










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiomédica
Print version ISSN 0120-4157
Biomédica vol.36 supl.2 Bogotá Aug. 2016
https://doi.org/10.7705/biomedica.v36i0.2990
ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2990
1 Grupo de Virología, Dirección de Redes en Salud Pública, Instituto Nacional de Salud, Bogotá, D.C., Colombia
2 Departamento de Ciencias Básicas, Universidad de La Salle, Bogotá, D.C., Colombia
3 Grupo de Enfermedades Transmitidas por Vector, Dirección de Vigilancia y Análisis de Riesgo en Salud Pública, Instituto Nacional de Salud, Bogotá, D.C., Colombia
4 Grupo de Entomología, Dirección de Redes en Salud Pública, Instituto Nacional de Salud, Bogotá, D.C., Colombia
Contribución de los autores:
Katherine Laiton-Donato y José A. Usme-Ciro: concepción y diseño del estudio
Todos los autores participaron en el análisis y la interpretación de los resultados, la revisión crítica del manuscrito, la aprobación de la versión final y de todos los aspectos del manuscrito.
Recibido: 21/07/15; aceptado: 22/10/15
Introducción. El virus del chikungunya, perteneciente al género Alphavirus de la familia Togaviridae, es un virus ARN de 11,8 kb, de cadena sencilla y polaridad positiva, transmitido por Aedes spp . Se han identificado tres genotipos a nivel mundial: el de Asia, el del este-centro-sur de África ( East/Central/South African, ECSA) y el de África occidental ( West African, WA). La fiebre del chikungunya es una enfermedad febril aguda, acompañada principalmente de inflamación en las articulaciones y erupción cutánea. Después de su aparición en las Américas en el 2013, los primeros casos en Colombia ocurrieron en septiembre de 2014 y hasta junio del 2015 se habían notificado 399.932 casos.
Objetivo. Identificar el genotipo o los genotipos responsables de la primera epidemia por el virus del chikungunya en Colombia y la variabilidad genética asociada a su dispersión en el territorio nacional.
Materiales y métodos. Se seleccionaron muestras de suero de pacientes con síntomas indicativos de fiebre del chikungunya durante 2014 y 2015. Se hizo una transcripción inversa seguida de reacción en cadena de la polimerasa del gen E1, así como su secuenciación, análisis filogenético y análisis de evolución adaptativa.
Resultados. Se demostró la presencia exclusiva del genotipo de Asia en Colombia. Se registró un promedio de 0,001 sustituciones de bases por sitio, una identidad de 99,7 a 99,9 % en los nucleótidos y de 99,9 % en los aminoácidos entre las secuencias colombianas y las secuencias de las Américas. Los análisis de evolución adaptativa indicaron una fuerte selección purificadora en el gen E1 .
Conclusiones. Se determinó la circulación del genotipo de Asia del virus del chikungunya como la causa de la primera epidemia en Colombia. Es necesario continuar con la vigilancia de genotipos, con el fin de detectar posibles cambios en la epidemiología, la eficacia ( fitness ) viral y la patogenia del virus.
Palabras clave: virus del chikungunya , vigilancia, genotipo, filogenia, Colombia .
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2990
Phylogenetic analysis of Chikungunya virus in Colombia: Evidence of purifying selection in the E1 gene
Introduction: Chikungunya virus (CHIKV) is a single-stranded positive sense RNA virus that belongs to the Alphavirus genus of the family Togaviridae. Its genome is 11.8 kb in length, and three genotypes have been identified worldwide: Asian, East/Central/South African (ECSA) and West African. Chikungunya fever is an acute febrile disease transmitted by Aedes spp . that usually presents with polyarthralgia and cutaneous eruption. Following introduction of the virus to the Americas in 2013, the first cases in Colombia occurred in September of 2014, and they reached a cumulative total of 399,932 cases by June of 2015.
Objective: To identify the genotype or genotypes responsible for the current epidemic in Colombia and to describe the genetic variability of the virus in the country.
Materials and methods: Serum samples from patients presenting with symptoms compatible with Chikungunya fever during 2014-2015 were selected for the study. RT-PCR products of the E1 gene from these samples were used for sequencing and subsequent phylogenetic and adaptive evolution analyses.
Results: The study identified only the presence of the Asian genotype in Colombia. Comparing the Colombian sequences with other sequences from the Americas revealed an average of 0.001 base substitutions per site, with 99.7% and 99.9% nucleotide identity and 99.9% amino acid identity. The adaptive evolution analysis indicated that the E1 gene is under strong purifying selection.
Conclusions: The first epidemic of Chikunguya fever in Colombia was caused by the circulation of the virus Asian genotype. Further genotypic surveillance of the virus in Colombia is required to detect possible changes in its epidemiology, fitness and pathogenicity.
Key words: Chikungunya virus, surveillance, genotype, molecular epidemiology, phylogeny, Colombia.
