










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.36 supl.1 Bogotá dez. 2016
https://doi.org/10.7705/biomedica.v36i3.2729
CASE PRESENTATION
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2729
Author´s contributions:
Kevin Escandón-Vargas conceived the manuscript idea, reviewed the literature, and drafted the manuscript.
Andrés Zorrilla-Vaca contributed in the data collection and literature review.
Raúl Helí Corral-Prado conceived the manuscript idea and contributed in the revision of the manuscript.
Received: 02/03/15; accepted: 22/02/16
Prion diseases are rare neurodegenerative disorders occurring worldwide and affecting both humans and animals. Herein, we present the case of a patient diagnosed with definite sporadic Creutzfeldt-Jakob disease in Cali, Colombia. Besides neurological examination, 14-3-3 and tau proteins were valuable tools supporting the diagnosis. We also present a brief perspective of the prion diseases reported in Colombia to date. Although the incidence of prion diseases is unknown in Colombia, our literature review revealed that one case of scrapie in 1981 and 29 human sporadic cases of Creutzfeldt-Jakob disease have been documented and published in our country.
Key words: Creutzfeldt-Jakob syndrome, prions, case reports.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2729
Proteínas 14-3-3 y tau positivas en un caso de enfermedad esporádica de Creutzfeldt-Jakob y una breve reseña de las enfermedades priónicas en Colombia
Las enfermedades priónicas son alteraciones neurodegenerativas raras que ocurren en todo el mundo y afectan tanto a humanos como a animales. En el presente artículo, se reporta un caso con diagnóstico confirmado de enfermedad esporádica de Creutzfeldt-Jakob. Además del examen neuropatológico, las proteínas 14-3-3 y tau fueron herramientas valiosas que ayudaron en el diagnóstico. También, se presenta una breve reseña de las enfermedades priónicas reportadas en Colombia hasta la fecha. Aunque en el país se desconoce la incidencia de las enfermedades priónicas, nuestra búsqueda en la literatura científica reveló informes publicados sobre un caso de tembladera de las ovejas ( scrapie o encefalopatía espongiforme ovina) en 1981 y 29 casos esporádicos de Creutzfeldt-Jakob en el país.
Palabras clave: síndrome de Creutzfeldt-Jakob, priones, informes de casos.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.2729
Prion diseases, also known as transmissible spongiform encephalopathies, are a family of rare, invariably fatal neurodegenerative disorders which occur worldwide and affect both humans and animals. Human transmissible spongiform encephalopathies currently include sporadic, familial, iatrogenic and variant Creutzfeldt-Jakob disease (s, f, i, and vCJD, respectively), and other syndromes such as Gerstmann-Sträussler-Scheinker syndrome, kuru, fatal familial insomnia, sporadic fatal insomnia and the recently discovered prion protein cerebral amyloid angiopathy and variably protease-sensitive prionopathy. Most of the transmissible spongiform encephalopathies are sporadic, whereas only a minority are hereditary or acquired (1-3).
The recognition of human prion diseases is a medical challenge due to the small number of cases; sCJD accounts for around 85% of cases of Creutzfeldt-Jakob disease and has a global incidence of about one case per million people per year (1,4). Herein, we present the case of a patient diagnosed with definite sCJD in Cali, Colombia. Besides neurological examination, 14-3-3 and tau proteins were tools supporting the diagnosis.
Additionally, we conducted a literature search on PubMed/Medline, Google Scholar, Bireme/Lilacs and SciELO databases from inception through 2015 using the English/Spanish terms for "Creutzfeldt-Jakob syndrome", "prion diseases" and "prions", to present a brief perspective of the prion diseases reported in Colombia.
Case report
In 2011, a 53-year-old Afro-Colombian man from Florida, Valle del Cauca, in southwestern Colombia, presented to a referral medical center in Cali with two months of dizziness, behavior changes, speech impairment, gait imbalance and disorientation. The patient worked as a cane cutter and had never traveled abroad. He was hypertensive and a smoker, and there was no personal or familial history of neurodegenerative diseases or neurosurgery.
