-tiocitosis hemofagocítica y coagulación intravascular diseminada, asociadas a histoplasmosis diseminada en un paciente con HIV/sida en estadio 3.
Se continuó, entonces, el tratamiento con anfo-tericina B hasta completar 14 días, al cabo de los cuales se cambió a 200 mg de itraconazol en solución oral cada ocho horas durante tres días y, después, cada 12 horas durante un año. Además, se inició la administración de prednisolona en dosis equivalente a 1 mg/kg/día durante tres semanas, la cual se suspendió gradualmente en el transcurso de un mes.
El paciente evolucionó satisfactoriamente y presentó mejoría de la falla en los diversos órganos afectados; asimismo, se controló la coagulación intravascular diseminada y se suspendió la hemodiálisis después de cuatro sesiones consecutivas. Al mes de haber iniciado el tratamiento antifúngico, se inició la administración de los antirretrovirales. El paciente completó todo su tratamiento antifúngico y al año de seguimiento se encontraba en buen estado general, asintomático y con función renal normal.
Consideraciones éticas
Los autores declaran que han seguido los protocolos del Hospital Pablo Tobón Uribe sobre la publicación de datos de pacientes y que el manuscrito contó con la aprobación del Comité de Ética de la institución.
Discusión
Se presenta el caso de un hombre joven con diagnóstico de infección por HIV en etapa de sida, quien presentó falla en múltiples órganos y coagulación intravascular diseminada secundaria a linfohistiocitosis hemofagocítica asociada a histo-plasmosis diseminada. El paciente fue atendido en la unidad de cuidados intensivos y recibió soporte hemodinámico, hemodiálisis, transfusiones, anfotericina B y esteroides, con muy buena respuesta al tratamiento.
La linfohistiocitosis hemofagocítica se produce por una alteración en la función de las células NK o T citotóxicas, lo cual lleva a una activación incontrolada e inefectiva del sistema inmunológico, que produce daño celular, y proliferación y activación de macrófagos benignos con hemofagocitosis en el sistema reticuloendotelial (1).
Los criterios diagnósticos de esta condición son: 1) fiebre; 2) esplenomegalia; 3) citopenias (leuco-penia, anemia o trombocitopenia); 4) ferritina de 500 ng/dl o más; 5) triglicéridos de 265 mg/dl o más, o fibrinógeno de 150 mg/dl o más; 6) hemofagocitos en médula ósea o bazo, o en ganglios linfáticos sin neoplasia maligna; 7) disminución o ausencia de citotoxicidad de células NK, y 8) aumento del CD25 soluble (6). En el contexto clínico adecuado, el diagnóstico se
hace ante la presencia de cinco de estos criterios (6). Cabe anotar que puede cursar con alteración del perfil hepático y coagulopatía (4).
En el caso de este paciente, se cumplían seis de los ocho criterios, sin incluir la medición del CD25 soluble ni de la actividad de las células NK, al no estar disponibles. Llama la atención que el nivel de ferritina del paciente estaba supremamente elevado (>200.000 ng/dl) sin otra causa aparente, lo que apoyaba este diagnóstico. En el diagnóstico de la linfohistiocitosis hemofagocítica, los niveles de ferritina por encima de 10.000 ng/dl parecen tener una sensibilidad y una especificidad de 90 y 96 %, respectivamente (7).
La linfohistiocitosis hemofagocítica puede clasi-ficarse como primaria, por alteración genética, o secundaria a infecciones, neoplasia maligna o enfermedad autoinmunitaria. Las infecciones contribuyen a la iniciación de la hemofagocitosis actuando como desencadenantes en pacientes con predisposición genética y enfermedades autoinmunitarias, o pueden ser el factor primario para el desarrollo del síndrome en pacientes sin alteraciones subyacentes (3).
Este síndrome aparece asociado a una amplia variedad de infecciones, entre ellas las fúngicas, que ocurren más frecuentemente en pacientes con sida, linfoma, uso crónico de esteroides y tras-plantes (3). El HIV, por su parte, actúa en la mayoría de los casos como condición predisponente para el desarrollo de infecciones oportunistas que desencadenan el síndrome; sin embargo, se han descrito casos en que el HIV es el único factor identificable, como en los casos de infección aguda y de síndrome de reconstitución inmunitaria (8).
