










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkBiomédica
versão impressa ISSN 0120-4157
Biomédica vol.36 supl.1 Bogotá dez. 2016
https://doi.org/10.7705/biomedica.v36i3.3011
ORIGINAL ARTICLE
doi: http://dx.doi.org/10.7705/biomedica.v36i3.3011
Production of recombinant proteins from Plasmodium falciparum in Escherichia coli
Ángela Patricia Guerra 1,2,3, Eliana Patricia Calvo 1, Moisés Wasserman 1, Jacqueline Chaparro-Olaya 3
1 Laboratorio de Investigaciones Básicas en Bioquímica, Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
2 Grupo de Bioquímica y Biología Celular, Instituto Nacional de Salud, Bogotá, D.C., Colombia
3 Laboratorio de Parasitología Molecular, Universidad El Bosque, Bogotá, D.C., Colombia
Author´s contributions:
Eliana Patricia Calvo, Moisés Wasserman and Jacqueline Chaparro-Olaya conceived the study.
Ángela Patricia Guerra and Eliana Patricia Calvo carried out laboratory procedures.
Ángela Patricia Guerra, Eliana Patricia Calvo and Jacqueline Chaparro-Olaya performed analysis and interpreted the data.
All authors participated in the writing of the paper.
Received: 31/07/15; accepted: 14/02/16
Introduction: The production of recombinant proteins is essential for the characterization and functional study of proteins from Plasmodium falciparum . However, the proteins of P . falciparum are among the most challenging to express, and when expression is achieved, the recombinant proteins usually fold incorrectly and lead to the formation of inclusion bodies.
Objective: To obtain and purify four recombinant proteins and to use them as antigens to produce polyclonal antibodies. The production efficiency and solubility were evaluated as the proteins were expressed in two genetically modified strains of Escherichia coli to favor the production of heterologous proteins (BL21-CodonPlus (DE3)-RIL and BL21-pG-KJE8).
Materials and methods: The four recombinant P. falciparum proteins corresponding to partial sequences of PfMyoA (Myosin A) and PfGAP50 (gliding associated protein 50), and the complete sequences of PfMTIP (myosin tail interacting protein) and PfGAP45 (gliding associated protein 45), were produced as glutathione S-transferase-fusion proteins, purified and used for immunizing mice.
Results: The protein expression was much more efficient in BL21-CodonPlus, the strain that contains tRNAs that are rare in wild-type E. coli , compared to the expression in BL21-pG-KJE8. In spite of the fact that BL21-pG-KJE8 overexpresses chaperones, this strain did not minimize the formation of inclusion bodies.
Conclusion: The use of genetically modified strains of E . coli was essential to achieve high expression levels of the four evaluated P . falciparum proteins and lead to improved solubility of two of them. The approach used here allowed us to obtain and purify four P . falciparum proteins in enough quantity to produce polyclonal antibodies in mice, and a fair amount of two pure and soluble recombinant proteins for future assays.
Key words: Plasmodium falciparum, recombinant proteins, Escherichia coli .
doi: http://dx.doi.org/10.7705/biomedica.v36i3.3011
Producción de proteínas recombinantes de Plasmodium falciparum en Escherichia coli
Introducción. La producción de proteínas recombinantes es fundamental para el estudio funcional de las proteínas de Plasmodium falciparum . Sin embargo, las proteínas recombinantes de P . falciparum están entre las más difíciles de expresar y, cuando lo hacen, usualmente se agregan dentro de cuerpos de inclusión insolubles.
Objetivo. Evaluar la producción de cuatro proteínas de P. falciparum usando como sistema de expresión dos cepas de Escherichia coli genéticamente modificadas para favorecer la producción de proteínas heterólogas y establecer una reserva de proteínas recombinantes puras y solubles, y producir anticuerpos policlonales a partir de ellas.
Materiales y métodos. Las proteínas recombinantes, las cuales correspondían a secuencias parciales de PfMyoA (Miosina-A) y PfGAP50 (proteína-asociada a glideosoma de 50 kDa) y a las secuencias completas de PfMTIP (proteína de interacción con miosina-A) y PfGAP45 (proteína asociada a glideosoma de 45 kDa), fueron expresadas como proteínas de fusión con la glutatión S-transferasa y luego purificadas y usadas para producir anticuerpos policlonales en ratón.
Resultados. La expresión de las proteínas recombinantes fue mucho más eficiente en la cepa BL21-CodonPlus (la cual expresa tRNAs escasos en las bacterias silvestres), que en la cepa BL21-pG-KJE8. Por el contrario, aunque la cepa BL21-pG-KJE sobreexpresa chaperonas, no redujo la formación de cuerpos de inclusión.
Conclusión. El uso de cepas de E . coli genéticamente modificadas fue fundamental para alcanzar altos niveles de expresión de las cuatro proteínas recombinantes evaluadas y permitió obtener dos de ellas en forma soluble. La estrategia utilizada permitió expresar cuatro proteínas recombinantes de P . falciparum en cantidad suficiente para inmunizar ratones y producir anticuerpos policlonales y, además, conservar proteína pura y soluble de dos de ellas para ensayos futuros.
Palabras clave: Plasmodium falciparum, proteínas recombinantes, Escherichia coli.
doi: http://dx.doi.org/10.7705/biomedica.v36i3.3011
Malaria is the most important parasitic infection worldwide and one of the greatest public health challenges in developing countries. Of the five species of Plasmodium that infect humans, P. falciparum is the most lethal and causes 198 million new cases each year and nearly 600,000 deaths (1). Despite the reduction in the incidence and mortality of malaria over the past decade, there is a global emergence and gradual spread of antimalarial drug resistance, which has prompted significant efforts to determine the molecular mechanisms governing the biology of the parasite, either to counter this resistance or to propose alternative therapeutic targets.