On admission, he had cognitive impairment, vision loss, aphasia, and ataxia. There were no signs of meningeal irritation. Metabolic, toxic, neoplastic and rheumatologic causes were screened but routine laboratory analyses were reported as normal. HIV, syphilis, HTLV, cytomegalovirus, Epstein-Barr virus, hepatitis B virus, and hepatitis C virus were ruled out. Initial brain magnetic resonance imaging (MRI) scan was normal. His functional and neurological status began declining and he entered a state of rapidly progressive dementia accompanied by generalized myoclonus and occasional tonic-clonic seizures. Soon after, the patient developed akinetic mutism and prostration. Cerebrospinal fluid (CSF) obtained in three lumbar punctures did not show abnormalities. During hospitalization the patient had several healthcare-associated infections including catheter-related Staphylococcus aureus bacteremia, pneumonia, urinary tract infection and an infected sacral pressure ulcer, all of which were treated appropriately.
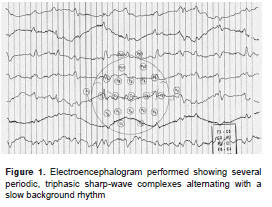
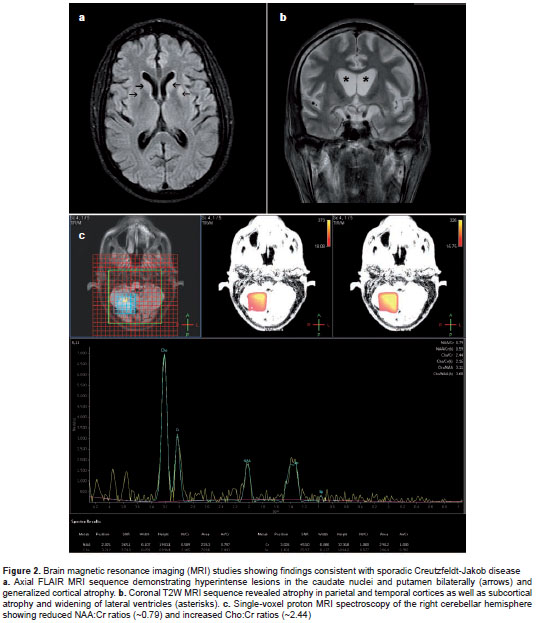
Four weeks after hospital admission, the patient underwent electroencephalogram (EEG) which revealed abundant periodic, triphasic sharp-wave complexes superimposed on a slow cerebral electrical rhythm (figure 1). During the second month of hospitalization, two new brain MRI studies showed progressive, symmetric, cortical-subcortical atrophy, and hyperintensive signals in the caudate nucleus, putamen, thalami, periaqueductal gray matter, right cerebellum, and parts of parietal and temporal cortices on T 2 -weighted (T 2 W), diffusion-weighted imaging (DWI), and fluid-attenuated inversion recovery (FLAIR) sequences. Proton magnetic resonance spectroscopy revealed reduced N-acetylaspartate to creatine (NAA:Cr) ratios and increased choline to creatine (Cho:Cr) ratios in the global cerebral parenchyma (figure 2).
A 14-3-3 protein test performed in CSF by immunoblot was positive. Also, an ELISA immuno-assay for the tau protein was reported as positive (21,789 pg/ml; decision point: 1,150 pg/ml). The patient died five months after disease onset. Autopsy was conducted for neuropathological examination which demonstrated the presence of spongiform changes, neuronal loss and gliosis. A definite sCJD diagnosis was made according to MRI-CJD Consortium (MCC) criteria (5).
Discussion
Sporadic CJD is the most frequent prion disease in humans and is secondary either to spontaneous conversion of the normal cellular prion protein (PrP C ) into a disease-causing protease-resistant prion protein (PrP Sc ) or to somatic mutation of the PrP-encoding gene PRNP (1,2,4).