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2990
El virus del chikungunya pertenece al género Alphavirus de la familia Togaviridae , y es un importante patógeno reemergente transmitido por mosquitos del género Aedes . Es un virus encapsulado de 60 a 70 nm de diámetro, el cual posee una cápside de simetría icosaédrica que alberga un genoma de ARN de cadena sencilla y polaridad positiva de 11,8 kb, aproximadamente (1). El genoma del virus codifica para cuatro proteínas no estructurales (nsP1 – nsP4), las cuales participan en la replicación del ARN viral y en la producción de ARN mensajero (ARNm) subgenómico, el cual codifica, a su vez, para las cinco proteínas estructurales C, E3, E2, 6K y E1. La proteína E1 forma heterodímeros con E2, los cuales son transportados a la membrana celular para hacer parte de los viriones maduros luego de la gemación de las nucleocápsides (2).
El virus fue aislado por primera vez e identificado como el agente etiológico responsable de un brote de enfermedad febril en Tanzania entre 1952 y 1953 (3).
La fiebre del chikungunya es una enfermedad febril aguda, con un periodo de incubación de dos a 12 días, cuyas principales manifestaciones clínicas incluyen fiebre alta, mialgias, artralgias o artritis, linfadenopatías, dolores multifocales y erupción cutánea (1). La fase aguda de la enfermedad puede resolverse en un periodo de siete a 14 días, en tanto que la fase crónica, caracterizada por manifestaciones reumatológicas, puede persistir durante meses e incluso años (4).
El virus aumentó su rango de distribución entre los años 60 y 70, con brotes en diversos países asiáticos y en el subcontinente indio. En el 2004 emergió nuevamente de forma epidémica en el este de África, y rápidamente se distribuyó en países tropicales y subtropicales de África, Asia y Oceanía (5,6). En 2013 llegó a la región de las Américas a través de la isla Saint Martin y en poco tiempo se diseminó por diversas islas del Caribe y varios países de la región (7-9).
En Colombia el primer caso de fiebre del chikungunya importado se registró en Cali el 19 de junio de 2014, y los primeros casos de transmisión autóctona se notificaron en septiembre de 2014, con un total de 399.932 casos y 39 muertes desde el inicio de la epidemia hasta la semana epidemiológica 25 de 2015 (10), lo cual es el resultado de su transmisión en una población humana vulnerable en zonas con gran densidad del vector Aedes aegypti .
El análisis filogenético de las cepas del virus del chikungunya que circulan a nivel mundial ha permitido la identificación de tres linajes denominados genotipo de Asia, genotipo del este-centro-sur de África ( East/Central/South African, ECSA) y genotipo de África occidental ( West African, WA) (11-13); se estima que el ancestro común más reciente de los tres genotipos pudo haberse originado hace 500 años, aproximadamente, y que la divergencia entre el genotipo de Asia y el de ECSA pudo haber ocurrido hace 150 años (14). En un análisis del gen E1 se encontró una tasa de sustitución de 1,4 x 10 -3 por sitio por año (15).
El genotipo de Asia ha sido el principal responsable de la enfermedad en las islas del Caribe y en Centroamérica y Suramérica (9,16); sin embargo, recientemente se reportó la presencia del genotipo ECSA en Brasil (17). Por ello, es de vital importancia determinar los genotipos de los virus responsables de la epidemia actual en Colombia y la variabilidad genética asociada a su diseminación en el territorio nacional, lo cual contribuye al conocimiento de la dinámica de circulación y transmisión del virus.
Materiales y métodos
Muestras clínicas
Se seleccionaron 29 muestras de suero remitidas al Laboratorio de Virología del Instituto Nacional de Salud entre 2014 y 2015, por sospecha clínica de fiebre del chikungunya en el marco del programa de vigilancia virológica del virus a nivel nacional.
Reacción en cadena de la polimerasa en tiempo real
En 18 de las 29 muestras, la detección del virus se hizo mediante reacción en cadena de la polimerasa (PCR) en tiempo real con el gen nsP4 y utilizando el estuche SuperScript III Platinum One-step ® (Invitrogen). Se utilizaron 0,4 mM de cada dNTP, 10 pmol de los oligonucleótidos CHIKV 6856/CHIKV 6981c y 5 µM de la sonda CHIKV 6919-FAM diseñados previamente, y 1 µl de la mezcla de enzimas (18), para un volumen final de la reacción de 25 µl. El perfil térmico incluyó un paso de transcripción inversa a 50 °C durante 30 minutos, la activación de la ADN polimerasa Taq Hot Start a 94 °C durante 15 minutos, 45 ciclos a 95 °C durante 15 segundos y a 60 °C durante un minuto para su hibridación y extensión.