Plasmodium spp. is an obligate intracellular para-site whose survival and proliferation depends on its ability to invade the host cell. Apicomplexan parasites actively enter host cells making use of a unique form of motility referred to as gliding, powered by an actomyosin-motor immersed into a protein complex termed the glideosome (2,3). Since the proteins in this complex are highly conserved and specific to all Apicomplexa (4), the glideosome has enormous potential as chemotherapeutic target. However, the characterization and functional study of proteins from extracts of parasites in culture is difficult because it is impossible to obtain them in pure form and in sufficient quantities. Thus, many of the current functional studies rely on recombinant protein production in heterologous expression systems, amongst which Escherichia coli is the most widely used. Its popularity is primarily a result of the abundant knowledge of the biology and biochemistry of this bacterium, the facility of its genetic manipulation and the low cost of culture maintenance (5). Despite these advantages, the expression of the recombinant proteins of Plasmodium is hampered by the high A/T content in its genome (80%) (6) and the dissimilar codon usage between these two organisms (7), with both issues leading to the production of truncated proteins or inefficient synthesis (8). Moreover, the recombinant proteins can fold incorrectly and lead to the formation of inclusion bodies (9), which hinder their purification.
The aim of this work was to assess two genetically modified strains of E. coli , BL21-CodonPlus (DE3)-RIL and BL21-pG-KJE8, on the expression of four P. falciparum glideosome proteins: Myosin A (PfMyoA), the myosin tail interacting protein (MTIP), the gliding-associated protein GAP45 and the gliding-associated protein GAP50. The first strain overexpresses four tRNAs that are typically rare in wild-type strains of E. coli, and the second strain overexpresses five chaperones ( dnaK, dnaJ, grpE, groEL, and groES ) that promote the correct folding of proteins. The recombinant proteins were obtained as glutathione S-transferase (GST) fusion proteins from the partial sequences of the PfmyoA and gap50 genes and complete sequences of the genes mtip and gap45 .
The results showed that the expression of the four recombinant proteins of P. falciparum was much more efficient in strains containing tRNAs normally scarce in wild-type strains of E. coli, than in strains that have elevated levels of chaperone proteins. Furthermore, the use of the latter did not diminish the formation of insoluble protein aggregates. Finally, the purified recombinant proteins were produced in suitable quantities for immunizing mice and generating antibodies, which recognized the four proteins of interest in P. falciparum protein extracts.
Materials and methods
Bioinformatics
The A/T content of the sequences coding for the recombinant proteins was assessed with the GC Content Calculator (http://www.biologicscorp.com/tools/GCContent). The theoretical molecular weight and isoelectric point (pI) of each recombinant protein were calculated with the ExPASy compute pI/Mw tool (http://web.expasy.org/compute_pi/), and the charge and number of acidic aminoacids of the proteins were determined using PEPSTATS (http://www.ebi.ac.uk/Tools/seqstats/emboss_pepstats/). Finally, as codons are unevenly used in different organisms, the codon usage bias between E. coli and P. falciparum was examined using the Codon Usage Database at http://www.kazusa.or.jp/codon/. To assess preferences for particular synonymous codons, the number and frequency of each codon in the recombinant sequences were calculated using Sequence Manipulation Suite (SMS) (http://www.bioinformatics.org/sms2/reference.html).
Cloning of genes
Based on the sequences of PfmyoA, mtip, gap45 and gap50 (PlasmoDB ID: PF3D7_1342600, PF3D7_1246400, PF3D7_1222700 and PF3D7_ 0918000, respectively), gene-specific primers were designed as described in table 1. Each fragment was amplified from genomic DNA of P. falciparum (3D7 strain) that was extracted by proteinase K lysis, phenol chloroform treatment and subsequent dialysis against TE buffer (10). The polymerase chain reaction (PCR) conditions were as follows: 200 µM dNTPs, 1 µM primers, 0.625 U Pfu DNA polymerase (Thermo scientific) and 1.5 mM MgSO 4 . The amplification program included an initial denaturation cycle of 94°C x 5 min; 35 cycles of 94°C x 30 s, 50°C x 30 s, and 72°C x 1 min; and a final extension of 72°C x 7 min. The amplicons were cloned into the vector pGEM-T easy (Promega) using the manufacturer´s recommendations.

Subcloning into pGEX-4T2
The cloned fragments in pGEM-T easy were released by digestion with the enzymes BamHI and NotI and ligated into the expression vector pGEX-4T2 (Amersham Pharmacia Biotech) in the presence of T4 DNA ligase (Promega) following the manufacturer´s recommendations. The ligation products were transformed into TOP10 bacteria (Invitrogen™) by heat shock. The colonies were confirmed by PCR and the presence of the fragment of interest was verified by sequencing.
Expression of recombinant proteins
The TOP10 (Invitrogen™), BL21-CodonPlus (DE3)-RIL (Agilent) and BL21-pG-KJE8 (Takara Bio Inc) strains were transformed with the recombinant plasmids. For expression assays, a positive colony for each construct in each strain was grown in LB medium that was supplemented with 100 µg/ml ampicillin and 50 µg/ml chloramphenicol overnight at 37° C with constant stirring at 200 rpm. The following day, each culture was diluted with fresh medium, and for the BL21-pG-KJE8 system, the medium was supplemented with 1 mg/ml arabinose and 10 ng/ml tetracycline (chaperone expression-inducing agents). The cultures were incubated until they reached an optical density (OD 600 ) between 0.6 and 0.8, time at which isopropyl thio-beta-galactopyranoside (IPTG, 0.1 mM) was added, and growth was allowed for 16 h at 37° C.