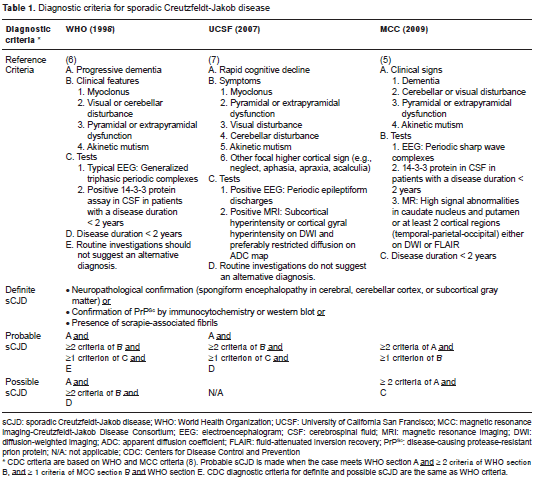
Physicians should consider sCJD as a differential diagnosis in patients with rapidly progressive dementia and be familiar with the available diagnosis tools according to the accepted criteria. sCJD can be diagnosed based on: (a) the World Health Organization (WHO) consultation group criteria published in 1998 (6); (b) the University of California San Francisco (UCSF) group criteria published in 2007 (only for probable sCJD diagnosis) (7), or (c) the MCC criteria published in 2009 (5). These diagnostic criteria are outlined in table 1. As noted, definite diagnosis of sCJD requires pathological studies of the brain. Probable diagnosis of sCJD requires neurological manifestations and at least one positive test. Among diagnostic tests, while WHO and MCC criteria include 14-3-3 protein test, UCSF and MCC criteria include MRI findings. Possible diagnosis of sCJD is made when there is absence of tests; however, it is not a diagnostic entity according to the UCSF criteria. Another diagnostic criteria set, that of the Centers for Disease Control and Prevention (CDC), is based on WHO and MCC criteria (8).
Based on the medical history, the clinical manifestations, typical EEG and MRI findings, positive 14-3-3 and tau proteins, and neuropathological examination, the patient presented herein was diagnosed with sCJD. The case met the criteria for definite diagnosis and did not present any relevant personal or familial antecedents. We consider this to be the first Colombian case report of sCJD using 14-3-3 protein, tau protein and proton MRI spectroscopy together as diagnostic tools.
Although sCJD spectrum of clinical manifestations is broad, it usually manifests as an irreversible course of rapidly progressing dementia with ataxia and myoclonus, often leading to death within less than one year after disease onset. sCJD has a mean illness duration of five months, which is consistent with our case report (2,4). Other clinical features of sCJD are vague symptoms including fatigue, disordered sleep and weight loss, as well as neurological manifestations such as ataxia, altered sensorium, language deficits, behavior changes, visual and auditory hallucinations, parkinsonism, seizures and akinetic mutism (2,9). Most of these findings were present in our patient.
Abnormal electroencephalographic tracing, when present, is suggestive for probable sCJD but is not pathognomonic, as some metabolic and toxic encephalopathies and other dementias could present with abnormal EEG as well. The cerebral electrical activity is usually normal or slow in the early course of the disease. Later in the course of the disease, periodic, biphasic or triphasic synchronous sharp-wave complexes may emerge and alternate with the background rhythm. These typical alterations were found in our patient. The sensitivity and specificity of a typical EEG for detection of sCJD are 64-67% and 86-91%, respectively (10,11).
Sensitivity of brain MRI in probable and definitive sCJD cases ranges from 58-71%, and specificity ranges from 82-90%; however, MRI may be normal in early stages of the disease. MRI sequences, such as DWI and FLAIR, show cerebral atrophy and prominent hyperintensities in both basal ganglia and cortex, which are common changes in sCJD as seen in our case (12). Furthermore, our patient had abnormalities on proton MRI spectroscopy, which according to the literature, might increase the diagnostic accuracy for sCJD. N-acetylaspartate to creatine ratios =1.21 have been associated to sCJD diagnosis with a sensitivity and a negative predictive value of 100% (13).
The 14-3-3 protein in the CSF has been reported to have a sensitivity between 85% and 96% and a specificity between 79% and 96%. As it may be present in other conditions such as viral encephalitis, metabolic encephalopathy and cerebral hemorrhages, the test is not appropriate as a general screening test for CJD, but is useful for diagnosis of truly suspect cases (14-16). In contrast to qualitative 14-3-3 test, ELISA immunoassay for the quantitative detection of tau protein in CSF has shown superior accuracy with less ambiguous results (17). Both the 14-3-3 and tau proteins in the CSF of the patient were positive and therefore supported the diagnosis of sCJD.