Aislamiento viral
Las 11 muestras restantes se diluyeron en medio esencial mínimo de Eagle (E-MEM) en proporción de uno a diez y, posteriormente, se utilizaron en el intento de aislamiento viral inoculando 200 µl en cultivos de células Vero. La fase de adhesión se llevó a cabo a 37 °C durante una hora, momento en el cual se añadieron 800 µl de E-MEM con suplemento de 2 % de suero fetal bovino (FBS) y la solución tampón HEPES. Los cultivos se incubaron a 37 °C en una atmósfera con CO 2 . Los sobrenadantes de los aislamientos se recolectaron cuando se observó el efecto citopático, o a los cinco días de infección en caso de no observarse dicho efecto.
Extracción de ARN
El ARN viral se extrajo a partir de 18 muestras de suero y de 11 sobrenadantes de cultivo, utilizando el siguiente protocolo recomendado por el fabricante del estuche comercial QIAamp Viral RNA Mini Kit ® (Qiagen Inc., Chatsworth, CA, USA): se mezclaron 140 µl de muestra con 560 µl de solución tampón AVL y 5,6 µl de ARN carrier ; luego, se agregaron 500 µl de etanol absoluto y el volumen total se pasó a través de una columna de sílice mediante centrifugación. Posteriormente, se adicionaron 500 µl de solución tampón AW1 y 500 µl de AW2 para eliminar contaminantes y, por último, se eluyó mediante la adición de 60 µl de solución tampón AVE con centrifugación.
Amplificación del gen E1 mediante transcripción inversa seguida de PCR
La PCR en tiempo real para la amplificación de un fragmento de 1.666 pares de bases (pb) que incluía el gen E1 completo, se hizo utilizando el estuche QIAGEN One-Step RT-PCR kit ® (Qiagen Inc., Chatsworth, CA, USA) y los oligonucleótidos diseñados en este estudio (cuadro 1).
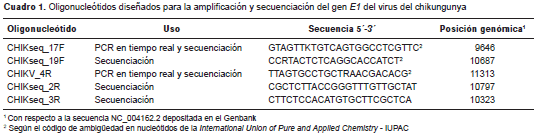
Se utilizaron 10 pmol de los oligonucleótidos CHIKseq_17F y CHIKV_4R, 5 µl de solución tampón 5X (que incluía 12,5 mM de MgCl 2 ), 1 µl de la mezcla de enzimas, 0,4 mM de cada dNTP y la solución Q en una concentración final de 1X para un volumen final de 25 µl. El perfil térmico consistió en un paso de transcripción inversa a 41 °C durante 45 minutos, un paso de activación de la ADN polimerasa Taq Hot Start a 94 °C durante 15 minutos, 40 ciclos a 94 °C durante 30 segundos para la desnaturalización, a 58 °C durante 30 segundos para la hibridación de oligonucleótidos, y a 72 °C durante dos minutos para la extensión, seguidos de una extensión final a 72 °C durante cinco minutos. Los amplicones se purificaron a partir de las reacciones de RT-PCR en tiempo real con el estuche comercial QIAquick PCR Purification Kit ® (Qiagen Inc., Chatsworth, CA, USA).
Secuenciación y edición de secuencias
Las secuencias se obtuvieron usando los oligo-nucleótidos descritos en el cuadro 1. Se agregaron 10 ng de cada amplicón a una mezcla de reacción que contenía 1 µl de BigDye Terminator Cycle Sequencing ® v3.1 (Applied Biosystems, Carlsbad, CA, USA), 3,2 pmol del oligonucleótido específico y 1,5 µl de solución tampón 5X, en un volumen final de 10 µl. El perfil térmico fue el siguiente: a 96 °C durante un minuto; 25 ciclos a 96 °C durante 10 segundos, a 50 °C durante 5 segundos y a 60 °C durante 4 minutos. Las reacciones se purificaron con el estuche Big Dye XTerminator ® (Applied Biosystems, Carlsbad, CA, USA) y posteriormente se procesaron mediante el analizador genético ABI3130 (Applied Biosystems, Carlsbad, CA, USA). Las secuencias se editaron y ensamblaron mediante el módulo SeqMan del programa LaserGene ® , v8.1 (DNASTAR Inc.).
Análisis filogenético
Con el fin de establecer la relación filogenética entre las cepas del virus del chikungunya circulantes en Colombia y las que han circulado a nivel mundial, se generó una matriz de secuencias correspondientes al gen E1 obtenidas en el presente estudio (posiciones de los nucleótidos 9994-11313 con respecto a la secuencia NC_ 004162.2), y las secuencias depositadas en el GenBank correspondientes a los tres genotipos, las cuales fueron alineadas mediante el programa Clustal X2.1 (19).
Se estimó el modelo de sustitución de nucleótidos que mejor se ajustara a la matriz de secuencias teniendo en cuenta el criterio de información de Akaike (AIC), mediante la herramienta jModelTest v2.1.3 (20).
Se hizo una inferencia bayesiana mediante el método de Monte Carlo vía cadenas de Markov (MCMC) con el programa MrBayes 3.2.6 (21); se utilizó el modelo de sustitución de nucleótidos GTR, y se evaluaron cuatro MCMC (tres frías, una caliente) en 1´000.000 de generaciones con muestreos cada 100 generaciones para un total de 10.000 árboles.