Aliquots of 1 ml were collected just before induction (time 0) and at 2, 4, and 16 h after induction. These aliquots were centrifuged at 1,000 x g for 5 min; the pellet was solubilized in 1X loading buffer for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and the proteins were denatured at 95° C for 5 min. The resulting extract was run on 10% denaturing acrylamide gels, and the recombinant proteins were detected by Coomassie blue staining and/or western blotting (WB). The expression from the pGEX-4T2 vector results in GST fusion proteins with the GST tag (26 kDa) at the amino terminus and the protein of interest at the carboxyl terminus; thus, a poly-clonal anti-GST antibody (Sigma) was used as the primary antibody to detect recombinants in WB. A signal amplification system that involved the use of a secondary anti-rabbit IgG antibody conjugated to biotin (Sigma) and alkaline phosphatase enzyme conjugated to streptavidin (Pierce TM ) was used.
Evaluation of the solubility of the recombinant proteins
The bacterial culture was centrifuged at 1,000 x g and the pellet was re-suspended in five volumes of buffer R (50 mM NaCl, 50 mM Tris HCl pH 7.5 and 5% glycerol), lysozyme (1 mg/ml) and 1:200 protease inhibitor cocktail (Sigma). Five cycles of freezing and thawing (liquid nitrogen x 45 s, water bath at 37° C x 2 min) were performed, and the suspension was then centrifuged at 15,000 x g for 15 min to separate the supernatant from the particulate fraction. The presence of the protein of interest in both fractions was monitored by SDS-PAGE and WB to determine its solubility.
Effect of temperature on the recovery of soluble proteins
In certain cases, temperature decrease may reduce the formation of protein aggregates and thus improve the solubility of the recombinant proteins (11). Therefore, the protocol previously described for the expression of proteins in E. coli BL21-pG-KJE8 was followed with the culture maintained at 30° C.
Purification of recombinant proteins
The soluble recombinant proteins were purified by affinity chromatography with glutathione-agarose resin (Thermo Scientific) from 1 L of the bacterial culture. The purification was performed in a batch with 300 µl of the resin bed per 6 ml extract. Binding was allowed for 30 min at 4° C with gentle shaking; the mixture was then centrifuged at 1,000 x g for 2 min and the supernatant was saved. The resin was washed 10 times each with 10 volumes of wash buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl) or until the absorbance of the wash solution at 280 nm was at or below 0.010. The recombinant protein was eluted with 300 µl elution buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl and 10 mM reduced glutathione). The resulting eluates were analyzed by SDS-PAGE and stored at -70° C.
The insoluble recombinant proteins were purified from the particulate fraction as follows: One gram of pellet was homogenized in wash buffer (100 mM Tris-HCl pH 7, 5 mM EDTA, 5 mM DTT, 2 M urea, 2% triton X-100) and centrifuged at 22,000 x g for 30 min at 4° C. This procedure was repeated twice, and the resulting inclusion bodies were then re-suspended in wash buffer without urea or Triton and centrifuged two times as mentioned above; 0.5 ml of extraction buffer (50 mM Tris-HCl pH 7, 5 mM EDTA, 5 mM DTT, 8 M guanidine-HCl), were added to the pellet and the suspension was then homogenized and centrifuged at 17,000 x g for 1 h at 4° C. The supernatant was dialyzed against 1 L of dialysis buffer (150 mM NaCl and 50 mM Tris-HCl pH 8.0) with two buffer changes. The resulting suspension after dialysis was run under denaturing conditions on a preparative gel. The region of the gel containing each recombinant protein was excised, triturated, and incubated with four volumes of ultrapure water at 37° C for 4 h (12). The purity and concentration of the protein in the eluate were analyzed by SDS-PAGE and WB.
Production of polyclonal antibodies
Each of the purified recombinant proteins was inoculated intraperitoneally into four Balb/c mice of 5-6 weeks of age. The immunization schedule consisted of three injections on days 1, 14 and 35, the first one with 100 µg of protein in Freund´s complete adjuvant and two boosts with 50 µg of antigen in incomplete adjuvant. The final bleeding was done on day 42 (13). Then, the four recombinant proteins (50-250 ng/lane) and P. falciparum protein extracts (Strain 3D7, 50-100 µg/lane) were separated by SDS-PAGE, transferred onto PVDF or nitrocellulose membranes and immunoblotted with the produced antibodies. The anti-MTIP, anti-GAP50 and anti-GAP45 sera were used at dilutions of 1:500, while the anti-MyoA serum was used at a dilution of 1:5000. The secondary antibody used was biotin goat-anti mouse antibody (Dako) at a dilution of 1:2000 or 1:5000, respectively, and streptavidin conjugated to alkaline phosphatase (1:3000) (Promega) or horseradish peroxidase (1:10000) enzymes (Vector).
Ethics
This study was approved by the Ethics Committees of the Colombian Instituto Nacional de Salud (approval 9, October 7 th, 2010) and of the Facultad de Ciencias of the Universidad Nacional de Colombia (approval 5, October 11 th, 2010).