The patient did not receive any treatment before death as there are currently no effective therapies for prion diseases (4). Unfortunately, it was not possible to know the sCJD subtype because the immunological analysis of PrP Sc and the PRNP sequencing were not performed.
The real incidence of prion diseases in Colombia is unknown, but it is suspected to be higher than believed (18). Our literature search on prion diseases revealed that one case of scrapie in 1981 and 29 human sporadic cases of CJD (18-32) have been documented and published in our country. To our knowledge, there are no records or evidence of vCJD or bovine spongiform encephalopathy in Colombia (28,33,34). We found other publications such as reviews and correspondences about transmissible spongiform encephalopathies (18,33-41). General features of the sCJD case reports from Colombia are shown in table 2. The majority of these sCJD cases occurred in persons between 50 and 70 years of age, which is a fact consistent with the global literature (1,9). sCJD has been reported in different regions of the country showing a random geographic distribution of the reported cases. Overall, there was no significant, personal or familial history which had indicated iatrogenic causes or suggested differential diagnosis of neurodegenerative diseases. The disease duration in most of the cases was reported to be less than eight months.
Transmissible spongiform encephalopathies have profound impacts on public health and economy, particularly bovine spongiform encephalopathy and vCJD because of their potential transmissibility. The World Organization for Animal Health (OIE, as it was formerly called Office International des Epizooties ) and the WHO have urged several countries to implement policies regarding the international trade of animals and their derived products, and to state the surveillance, notification, data collection, analysis, and confirmation of suspected human cases of prion disease as mandatory (34,39) (Sierra-Zuleta UE. Avance y estado actual de la prevención y vigilancia de la encefalopatía espongiforme bovina (EEB) en Colombia. Jornada Nacional de Zoonosis y Enfermedades Emergentes y Re-emergentes, August 21 and 22, 2008. Accessed: June 15, 2015. Available from: http://sites.google.com/site/saludpublicaveterinaria).
Colombia was initially considered by the OIE to be a country with controlled bovine spongiform encephalopathy risk because of the antecedents of livestock importation in the 1980s and 1990s from countries where bovine spongiform encephalopathy was documented (28). Since 2001, the Instituto Colombiano Agropecuario (ICA) has been responsible for the national program for prevention of bovine spongiform encephalopathy. Moreover, in 2005, the Instituto Nacional de Salud gathered efforts with the ICA, the Ministry of Social Protection and the Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) to set up a surveillance system for vCJD in the framework of the Sistema Nacional de Vigilancia en Salud Pública (Sivigila) (34,39) (Sierra-Zuleta UE. Avance y estado actual de la prevención y vigilancia de la encefalopatía espongiforme bovina (EEB) en Colombia. Jornada Nacional de Zoonosis y Enfermedades Emergentes y Re-emergentes, August 21 and 22, 2008. Accessed: June 15, 2015. Available from: http://sites.google.com/site/saludpublicaveterinaria). Thanks to these initiatives, Colombia is officially regarded as having a negligible bovine spongiform encephalopathy risk status since May 2012 (28).
Although Colombia is currently considered to be free of bovine spongiform encephalopathy and the risk of vCJD cases is consequently low, emergence of prion disease forms yet not reported in Colombia could occur, with devastating consequences in the health and farming industry sectors. Epidemiological surveillance of prion diseases must continue and confirmation procedures of suspected cases must be encouraged to keep the risk of zoonotic transmission of transmissible spongiform encephalopathies to a minimum (28,34,39).
The authors declare no conflict of interests.