Las infecciones desencadenantes más frecuentes en estos pacientes son las causadas por virus herpes, como el citomegalovirus y el virus de Epstein-Barr, y por micobacterias (8,9). En el pre-sente caso, el paciente se encontraba seriamente inmunosuprimido debido a la infección por HIV y se logró confirmar la presencia de histoplasmosis diseminada como causa de la linfohistiocitosis hemofagocítica, hallazgos similares a los repor-tados por otros autores (10).
En un reporte reciente de 19 casos de linfohistio-citosis hemofagocítica asociada a histoplasmosis en pacientes con HIV (10), se encontró que el sexo masculino, la enfermedad avanzada y un recuento bajo de CD4 eran los principales factores predisponentes. Los hallazgos más importantes en esta serie fueron la fiebre (100 %), la hiperferriti-nemia (95 %) y la presencia de hemofagocitos (95 %) (10). La mayoría de los pacientes recibió tratamiento con anfotericina B y cinco de ellos recibieron, además, inmunoglobulina G; ninguno recibió quimioterapia, tratamiento con anticuerpos monoclonales ni esteroides. La mortalidad fue de 44.4 %, y la fungemia y la falta de tratamiento antifúngico se asociaron con una mayor mortalidad. También, se han reportado casos de linfohistioci-tosis hemofagocítica secundaria a histoplasmosis en pacientes con otro tipo de inmunosupresión, por ejemplo, en aquellos con trasplante renal; dos de tales casos ya fueron reportados por este equipo de investigación, con una mortalidad de 50 % (11,12).
El tratamiento de la linfohistiocitosis hemofagocítica primaria es la quimioterapia. El protocolo más usado incluye etopósido, ciclosporina A y esteroides. Otros medicamentos empleados son la inmunoglobulina antitimocito y los anticuerpos monoclonales (13). En la forma secundaria, el tratamiento se fundamenta en la identificación y el manejo de la enfermedad subyacente, lo cual en algunos casos puede ser suficiente para controlarla.
La eliminación de factores precipitantes como la inmunosupresión, debe basarse en la evaluación individual de cada paciente. La elección del mejor tratamiento adicional no está estandarizada y los esquemas de quimioterapia similares a los empleados en los casos de origen genético se usan principalmente en los casos asociados con el virus de Epstein-Barr. El uso de esteroides depende de la causa desencadenante, principalmente en las enfermedades autoinmunitarias, en las condi-ciones inflamatorias graves y en linfomas asociados con la quimioterapia (3,4).
A pesar de los avances en el tratamiento, la tasa de cura, especialmente cuando hay falla de varios órganos, es baja. La mayoría de los estudios se han enfocado en el manejo de la linfohistiocitosis hemofagocítica primaria en niños y son pocos los que evalúan adultos con formas secundarias, en quienes las tasas de supervivencia son menores, y el pronóstico empeora en los casos secundarios a neoplasia maligna (14).
En un estudio retrospectivo reciente en 103 pacientes adultos (2) (47,6 % con neoplasia hematológica maligna y 23,3 % con infección), se encontró una mortalidad global de 74,8 %. Los pacientes que fallecieron eran mayores de 30 años, de sexo masculino, y tenían esplenomegalia y trombocitopenia.
Se ha descrito que tener más de 30 años, hiper-ferritinemia, coagulación intravascular diseminada, ictericia, elevación de la β2-microglobulina, y empeoramiento de la anemia y la trombocitopenia, son factores de mal pronóstico (15). En un estudio en pacientes pediátricos, se encontró que la caída rápida en los niveles de ferritina después de iniciar el tratamiento se asoció con disminución de la mortalidad, por lo que este parámetro podría usarse en el seguimiento de los pacientes (16).
En este caso, el paciente tuvo una evolución favorable, probablemente porque se sospechó tempranamente el diagnóstico, y se inició el trata-miento empírico con anfotericina B y esteroides sistémicos. Durante la evolución se observó cómo los niveles de ferritina disminuyeron notoriamente, lo que coincidió con la mejoría clínica, por lo que se evidenció que puede ser un parámetro importante de seguimiento, como lo han sugerido otros autores (figura 1).