Results
Bioinformatics
The A/T content of the sequences coding for the recombinant proteins was about 65% in all the cases. The theoretical molecular weight and pI of the four recombinants: GST-PfMyoA, GST-PfMTIP, GST-PfGAP45 and GST-PfGAP50, were 54.6 kDa/5.67, 49.8 kDa/4.71, 49.9 kDa/4.83 and 43.6 kDa/6.33, respectively. The PEPSTATS tool found 70, 77, 90 and 51 acidic amino acids equivalent to 15%, 18%, 21% and 13% of the total amino acids in the named proteins. As stated above, BL21-CodonPlus (DE3)-RIL strain overexpresses four tRNAs that are typically rare in wild-type strains of E. coli; then , the Codon Usage Database was used to get the frequency of use of AGA, AGG, AUA and CUA codons in E. coli and P. falciparum . The relative abundance of these codons in the expressed sequences was also calculated (table 2).
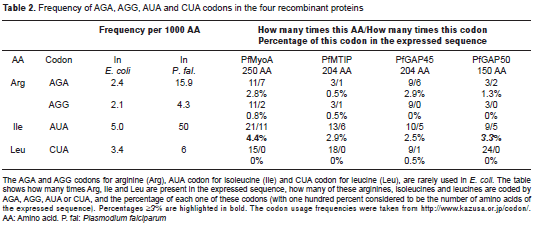
Effect of host strain on the production of recombinant proteins
This study evaluated the expression of four proteins from P. falciparum in three different strains of E. coli : TOP10, BL21-CodonPlus (DE3)-RIL (hereinafter called CodonPlus), and BL21-pG-KJE8 (hereinafter called pG-KJE8). The IPTG-induced expression of each protein was evaluated by SDS-PAGE and WB with an anti-GST antibody; GST-PfMyoA, GST-PfMTIP, GST-PfGAP45 and GST-PfGAP50 proteins were detected as bands of ~ 52 kDa, ~ 55 kDa, ~ 60 kDa and ~ 43 kDa, respectively. The recombinant GST-PfGAP45 had the same molecular weight ( ~ 60 kDa) as one of the molecular chaperones (groEL) expressed in the pG-KJE8 strain, resulting in bands overlapping; thus, at time zero, a band was observed that corresponded to the chaperone groEL. Expression of this recombinant was confirmed by WB that showed a weak expression in this strain. In contrast to the other three proteins, GST-PfMTIP was not successfully expressed in E. coli pG-KJE8 (figure 1).
Regarding the incubation time for the CodonPlus system, GST-PfGAP45 was expressed from 2 h post incubation; however, at 16 h a decreased signal was observed, which suggests proteolytic degradation. GST-PfMTIP was expressed quite well from 2 h after incubation and increased until 16 h. For GST-PfGAP50, expression was significantly higher compared to that of the other two recombinant proteins, and it was produced in large amounts beginning at 2 h after incubation and continuing until 16 h (figure 1). The expression of GST-PfMyoA did not increase in the CodonPlus strain and was almost undetectable by Coomassie blue staining; conversely, in the pG-KJE8 strain, protein synthesis was evident between 2 and 4 h of incubation (figure 1).
Effect of host strain on the solubility of the recombinant proteins
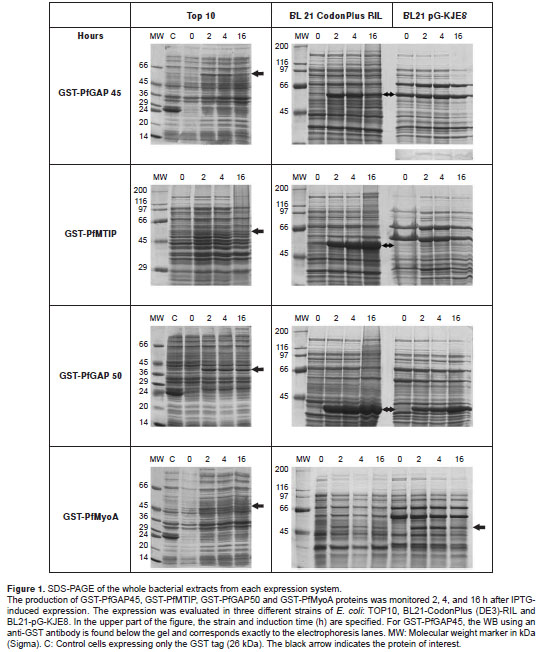
Once the optimal conditions for the production of each recombinant protein had been established, solubility was assessed. For this test, protein extracts were prepared under non-denaturing conditions, and the presence of the protein of interest in both the cytosolic extract and pellet was evaluated. GST-PfGAP45 and GST-PfMTIP were found in both the cytosolic fraction and pellet, whereas GST-PfGAP50 and GST-PfMyoA were found in the pellet (figure 2). These two proteins were only soluble in the presence of strong chaotropic agents such as 8 M guanidine, which indicated that insoluble protein aggregates or inclusion bodies (IB) had formed. No differences in solubility were observed between the host strain that co-expressed the chaperones and strains that only had basal levels of these proteins.
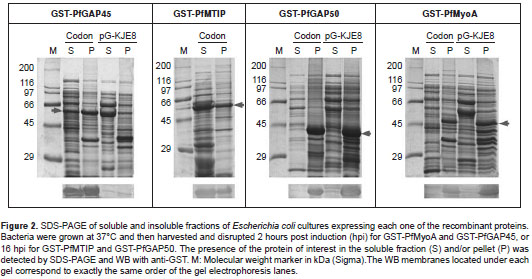
Effect of temperature on the solubility of recombinant proteins expressed in the strain pG-KJE8
Because the activity of certain chaperones of E. coli is increased at temperatures of approximately 30° C, and temperature reduction at the time of induction has been used favorably to improve the solubility of the recombinant proteins (11,14,15), an expression assay was conducted by incubating at 30° C proteins GST-PfMyoA and GST-PfGAP50 producing pG-KJE8 clones. Despite this modification, no increase in the solubility of the two proteins was observed (figure 3).