La convergencia de los parámetros estadísticos se determinó usando el programa Tracer 1.6 (http://tree.bio.ed.ac.uk/software/tracer/). Se descartaron 2.000 generaciones y los valores de probabilidades a posteriori se obtuvieron a partir de los 32.000 árboles resultantes.
La inferencia filogenética se hizo con el programa MEGA 6.0 (22) mediante los métodos de neighbor-joining , el modelo de sustitución de nucleótidos Kimura 2, parámetros con 1.000 réplicas bootstrap de máxima verosimilitud, y el modelo de sustitución de nucleótidos GTR con 1.000 réplicas bootstrap .
Los árboles de consenso se visualizaron mediante el programa FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) y se editaron en el programa MEGA 6.0 (22).
Análisis de diversidad genética y evolución adaptativa
Se estimaron las distancias genéticas dentro del genotipo, entre genotipos y para el grupo representado por las secuencias obtenidas en el estudio utilizando el modelo evolutivo Kimura 2 y los parámetros en el programa MEGA 6.0 (22).
Con el fin de establecer las posibles fuerzas que han moldeado la evolución del virus, se hizo un primer análisis utilizando diferentes pruebas de neutralidad: la prueba estadística D de Tajima (23) y los tests F* y D* de Fu y Li (24) utilizando el programa DnaSP, versión 5.10.01 (25).
Para determinar los codones específicos de la secuencia que podrían estar sujetos a selección, se analizó la matriz por medio del servidor Datamonkey (http://www.datamonkey.org) (26).
Se determinó la razón del número de sustituciones no sinónimas por sitio no sinónimo sobre el número de sustituciones sinónimas por sitio sinónimo ( divergence at nonsynonimous and synonimous sites , dN/dS).
En todos los análisis se usó el modelo de sustitución GTR ( generalised time reversible ) y un nivel de significación de p<0,1. Se seleccionaron los sitios que presentaron significación estadística mediante los métodos de máxima verosimilitud de probabilidad de efectos fijos ( Fixed Effects Likelihood , FEL), de probabilidad interna de efectos fijos ( Internal FEL , IFEL) y de single likelihood ancestor counting (SLAC).
Resultados
El virus del chikungunya en Colombia
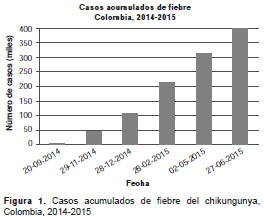
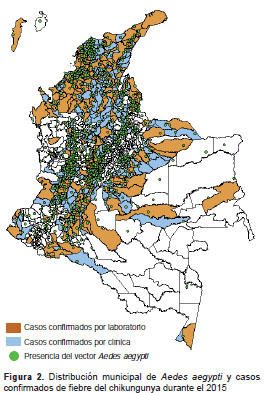
En Colombia hubo varias alertas por casos importados del virus del chikungunya antes del reporte de los primeros casos autóctonos en el departamento de Bolívar, en septiembre de 2014 (27). Después de su establecimiento, el virus ha tenido un comportamiento epidémico durante el periodo 2014-2015 (figura 1 ), y logró colonizar una gran proporción de los municipios con presencia del vector A. aegypti (figura 2 ).
Circulación del genotipo de Asia del virus del chikungunya en Colombia
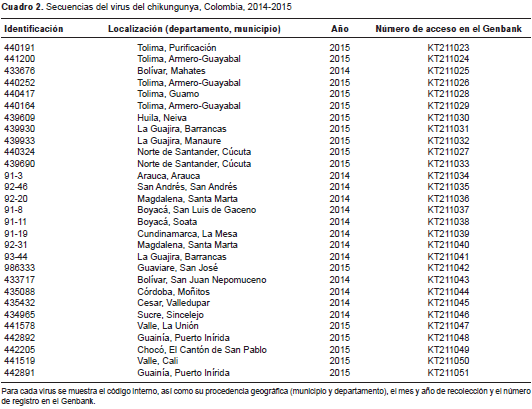
En el presente estudio se secuenció el gen E1 completo en un total de 29 sueros o sobrenadantes de cultivo celular provenientes de 17 departamentos colombianos (cuadro 2), es decir, 53 % de los departamentos con reporte de casos confirmados de la enfermedad.