Conclusión
La linfohistiocitosis hemofagocítica es un síndrome raro, de causa primaria o secundaria, que com-promete múltiples órganos y puede llevar a la muerte en un porcentaje elevado de los casos. Una de las principales causas de la linfohistiocitosis hemofagocítica son las infecciones oportunistas en los pacientes inmunosuprimidos, entre ellas, la histoplasmosis diseminada. La identificación temprana y el tratamiento oportuno, son los pilares fundamentales para evitar las complicaciones y la muerte de los pacientes por esta causa.
Los autores declaran no tener ningún conflicto de intereses con respecto al contenido de este artículo.
El manuscrito contó con el apoyo del Hospital Pablo Tobón Uribe.
Correspondencia:
Lina María Serna, Calle 78b N° 69-240, Medellín, Colombia
Teléfono: (574) 445 9902
1. Janka GE, Lehmberg K. Hemophagocytic syndromes - An update. Blood Rev. 2014;28:135-42. http://dx.doi.org/10.1016/j.blre.2014.03.002 [ Links ]
2. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-22. http://dx.doi.org/10.1016/S1473-3099(07)70290-6 [ Links ]
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: Diagnosis and treatment. Jt Bone Spine. 2012;79:356-61. http://dx.doi.org/10.1016/j.jbspin.2011.10.015 [ Links ]
4. Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang W, et al . Hemophagocytic lymphohistiocytosis: Clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-5. http://dx.doi.org/10.1097/MD.0000000000000022. [ Links ]
5. Foundation KDIGO. Clinical Guidelines. National Kidney. Section 2: AKI Definition. Kidney Int Suppl. 2012;2:19-36. [ Links ]
6. Henter J, Horne A, Aricó M, Egeler R, Filipovich A, Imashuku S, et al . HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-31. http://dx.doi.org/10.1002/pbc.21039 [ Links ]
7. Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:1227-35. http://dx.doi.org/10.1002/pbc.21423 [ Links ]
8. Doyle T, Bhagani S, Cwynarski K. Haemophagocytic syndrome and HIV. Curr Opin Infect Dis. 2009;22:1-6. http://dx.doi.org/10.1097/QCO.0b013e32832180b0 [ Links ]
9. Fardet L, Lambotte O, Meynard J-L, Kamouh W, Galicier L, Marzac C, et al . Reactive haemophagocytic síndrome in 58 HIV-1-infected patients: Clinical features, underlying diseases and prognosis. AIDS. 2010;24:1299-306. http://dx.doi.org/10.1097/QAD.0b013e328339e55b. [ Links ]
10. Subedee A, van Sickels N. Hemophagocytic syndrome in the setting of AIDS and disseminated histoplasmosis: Case report and a review of literature. J Int Assoc Provid AIDS Care. 2015;1-7. http://dx.doi.org/10.1177/2325957415570740 [ Links ]
11. Nieto-Ríos JF, Aristizábal-Alzate A, Ocampo C, Serrano-Gayubo AK, Serna-Higuita LM, Zuluaga-Valencia G. Disseminated histoplasmosis and haemophagocytic syndrome in two kidney transplant patients. Nefrologia. 2012;32:683-4. http://dx.doi.org/10.3265/Nefrologia.pre2012.Jun.11508 [ Links ]
12. Lo MM, Mo JQ, Dixon BP, Czech KA. Disseminated histoplasmosis associated with hemophagocytic lympho-histiocytosis in kidney transplant recipients. Am J Transplant. 2010;10:687-91. http://dx.doi.org/10.1111/j.1600-6143.2009. 02969.x. [ Links ]
13. Mehta R, Smith R. Hemophagocytic lymphohistiocytosis (HLH): A review of literature. Med Oncol. 2013;30:740. http://dx.doi.org/10.1007/s12032-013-0740-3 [ Links ]
14. Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control. 2014; 21:301-12. [ Links ]
15. Kaito K, Kobayashi M, Katayama T, Otsubo H, Ogasawara Y, Sekita T, et al. Prognostic factors of hemophagocytic syndrome in adults: Analysis of 34 cases. Eur J Haematol. 1997;59:247-53. [ Links ]
16. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, Mcclain KL. Rate of decline of ferritin in patients with hemo-phagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56:154-5. http://dx.doi.org/10.1002/pbc.22774 [ Links ]