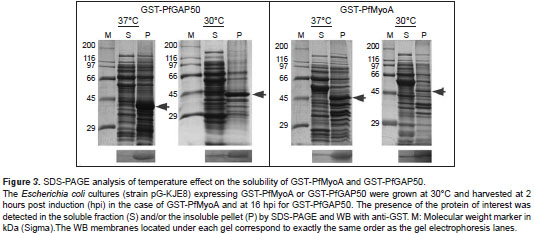
Purification of recombinant proteins
The culture was scaled up to 1 L to obtain sufficient raw material to begin the purification process. The soluble GST-PfMTIP protein was purified by affinity chromatography (Pierce TM Glutathione Agarose), and although other lower-weight bands were found using SDS-PAGE, GST-PfMTIP was always found in greater amounts than the others (figure 4). The bands of different molecular weights were most likely degraded or truncated proteins whose GST tag at the amino terminus allowed their co-purification by affinity chromatography and recognition by the anti-GST antibody. Furthermore, although GST- PfGAP45 was detected in the cytosolic extract, it was not purified by affinity with glutathione-agarose resin and had to be excised from acrylamide gels. Seven milligrams of GST-PfMTIP were obtained, whereas 1 mg was recovered for GST-PfGAP45.
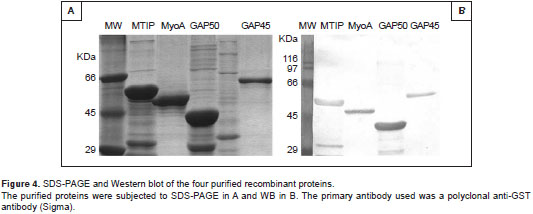
GST-PfMyoA and GST-PfGAP50 were solubilized from inclusion bodies with guanidinium hydro-chloride (8 M) and analyzed by SDS-PAGE. GST-PfMyoA, unlike GST-PfGAP50, presented several contaminating proteins; therefore, it was necessary to extract it from acrylamide gels. Approximately 3.2 mg of GST-PfGAP50 was recovered, whereas 800 µg of GST-PfMyoA was recovered. Once purified, all of the proteins were confirmed by WB with anti-GST (figure 4).
Immunoblotting
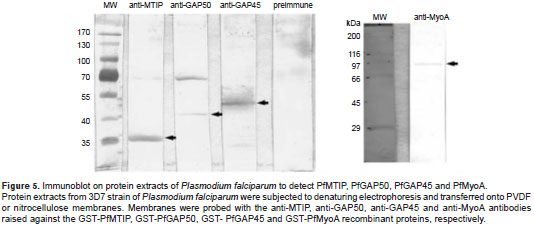
The MTIP ( ~ 32 kDa), GAP50 ( ~ 45 kDa), GAP45 ( ~ 50 kDa) and PfMyoA ( ~ 90 kDa) proteins were identified in immunoblots of parasite protein extracts by the antibodies raised against GST-PfMTIP, GST-PfGAP50, GST-PfGAP45 and GST-PfMyoA respectively (figure 5).
Discussion
The expression of P. falciparum proteins in heterologous systems typically results in a lack of expression or in insoluble expression. This problem was highlighted by a large-scale study where 1,000 P. falciparum open reading frames were selected for expression in E. coli (16). Of these ORFs, only 30% were expressed and only 6.3% of the resulting proteins were soluble. Higher molecular weight and protein disorder were negatively correlated with expression, hydrophobicity did not correlate with either expression or solubility, and pI was the only one factor associated with both expression and solubility. Notably, the high A/T content and the codon usage of P. falciparum , both usually correlated with lack of expression, did not affect significantly either the expression or solubility (16).
Later, Vedadi, et al. (17) selected 1,008 apicomplexan genes to be expressed in codon-enhanced E. coli strains. These genes were selected using P. falciparum as a model and looking for orthologous genes from P. vivax , P. yoelii , P. berghei , P. knowlesi , Toxoplasma gondii and Cryptosporidium parvum . Of these 1,008 genes, 304 (30.2%) were produced as soluble proteins, and when the seven genomes were analyzed as a "super Apicomplexan genome", 229 (49%) out of the 468 target genes were produced as soluble proteins. Vedadi, et al. (17) found that protein size, A/T content or pI do not have any impact in the soluble expression and concluded that E. coli is in effect a suitable expression system for Apicomplexan proteins, including those from Plasmodium .
The present study evaluated the expression of four proteins from P. falciparum in three different strains of E. coli : TOP10, CodonPlus and pG-KJE8. TOP10 is a strain that allows for stable replication and a high copy number of plasmids; the other two strains are used extensively in protein expression and not only have a decreased protease activity but also facilitate translation (CodonPlus) or protein folding (pG-KJE8). By using this approach, the four recombinant proteins were successfully expressed (figure 1). GST-PfMyoA and GST-PfGAP50 proteins were detected in SDS-PAGE and immunoblotting experiments about the expected sizes. On the other hand, GST-PfMTIP and GST-PfGAP45 migrated with apparent molecular masses of ~ 55 kDa and ~ 60 kDa, although they were predicted to encode proteins of ~ 50 kDa (figure 4). This behavior can be explained by their content of acidic amino acids, whose interactions with SDS have been reported to slow down the rate of migration (18-20).