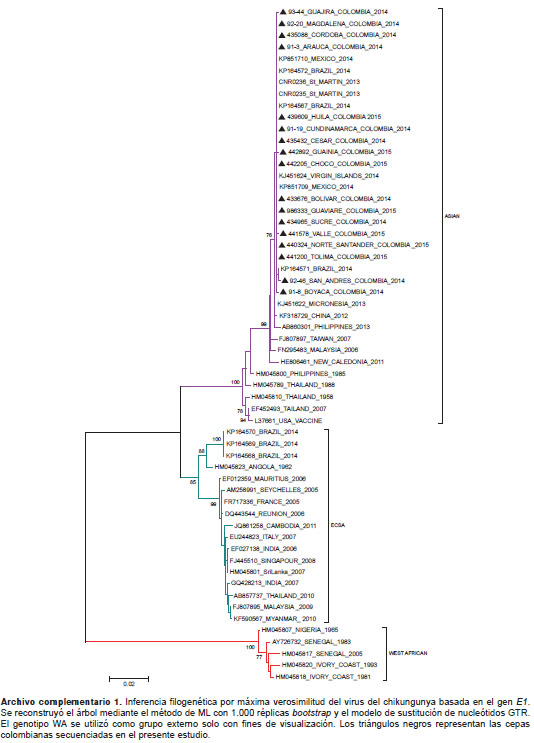
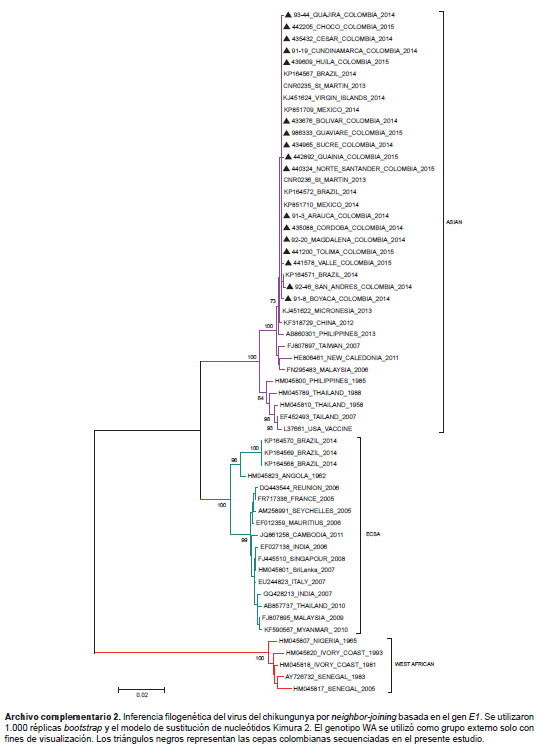
Las topologías obtenidas mediante inferencia bayesiana (figura 3 ) y los métodos de máxima verosimilitud y de neighbor joining (archivo complementario 1 y archivo complementario 2, disponibles en: doi: http://dx.doi.org/10.7705/biomedica.v36i0.2990), evidenciaron concordancia en los soportes bootstrap (mayores de 85 %) para las ramas correspondientes a los genotipos y los principales linajes dentro de cada genotipo. Las secuencias colombianas mostraron una estrecha relación filogenética con cepas del genotipo de Asia provenientes de la isla Saint Martin, las Islas Vírgenes, México y Brasil.
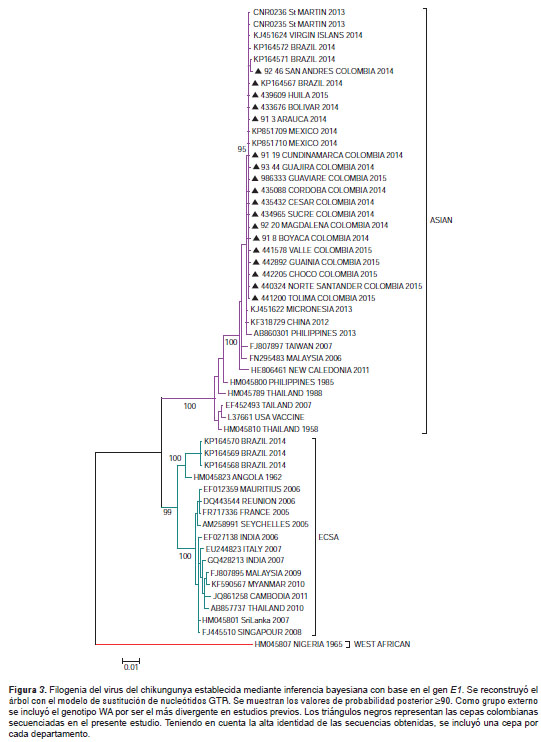
Diversidad genética y evolución adaptativa
Al estimar la diversidad genética, se evidenció un promedio de 0,001 sustituciones de bases por sitio entre cada par de secuencias colombianas del virus y las secuencias reportadas en el continente americano (Saint Martin, Islas Vírgenes, Brasil, México), con una identidad de 99,7 a 99,9 % a nivel de nucleótidos y de 99,9 % a nivel de aminoácidos. Entre las secuencias colombianas y las demás secuencias pertenecientes al genotipo de Asia se estimó un promedio de 0,005 sustituciones de bases por sitio entre cada par de secuencias.