None
Corresponding author:
Kevin Escandón-Vargas, Escuela de Medicina, Facultad de Salud, Universidad del Valle, Calle 4B N° 36-00, Cali, Colombia
Phone number: (572) 518 5652
kevin.escandon@correounivalle.edu.co
1. Prusiner SB. Shattuck lecture: Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516-26. http://dx.doi.org/10.1056/NEJM200105173442006 [ Links ]
2. Johnson RT. Prion diseases. Lancet Neurol. 2005;4:635-42. http://dx.doi.org/10.1016/S1474-4422(05)70192-7 [ Links ]
3. Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: Prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol. 2012;38:296-310. http://dx.doi.org/10.1111/j.1365-2990.2012.01265.x [ Links ]
4. Kim MO, Geschwind MD. Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol. 2015;28:302-10. http://dx.doi.org/10.1097/WCO.0000000000000197 [ Links ]
5. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al . Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132:2659-68. http://dx.doi.org/10.1093/brain/awp191 [ Links ]
6. World Health Organization. Global surveillance, diagnosis, and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation. Geneva: WHO; 1998. Accessed: December 2, 2015. Available from: http://www.who.int/csr/resources/publications/bse/whoemczdi989.pdf [ Links ]
7. UCSF Memory and Aging Center. 2007 UCSF criteria for probable sporadic Jakob-Creutzfeldt disease. 2007. Accessed: December 2, 2015. Available from: http://memory.ucsf.edu/cjd/medical/criteria [ Links ]
8. Centers for Disease Control and Prevention. CDC´s diagnostic criteria for Creutzfeldt-Jakob disease (CJD), 2010. 2010. Accessed: December 2, 2015. Available from: http://www.cdc.gov/prions/cjd/diagnostic-criteria.html [ Links ]
9. Johnson RT, Gibbs CJ Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. http://dx.doi.org/10.1056/NEJM199812313392707 [ Links ]
10. Steinhoff BJ, Räcker S, Herrendorf G, Poser S, Grosche S, Zerr I, et al . Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:162-6. http://dx.doi.org/10.1001/archneur.1996.00550020074017 [ Links ]
11. Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004;56:702-8. http://dx.doi.org/10.1002/ana.20261 [ Links ]
12. Tschampa HJ, Kallenberg K, Urbach H, Meissner B, Nicolay C, Kretzschmar HA, et al . MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: A study on inter-observer agreement. Brain. 2005;128:2026-33. http://dx.doi.org/10.1093/brain/awh575 [ Links ]
13. Lodi R, Parchi P, Tonon C, Manners D, Capellari S, Strammiello R, et al . Magnetic resonance diagnostic markers in clinically sporadic prion disease: A combined brain magnetic resonance imaging and spectroscopy study. Brain. 2009;132:2669-79. http://dx.doi.org/10.1093/brain/awp210 [ Links ]
14. Sánchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Sánchez-Valle R, Mitrováa E, et al . CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2006;67:637-43. http://dx.doi.org/10.1212/01.wnl.0000230159.67128.00 [ Links ]
15. Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med. 1996;335:924-30. http://dx.doi.org/10.1056/NEJM199609263351303 [ Links ]
16. Zerr I, Bodemer M, Gefeller O, Otto M, Poser S, Wiltfang J, et al . Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol. 1998;43:32-40. http://dx.doi.org/10.1002/ana.410430109 [ Links ]
17. Hamlin C, Puoti G, Berri S, Sting E, Harris C, Cohen M, et al . A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2012;79:547-52. http://dx.doi.org/10.1212/WNL.0b013e318263565f [ Links ]
18. Toro-González G. Demencia, priones y enfermedades priónicas. Referencia especial a las "vacas locas". Rev Acad Colomb Cienc. 1997;21:229-36. [ Links ]
19. Roselli A, Potes J, Kattah J, Palacios E, Ordóñez N. Encefalopatía espongiforme subaguda (enfermedad de Creutzfeldt-Jakob). Tribuna Médica. 1973;554:A7-10. [ Links ]