In fact, GST-PfMTIP and GST-PfGAP45 exhibited the highest contents of acidic amino acids (18% and 21%), the lowest pI values (4.71 and 4.83) and the most negative charge (-25 and -26). In contrast, the charge and pI values were estimated at -4.5 / 5.67 for GST-PfMyoA and 1.5 / 6.33 for GST-PfGAP50. In spite of their anomalous migration, the GST-PfMTIP and GST-PfGAP45 identity was validated by sequencing the recombinant plasmids, and because the antibodies raised against these two recombinant proteins recognized their target proteins on P. falciparum extracts. The PfMyoA, MTIP, GAP45 and GAP50 proteins have theoretical masses of ~ 92, ~ 23.5, ~ 23.6 and ~ 45 kDa, respectively, and they were detected at ~ 90, ~ 32, ~ 50 and ~ 45 kDa in WB assays using protein extracts of P. falciparum (3D7 strain) (figure 5). The anomalous migration of MTIP and GAP45 was expected since both proteins typically run at molecular weights greater than would be predicted by their sequence. This phenomenon has been extensively reported and thought to result from post-translational modifications of the proteins (21,22). The GST-PfMyoA, GST-PfGAP45 and GST-PfGAP50 proteins were expressed in all three strains of E. coli , GST-PfMTIP could only be expressed by one strain (CodonPlus), and among the three strains tested, CodonPlus showed the most efficient expression for three of the four recombinant proteins (GST-PfMTIP, GST-PfGAP45, and GST-PfGAP50).
This CodonPlus strain has a plasmid that contains extra copies of the argU , ileY , and leuW genes that code for tRNA with specific codons for arginine (AGA/AGG), isoleucine (AUA), and leucine (CUA) that are rare in E. coli . Since in P. falciparum the AGA and AUA codons are used 6.6-fold and 10-fold more often than in E. coli , the co-expression of this plasmid provides the bacteria with a supplement of specific tRNAs for the codons of the parasite. In fact, an analysis of the cloned sequences revealed that AGA is the most used codon for arginine and AUA/AUU are the most used codons for isoleucine (table 2). These differences in codon usage can explain why the expression of the GST-PfMTIP, GST-PfGAP45, and GST-PfGAP50 proteins in the CodonPlus strain is increased compared with the expression in the TOP10 and pG-KJE8 strains, which only have basal levels of these tRNAs. These results are consistent with what has been reported in other studies (23,24).
However, in the case of GST-PfMyoA, a significant increase in expression in the CodonPlus strain was not observed. Carstens (25) has proposed that there may be low or no expression if the frequency of at least one type of rare codon in the bacterium is =3% in the protein to be expressed. For GST-PfMyoA, three rare codons, AGG, AGA and AUA, were found, and they reached frequencies of 0.8, 2.8 and 4.4%, respectively (table 2). Carstens (25) also suggested that the consecutive presence of rare codons is the most important factor affecting the expression of a heterologous protein in E. coli . In GST-PfMyoA, three cases were found: two doublets (AUAAGA) and a triplet (AUAAUAAGA); in GST-PfGAP45, a single case was found (the doublet AGAAGA) and in GST-PfGAP50 and GST-PfMTIP, no cases were found. Goldman, et al. (26) reported that the effect of successive rare codons on expression is more noticeable when they are located near the amino terminus of the protein, especially when the codons code for arginine. Chen, et al. (27) explain that this result occurs because of instability in the protein synthesis machinery near the beginning of the message. Upon analyzing the four sequences, only GST-PfMyoA (250 amino acids) presented this condition at codons 14-15 and 36-37. All of the above characteristics of the GST-PfMyoA sequence can explain why despite the use of three genetically different strains, an important increase in its production was not achieved.
GST-PfMyoA and GST-PfGAP50 were produced as protein aggregates, while GST-PfMTIP and GST-PfGAP45 were found in the cytoplasmic fraction. The results show that, for these proteins, A/T content, size protein and pI do not have noticeable effect on expression or solubility. Although the CodonPlus strain promotes expression of certain proteins of P. falciparum , the insolubility of the recombinant protein has been reported. Because translation is more efficient, the set of available chaperones to assist in protein folding becomes saturated and this may cause the accumulation and aggregation of misfolded proteins in the cytoplasm (28). The E. coli pG-KJE8 strain overexpresses both multi-component chaperone complexes present in E. coli : the gro proteins EL (60 kDa) and ES (10 kDa), and the KJE complex, which is formed by chaperones dnaK , dnaJ and grpE (29). In this way, if there is poor folding because of the saturation of the chaperone system itself, the presence of additional molecules will facilitate the proper folding of the protein, and it would be reflected in an increase in solubility (30-32). In our case, the evaluation of the solubility of the recombinant proteins produced in CodonPlus and pG-KJE8 strains showed similar results that were independent of the adjusted conditions for expression (temperature, time of incubation and IPTG concentration). Thus, the use of a bacterial system that provides extra chaperones did not prevent the formation of insoluble aggregates of GST-PfMyoA or GST-PfGAP50, indicating that additional factors other than the availability of the folding machinery determined the solubility of these recombinant proteins.
According to Wall, et al. (33), the specific role of the co-production of chaperones on the expression of a gene in E. coli is unclear and appears to be specific to each protein. In fact, undesirable side effects derived from chaperone gene co-expression, such as growth inhibition, proteolysis, reduced yield, reduced solubility or reduced specific activity, have been reported (34).