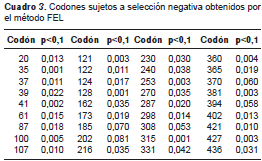
En los análisis de evolución adaptativa, las pruebas estadísticas de neutralidad presentaron las siguientes estimaciones: en la D deTajima, -0,28665 (p>0,10, no significativo), y en el test D* de Fu y Li, 0,93863 (p>0,10, no significativo) y en el test F*, 0,52645 (p>0,1, no significativo). El valor global de dN/dS fue 0,0517; el codón 145 fue el único seleccionado positivamente mediante los métodos FEL e IFEL, con un valor de 0,053 (p<0,1, significativo). Mediante los métodos SLAC, FEL e IFEL (p<0,1), se demostró la existencia de un gran número de codones sometidos a una fuerte selección negativa (cuadro 3).
Discusión
Desde la confirmación de la circulación autóctona del virus de chikinguña en septiembre de 2014, su transmisión en Colombia ha mostrado un patrón epidémico en el cual han contribuido de forma determinante diversos factores. En primer lugar, el virus encontró una población completamente vulnerable ante la infección, sin historia de retos previos frente a este virus o cualquier otro estrechamente relacionado. Al igual que el virus Mayaro detectado en Colombia hace más de 50 años (28), el virus del chikungunya pertenece al complejo del virus del bosque de Semliki (29).
Otros alfavirus que han circulado en las últimas décadas en Colombia, como el virus de la encefalitis equina venezolana y el virus de la encefalitis equina del este, pertenecen a otros serocomplejos y su incidencia en el humano ha sido limitada a nivel geográfico (30), por lo que es improbable que confieran inmunidad cruzada frente al virus del chikungunya. En segundo lugar, el virus encontró una alta densidad y una amplia distribución del mosquito vector A. aegypti doméstico, lo cual le permitió completar su ciclo de transmisión de forma eficiente e invadir rápidamente los 33 departamentos, ocasionando un aumento exponencial de los casos de fiebre del chikungunya durante el 2014 y el primer semestre del 2015 (31,32).
A nivel filogenético, las secuencias de las cepas colombianas mostraron una estrecha relación con las secuencias de cepas de la Isla Saint Martin, de Islas Vírgenes y de las otras reportadas en la región de las Américas, las cuales pertenecen al genotipo de Asia, y cuya dinámica de dispersión espacial y temporal a través de las islas del Caribe, de Centroamérica y de Suramérica sugiere la presencia exclusiva de dicho genotipo en la región. Recientemente se reportó la presencia del genotipo ECSA en Brasil (17), sin embargo, su impacto a nivel epidemiológico solo podrá determinarse en la medida en que se conozcan los resultados de otros estudios moleculares realizados en los diversos países de la región.
A pesar de que se espera un incremento en la diversidad genética de las cepas que circulan en la región debido a las múltiples rondas de replicación y a la alta tasa de error reportada para este virus (15), la posibilidad de rastrear el origen reciente de las cepas que hayan ingresado y se hayan diseminado por el territorio nacional se ve limitada por el bajo número de secuencias genómicas del virus disponibles hasta la fecha y responsables de los brotes o epidemias en los países de la región (33,34), así como por la gran estabilidad del genotipo, reflejada en un alto porcentaje de identidad entre las secuencias de las cepas provenientes de diferentes países de la región.
En el análisis de evolución adaptativa, aunque los valores obtenidos por las pruebas de neutralidad a nivel global (longitud total) para la región genómica analizada sugieren la ausencia de selección natural, la relación entre los dN y los dS y un análisis más sólido de los codones mediante los métodos FEL, IFEL y SLAC, demostraron la existencia de una fuerte selección purificadora para el gen E1 . La existencia de selección purificadora se sustenta en estudios con otros virus transmitidos por vectores artrópodos, en los cuales se ha demostrado la existencia de compromisos ( trade offs ) en su evolución, debido a que durante el ciclo de trasmisión deben preservar la función proteica que les permita replicarse en dos huéspedes filogenéticamente distantes (artrópodo y vertebrado) (33,35). Estos compromisos, con un efecto directo en la eficacia biológica ( fitness viral), comúnmente resultan en la eliminación de gran parte de la variabilidad a largo plazo y, por ende, en una mayor estabilidad genética, lo cual se ve reflejado en la poca acumulación de mutaciones en el gen E1 de las cepas que circulan en las Américas, a pesar de haber alcanzado poblaciones de gran tamaño y ser responsable de múltiples brotes epidémicos.
Durante la epidemia de 2005 a 2006 ocasionada por el genotipo ECSA del virus del chikungunya en la isla La Reunión, se encontró la sustitución A226V localizada en la proteína E1 y se la relacionó con la capacidad del virus de infectar y replicarse en el vector Aedes albopictus (34,36,37); sin embargo, esta sustitución no se ha encontrado en cepas pertenecientes al genotipo de Asia ni tampoco se evidenció en las secuencias colombianas analizadas. En el presente estudio la secuencia 93-44_Guajira_2014 presentó la sustitución de aminoácido T155I, la cual no se ha asociado al comportamiento epidemiológico o a la virulencia.