20. Zaninovic V, Dueñas A. Un caso de enfermedad de Creutzfeldt-Jakob. Acta Med Valle. 1979;10:131-6. [ Links ]
21. Zaninovic V, Dueñas A. Demencia viral transmisible. Confirmación de un caso de enfermedad de Creutzfeldt-Jakob. Colombia Médica. 1983;14:40-2. [ Links ]
22. Díez J, Jiménez A, Roselli A, Jiménez E, Ruiz N, Morales S. Enfermedad de Creutzfeldt-Jakob. Reporte de un caso. Neurocien Colom. 1999;7:49-52. [ Links ]
23. Colegial C, Silva F, Pérez C, Saavedra M, Fernández W, Pardo R, et al . Encefalopatía por priones, caso clínico-patológico. Revista de la Facultad de Medicina. 1999;47:13-20. [ Links ]
24. Villamil W, Gonzáles J, Arrieta JA, Álvarez C, Borja G, Vergara JC, et al . Creutzfeldt-Jakob disease: Case report. Infectio. 2007;11:124-8. [ Links ]
25. Villegas VE, Velandia F, Payán C. Sporadic Creutzfeldt-Jakob disease: Clinical, pathological and molecular study. Rev Cienc Salud. 2008;6:36-47. [ Links ]
26. Rincón OL, Vélez A, King LM, Hernández DL, Isaza R, Vagner B, et al . Sporadic Creutzfeldt-Jakob disease: Neuropsychiatric symptoms followed by a progressive cognitive decline: A case report. Medicina UPB. 2008;27: 59-63. [ Links ]
27. Díaz-Martínez JC, Takeuchi-Tan Y. Creutzfeldt-Jakob disease: Clinical, electroencephalographic, pathologic, and imaging characteristics. Acta Neurol Colomb. 2008;24: 118-23. [ Links ]
28. Paredes A, Castro A, Toro G, Parra E. Vigilancia epidemiológica de enfermedades priónicas en Colombia, 2005 a 2012. Inf Quinc Epidemiol Nac. 2013;18:32-41. Accessed: August 1, 2015. Available from: http://www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/IA/INS/IQEN%20vol%2018%202013%20num%203.pdf [ Links ]
29. Duque-Velásquez C, Garzón Á, Villegas A, Escobar LM, Zea J, Lopera F, et al . Human transmissible spongiform encephalopathy: Case report. Iatreia. 2014;27:330-6. [ Links ]
30. Tramontini-Jens C, Mora-Salazar JA, Castaño-Restrepo NE. Creutzfeldt-Jakob disease, MRI findings: A case report. Rev Colomb Radiol. 2014:25;4021-5. [ Links ]
31. Cadena-Sanabria M, Ardila-Báez M, Rueda-Prada L. Status epilepticus and rapidly progressive dementia in an elderly woman. Sporadic Creutzfeldt-Jakob disease. Rev Esp Geriatr Gerontol. 2015;50:103-4. http://dx.doi.org/10.1016/j.regg.2014.10.009 [ Links ]
32. Triana JD, Pérez-Rangel A. Creutzfeldt-Jakob disease. Hospital de San José, Bogotá DC, Colombia. Repertorio de Medicina y Cirugía. 2015;24:298-302. [ Links ]
33. Toro G, Díaz A, Saad C. Ayer, hoy y mañana, la teoría del prion. Revista Medicina. 2004;26:7-20. [ Links ]
34. Duque JC, Villegas A, Rodas JD. Transmissible spongiform encephalopathies: Biology of the prion and current state of the epidemiologic surveillance in Colombia. Rev Colomb Cienc Pecu. 2010;23:240-9. [ Links ]
35. Ossa JE, Machado G, Giraldo MA, McEwen JG. Prion plaques: Molecular tumors. A hypothesis on the etiopathogenesis of prion diseases. Med Hypotheses. 1995; 44:124-6. http://dx.doi.org/10.1016/0306-9877(95)90084-5 [ Links ]
36. Corral RH, Zapata JI. Priones como enfermedad en humanos. SIEI. 1998;4:6-17. [ Links ]
37. Zaninovic V. Correspondence. Colomb Med. 1998;29: 40-2. [ Links ]
38. Lopera-Restrepo F. La enfermedad de las vacas locas: efectos en el ser humano, su dominio y diagnóstico por el laboratorio. Medicina & Laboratorio. 2002;10:159-69. [ Links ]
39. Toro G, Pacheco OE, Sierra UE, Beltrán M, Díaz A, Parra EA, et al . Encefalopatías subagudas espongiformes transmisibles (ESET). La teoría prion-enfermedades priónicas. Acta Neurol Colomb. 2005;21:134-62. [ Links ]
40. Villegas-Lanau CA. Prion diseases: From molecular biology to clinical practice. Acta Neurol Colomb. 2010;26:87-111. [ Links ]
41. Barashi NS, Vargas-Acevedo C, Zarco LA. Enfermedades priónicas humanas. Univ Med. 2013;54:495-516. [ Links ]