With respect to the purification of GST-PfGAP45, we suggest that the protein adopts a three-dimensional structure that prevents free exposure of the GST tag, thus preventing the recombinant protein from interacting with the glutathione agarose resin. This may explain why GST-PfGAP45 could not be purified by affinity chromatography. The detection of GST-PfGAP45 with anti-GST in denaturing conditions allows us to state that the GST tag was present.
In this study, the main purpose of producing the four recombinant proteins was to achieve suitable quantities of proteins for immunizing animals and generating antibodies, and to obtain large quantities of soluble proteins in sight of future in vitro protein-protein interaction studies. To achieve this goal, we used different host cells to establish the conditions in which the most efficient expression might be obtained and sought conditions that would allow us to express the protein in its native conformation. Although it was not possible to obtain all the proteins in soluble form and the use of denaturing agents for solubilization was required in two cases, the four proteins were obtained in high quantity.
Our results indicate that the use of genetically modified strains for the expression of P. falciparum proteins was necessary to improve the synthesis and achieve higher expression levels detectable by Coomassie blue staining, but their use did not necessarily have a positive effect on the solubility of the proteins. The CodonPlus strain favored the expression of three proteins, but the solubility varied from a completely soluble protein such as GST-PfMTIP to a completely insoluble protein such as GST-PfGAP50.
Although it is not possible to generalize about heterologous expression strategies, the approach used here led us to obtain four P. falciparum proteins corresponding to partial sequences of PfMyoA and GAP50, and the complete sequences of MTIP and GAP45. From 1 L of culture, we were able to produce and purify enough material for immunizing mice and for keeping a large supply for future uses.
Conflicts of interest
None declared
Financing
This work was supported by Colciencias (projects 110152128729 and 130834319109, and Contrato de recuperación contingente FP44842-334-2014 (2014-0401 Colciencias-MW)), by División de Investigaciones Bogotá - Universidad Nacional de Colombia (Project 15828), by the Instituto Nacional de Salud (CTIN 17-2010) and by Universidad El Bosque (PCI 2011-264). Funding organizations had no role in the study design, data analysis, decision to publish or preparation of the manuscript.
Corresponding author: Jacqueline Chaparro-Olaya, Laboratorio de Parasitología Molecular, Vicerrectoría de Investigaciones, Universidad El Bosque, Avenida Carrera 9 N° 131A-02, Edificio Biblioteca, segundo piso, Bogotá, D.C., Colombia
Telephone: (571) 648 9000, extension 1522; fax: (571) 648 9066 chaparrojacqueline@unbosque.edu.co
1. World Health Organization. World Malaria Report 2014: Summary. Accessed: April 1 st, 2015. Available from: http://apps.who.int/iris/bitstream/10665/160458/1/WHO_HTM_GMP_2015.2_eng.pdf?ua=1. [ Links ]
2. Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755-66. http://dx.doi.org/10.1016/j.cell.2006.02.006 [ Links ]
3. Frénal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe. 2010;8:343-57. http://dx.doi.org/10.1016/j.chom.2010.09.002 [ Links ]
4. Baum J, Richard D, Healer J, Rug M, Krnajski Z, Gilberger TW, et al . A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J Biol Chem. 2006;281:5197-208. http://dx.doi.org/10.1074/jbc.M509807200 [ Links ]
5. Overton TW. Recombinant protein production in bacterial hosts. Drug Discov Today. 2014;19:590-601. http://dx.doi.org/10.1016/j.drudis.2013.11.008 [ Links ]
6. Horrocks P, Bowman S, Kyes S, Waters AP, Craig A. Entering the post-genomic era of malaria research. Bull WHO. 2000;78:1424-37. [ Links ]
7. Yadava A, Ockenhouse CF. Effect of codon optimisation on expression levels of a functionally folded malaria vaccine candidate in prokaryotic and eukaryotic expression system. Infect Immun. 2003;71:4961-9. http://dx.doi.org/10.1128/IAI.71.9.4961-4969.2003 [ Links ]
8. Flick K, Ahuja S, Chene A, Bejarano MT, Chen Q. Optimized expression of Plasmodium falciparum erythrocyte membrane protein 1 domains in Escherichia coli . Malar J. 2004;3:50. http://dx.doi.org/10.1186/1475-2875-3-50 [ Links ]
9. Villaverde A, Carrio MM. Protein aggregation in recombinant bacteria: Biological role of inclusion bodies. Biotechnol Lett. 2003;25:1385-95. http://dx.doi.org/10.1023/A:1025024104862 [ Links ]
10. Wasserman M, Contreras J, Pinilla G, Rojas MO, Páez A, Caminos E. Plasmodium falciparum : Characterization of a 0.7-kbp, moderately repetitive sequence. Exp Parasitol. 1995;81:165-71. http://dx.doi.org/10.1006/expr.1995.1105 [ Links ]
11. Sørensen HP, Mortensen KK. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli . Microb Cell Fact. 2005;4:1. http://dx.doi.org/10.1186/1475-2859-4-1 [ Links ]
12. Scheer JM, Ryan CA. A method for the quantitative recovery of proteins from polyacrylamide gels. Anal Biochem. 2001; 298:130-2. http://dx.doi.org/10.1006/abio.2001.5384 [ Links ]