Considerando la presencia en el país de A. aegypti y de A. albopictus y la circulación simultánea de los genotipos de Asia y ECSA del virus del chikungunya en un país vecino (Brasil), es necesario establecer una vigilancia de genotipos, con el fin de detectar la potencial introducción de un nuevo genotipo, su interacción con el genotipo existente y su contribución a la transmisibilidad o virulencia del virus.
Se concluye que el patrón de dispersión del virus del chikungunya fue de alcance epidémico en Colombia, debido a la amplia distribución del vector A. aegypti y a la presencia de una población vulnerable ante la infección. Además, se identificó el genotipo de Asia como el único presente en la muestra de estudio, lo cual sugiere que ha sido el predominante, si no el único, durante la epidemia en el país. Asimismo, los patrones de diversidad genética y evolución adaptativa indican que el gen E1 está sometido a una fuerte selección purificadora.
Los datos presentados en el presente manuscrito son los primeros sobre la caracterización genética del virus del chikungunya en Colombia, y su importancia para la salud pública es enorme, ya que nunca antes había habido transmisión de este virus en el país y, seguramente, la enfermedad se hará endémica. Por ello, es necesario continuar con la vigilancia de los genotipos del virus, con el fin de detectar posibles cambios en su epidemiología, su eficacia viral ( fitness ), su habilidad para colonizar nuevos vectores y su patogenia.
Los autores desean agradecer a las secretarías y laboratorios departamentales de salud pública por la recolección y el envío de las muestras al Grupo de Virología del Instituto Nacional de Salud. Igualmente, los autores desean agradecer a Martha González Pinilla del Grupo de Virología, por los aislamientos virales. J. A. Usme-Ciro fue beneficiario del Programa de Doctorados Nacionales de Colciencias.
Los autores manifiestan que no tienen conflicto de intereses con respecto al manuscrito.
Este estudio fue financiado por la Dirección de Redes en Salud Pública del Instituto Nacional de Salud, Bogotá, D.C., Colombia.
Correspondencia:
Andrés Páez, Laboratorio de Virología, Instituto Nacional de Salud, Avenida Calle 26 N° 51-20, Bogotá, D.C., Colombia
Teléfono: (571) 220 7700, extensión 1405
apaezm@ins.gov.co y apaezm@ins.gov.co
1. Powers AM. Chikungunya. Clin Lab Med. 2010;30:209-19. http://dx.doi.org/10.1016/j.cll.2009.10.003 [ Links ]
2. Knipe D, Howley P. Field´s Virology. Sixth edition. Philadelphia: Lippincott Williams & Wilkins; 2013. p. 651-86. [ Links ]
3. Ross RW. The Newala epidemic. III. The virus: Isolation, pathogenic properties and relationship to the epidemic. J Hyg (Lond). 1956;54:177-91. [ Links ]
4. Dupuis-Maguiraga L, Noret M, Brun S, Le Grand R, Gras G, Roques P. Chikungunya disease: Infection-associated markers from the acute to the chronic phase of arbovirus-induced arthralgia. PLoS Negl Trop Dis. 2012;6:e1446. http://dx.doi.org/10.1371/journal.pntd.0001446 [ Links ]
5. Rougeron V, Sam IC, Caron M, Nkoghe D, Leroy E, Roques P. Chikungunya, a paradigm of neglected tropical disease that emerged to be a new health global risk. J Clin Virol. 2015;64:144-52. http://dx.doi.org/10.1016/j.jcv.2014.08.032 [ Links ]
6. Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. Chikungunya: A re-emerging virus. Lancet. 2012;379:662-71. http://dx.doi.org/10.1016/s0140-6736(11)60281-x [ Links ]
7. Weaver SC. Arrival of chikungunya virus in the new world: Prospects for spread and impact on public health. PLoS Negl Trop Dis. 2014;8:e2921. http://dx.doi.org/10.1371/journal.pntd.0002921 [ Links ]
8. Mowatt L, Jackson ST. Chikungunya in the Caribbean: An epidemic in the making. Infect Dis Ther. 2014;3:63-8. http://dx.doi.org/10.1007/s40121-014-0043-9 [ Links ]
9. Morrison TE. Reemergence of chikungunya virus. J Virol. 2014;88:11644-7. http://dx.doi.org/10.1128/jvi.01432-14 [ Links ]
10. Instituto Nacional de Salud. Boletín epidemiológico semanal. Semana epidemiológica número 25 de 2015. Fecha de consulta: 15 de julio de 2015. Disponible en: http://www.ins.gov.co/boletin-epidemiologico/Paginas/default.aspx [ Links ]
11. Lo Presti A, Lai A, Cella E, Zehender G, Ciccozzi M. Chikungunya virus, epidemiology, clinics and phylogenesis: A review. Asian Pac J Trop Med. 2014;7:925-32. http://dx.doi.org/10.1016/s1995-7645(14)60164-4 [ Links ]