13. Harlow E, Lane D. Antibodies: A laboratory manual. First edition. New York: Cold Spring Harbor Laboratory Press; 1988. p. 67, 92-120. http://dx.doi.org/10.1002/jobm.3620300304 [ Links ]
14. Jonasson P, Liljeqvist S, Nygren PA, Ståhl S. Genetic design for facilitated production and recovery of recombinant proteins in Escherichia coli . Biotechnol Appl Biochem. 2002;35:91-105. http://dx.doi.org/10.1042/BA20010099 [ Links ]
15. Donovan RS, Robinson CW, Glick BR. Optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter. J Ind Microbiol. 1996;16:145-54. http://dx.doi.org/10.1007/BF01569997 [ Links ]
16. Mehlin C, Boni E, Buckner FS, Engel L, Feist T, Gelb MH, et al . Heterologous expression of proteins from Plasmodium falciparum : Results from 1000 genes. Mol Biochem Parasitol. 2006;148:144-60. http://dx.doi.org/10.1016/j.molbiopara.2006.03.011 [ Links ]
17. Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong A, et al . Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol. 2007;151:100-10. http://dx.doi.org/10.1016/j.molbiopara.2006.10.011 [ Links ]
18. Armstrong DJ, Roman A. The anomalous electrophoretic behavior of the human papillomavirus type 16 E7 protein is due to the high content of acidic amino acid residues. Biochem Biophys Res Commun. 1993;192:1380-7. http://dx.doi.org/10.1006/bbrc.1993.1569 [ Links ]
19. Iakoucheva LM, Kimzey AL, Masselon CD, Smith RD, Dunker AK, Ackerman EJ. Aberrant mobility phenomena of the DNA repair protein XPA. Protein Sci. 2001;10:1353-62. http://dx.doi.org/10.1110/ps.ps.40101 [ Links ]
20. Shi W, Huang Y, Sutton-Smith M, Tissot B, Panico M, Morris HR, et al . A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions. J Virol. 2008;82:6190-9. http://dx.doi.org/10.1128/JVI.02731-07 [ Links ]
21. Jones ML, Kitson EL, Rayner JC . Plasmodium falciparum erythrocyte invasion: A conserved myosin associated complex. Mol Biochem Parasitol. 2006;147:74-84. http://dx.doi.org/doi:10.1016/j.molbiopara.2006.01.009 [ Links ]
22. Rees-Channer RR, Martin SR, Green JL, Bowyer PW, Grainger M, Molloy JE, et al . Dual acylation of the 45 kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol Biochem Parasitol. 2006;149:113-6. http://dx.doi.org/doi:10.1016/j.molbiopara.2006.04.008 [ Links ]
23. Baca AM, Hol WG. Overcoming codon bias: A method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli . Int J Parasitol. 2000;30:113-8. http://dx.doi.org/10.1016/S0020-7519(00)00019-9 [ Links ]
24. Karmodiya K, Srivastav RK, Surolia N. Production and purification of refolded recombinant Plasmodium falciparum beta-ketoacyl-ACP reductase from inclusion bodies. Protein Expr Purif. 2005;42:131-6. http://dx.doi.org/10.1016/j.pep.2005.02.008 [ Links ]
25. Carstens P. Use of tRNA-supplemented host strains for expression of heterologous genes in E . coli . In: Vaillancourt PE, editor. E . coli gene expression protocols. Totowa, NJ: Humana Press; 2003. p. 225-33. http://dx.doi.org/10.1385/1-59259-301-1:225 [ Links ]
26. Goldman E, Rosenberg AH, Zubay G, Studier FW . Consecutive low-usage leucine codons block translation only when near the 5´ end of a message in Escherichia coli . J Mol Biol. 1995;245:467-73. http://dx.doi.org/10.1006/jmbi.1994.0038 [ Links ]
27. Chen GF, Inouye M. Suppression of the negative effect of minor arginine codons on gene expression; preferential usage of minor codons within the first 25 codons of the Escherichia coli genes. Nucleic Acids Res. 1990;18:1465-73. http://dx.doi.org/10.1093/nar/18.6.1465 [ Links ]
28. Rosano GL, Ceccarelli EA. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb Cell Fact. 2009;8:41. http://dx.doi.org/10.1186/1475-2859-8-41 [ Links ]
29. Schumann W, Ferreira LC. Production of recombinant proteins in Escherichia coli . Genet Mol Biol. 2004;27:442-53. http://dx.doi.org/10.1590/S1415-47572004000300022 [ Links ]
30. Barth S, Huhn M, Matthey B, Klimka A, Galinski EA, Engert A . Compatible-solute-supported periplasmic expres-sion of functional recombinant proteins under stress conditions. Appl Environ Microbiol. 2000;66:1572-9. http://dx.doi.org/10.1128/AEM.66.4.1572-1579.2000 [ Links ]
31. Schäffner J, Winter J, Rudolph R, Schwarz E. Cosecretion of chaperones and low-molecular-size medium additives increases the yield of recombinant disulfide-bridged proteins. Appl Environ Microbiol. 2001;67:3994-4000. http://dx.doi.org/10.1128/AEM.67.9.3994-4000.2001 [ Links ]
32. Choi JH, Lee SY. Secretory and extracellular production of recombinant proteins using Escherichia coli . Appl Microbiol Biotechnol. 2004;64:625-35. http://dx.doi.org/10.1007/s00253-004-1559-9 [ Links ]
33. Wall JG, Plückthun A. Effects of overexpressing folding modulators on the in vivo folding of heterologous proteins in Escherichia coli . Curr Opin Biotechnol. 1995;6:507-16. http://dx.doi.org/10.1016/0958-1669(95)80084-0 [ Links ]
34. Martínez-Alonso M, García-Fruitós E, Ferrer-Miralles N, Rinas U, Villaverde A. Side effects of chaperone gene co-expression in recombinant protein production. Microb Cell Fact. 2010;9:64. http://dx.doi.org/10.1186/1475-2859-9-64. [ Links ]