12. Powers AM, Brault AC, Tesh RB, Weaver SC. Re-emergence of Chikungunya and O´nyong-nyong viruses: Evidence for distinct geographical lineages and distant evolutionary relationships. J Gen Virol. 2000;81:471-9. http://dx.doi.org/10.1099/0022-1317-81-2-471 [ Links ]
13. Tsetsarkin KA, Chen R, Sherman MB, Weaver SC. Chikungunya virus: Evolution and genetic determinants of emergence. Curr Opin Virol. 2011;1:310-7. http://dx.doi.org/10.1016/j.coviro.2011.07.004 [ Links ]
14. Volk SM, Chen R, Tsetsarkin KA, Adams AP, García TI, Sall AA, et al . Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J Virol. 2010;84:6497-504. http://dx.doi.org/10.1128/jvi.01603-09 [ Links ]
15. Lo Presti A, Ciccozzi M, Cella E, Lai A, Simonetti FR, Galli M, et al . Origin, evolution, and phylogeography of recent epidemic CHIKV strains. Infect Genet Evol. 2012;12:392-8. http://dx.doi.org/10.1016/j.meegid.2011.12.015 [ Links ]
16. Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. Chikungunya in the Americas. Lancet. 2014;383:514. http://dx.doi.org/10.1016/s0140-6736(14)60185-9 [ Links ]
17. Nunes MR, Faria NR, de Vasconcelos JM, Golding N, Kraemer MU, de Oliveira LF, et al . Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015;13:102. http://dx.doi.org/10.1186/s12916-015-0348-x [ Links ]
18. Lanciotti RS, Kosoy OL, Laven JJ, Panella AJ, Vélez JO, Lambert AJ, et al . Chikungunya virus in US travelers returning from India, 2006. Emerg Infect Dis. 2007;13:764-7. http://dx.doi.org/10.3201/eid1305.070015 [ Links ]
19. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947-8. http://dx.doi.org/10.1093/bioinformatics/btm404 [ Links ]
20. Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9. United States2012. p. 772. http://dx.doi.org/10.1038/nmeth.2109 [ Links ]
21. Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8): 754-5. [ Links ]
22. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725-9. http://dx.doi.org/10. 1093/molbev/mst197 [ Links ]
23. Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3): 585-95. [ Links ]
24. Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693-709. [ Links ]
25. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451-2. http://dx.doi.org/10. 1093/bioinformatics/btp187 [ Links ]
26. Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531-3. http://dx.doi.org/10.1093/bioinformatics/bti320 [ Links ]
27. Instituto Nacional de Salud. Transmisión autóctona de Chikungunya en Colombia. IQEN Informe Quincenal Epidemiológico Nacional. 2014;19:311-38. Fecha de consulta: 6 de julio de 2015. Disponible en: http://www.ins.gov.co/iqen/Paginas/default.aspx [ Links ]
28. Groot H. Estudio sobre virus transmitidos por artrópodos en Colombia. Revista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales. 1964;12:197-217. [ Links ]
29. Powers AM, Brault AC, Shirako Y, Strauss EG, Kang W, Strauss JH, et al . Evolutionary relationships and systematics of the Alphaviruses. J Virol. 2001;75:10118-31. http://dx.doi.org/10.1128/jvi.75.21.10118-10131.2001 [ Links ]
30. Mesa F, Cárdenas J, Villamil L. Las encefalitis equinas en la salud pública. Bogotá: UNAL; 2005. p. 1-124. [ Links ]
31. Instituto Nacional de Salud. Boletín epidemiológico semanal. Semana epidemiológica número 19 de 2015. Fecha de consulta: 15 de julio de 2015. Disponible en: http://www.ins.gov.co/boletin-epidemiologico/Paginas/default.aspx. [ Links ]
32. Instituto Nacional de Salud. Boletín epidemiológico semanal. 2014 - 2015. Fecha de consulta: 15 de julio de 2015. Disponible en: http://www.ins.gov.co/boletin-epidemiologico/Paginas/default.aspx. [ Links ]
33. Weaver SC, Rico-Hesse R, Scott TW. Genetic diversity and slow rates of evolution in New World alphaviruses. Curr Top Microbiol Immunol. 1992;176:99-117. [ Links ]
34. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007;3:e201. http://dx.doi.org/10.1371/journal.ppat.0030201 [ Links ]
35. Weaver SC, Brault AC, Kang W, Holland JJ. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J Virol. 1999;73:4316-26. [ Links ]
36. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, et al . Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3:e263. http://dx.doi.org/10.1371/journal.pmed.0030263 [ Links ]
37. Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M, et al . Two Chikungunya isolates from the outbreak of La Réunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus . PLoS One. 2007;2:e1168. http://dx.doi.org/10.1371/journal.pone.0001168 [ Links ]

