











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkCerebrovascular disease (CVD) represents a critical public health problem and it is considered the first cause of mental and physical disabilities around the world 1,2. Ischemic stroke is a CVD, which mainly generates a focal ischemia by the occlusion of the middle cerebral artery characterized by a core and a penumbra zone 3. In the ischemic core, cellular necrotic events occur that make it impossible to rescue cells after the insult; however, the apoptotic cell death caused by nutrient deprivation, the oxidative stress, and the excitotoxicity occurring in the penumbra zone represent a key target for therapeutic intervention in cerebral ischemia 4,5.
During cerebral infarction, various affected molecular actors are involved in neurotransmission, cell adhesion, and cytoskeletal remodelling, among others 6. CDK5 is a cyclin dependent kinase implied in neurodevelopment and neurotransmission, but its over-activation is associated with degeneration. Given that an increased cleavage of p35 (activator) to p25 generates an abnormal production of the CDK5/p25 complex with a sustained activation of the enzyme, an unbalancing phosphorylation of tau and of several substrates related to the loss of plasticity and neurovascular unit, as well as dementia 6-9, it has been suggested that the prevention of a sustained activity of CDK5 is neuroprotective 10-12.
In this context, it is necessary to gain a deeper understanding of CDK5 RNAi-based therapy for future translational studies in humans, which are in great clinical demand and have a significant social impact 13. In this study, we analyzed the role of CDK5miR on neurogenesis and gliogenesis after an ischemic stroke.
Materials and methods
Animal model
Procedures in animals were performed according to the ARRIVE guidelines, the “Guide for the care and use of laboratory animals”, 8th edition, published by the National Institutes of Health (NIH), and the Colombian standards (Law 84/1989 and Resolution 8430/1993), taking special care to minimize suffering and reduce their number. The study was approved by the Ethics Committee for Animal Experimentation of the University of Antioquia, Medellín, Colombia.
Animals were obtained from a specific pathogen-free colony of the vivarium at the Sede de Investigación Universitaria (SIU), University of Antioquia, Medellín, Colombia. Rats were maintained in groups of five with food and water ad libitum and on a 12:12-hour dark:light cycle. We used 40 eight-week-old male Wistar rats (Rattus novergicus albinus) of average size and an average weight of 350 g. Ten rats per experimental group were used for neurological scoring. In addition, we used 5 per experimental group for the histologic assessments.
Surgical procedure
We performed a transient middle cerebral artery occlusion (t-MCAO) and reperfusion as described previously 12,14. Initially, animals were anaesthetized with a mixture of ketamine via IP (60 mg/kg; Holliday Scott S.A. Int. Neyer, Buenos Aires, Argentina), xylazine (5 mg/kg; Synthesis Ltda. & Cia., S.C.A, Bogotá, Colombia), subcutaneous atropine (100 mg/kg; Erma S.A., Bogotá, Colombia); they also received an inhalational mixture of 2% to 4% of isoflurane (Baxter, Deerfield, IL, USA) and 96% oxygen using an inhalation anaesthesia machine (VMC™ Table top gas anaesthesia Instrument).
The right common carotid artery (CCA) was exposed and carefully isolated from the vagus nerve (VN); the right external and internal carotid arteries (ECA and ICA) were exposed and isolated. ECA ascending pharyngeal artery, superior thyroid and occipital branches were cauterized by electrocoagulation (AARON Bipolar Cautery™, Albany, NY, USA). The ICA artery pterygopalatine first branch was isolated. The distal portion of the ECA was tied with a 6/0 nylon and then cut. A 4-0 monofilament nylon (Corpaul, Bogotá, Colombia), previously rounded at one end by flaming and coated with poly-L-lysine solution (0.1% w/v in deionized water; Sigma, St Louis, MO, USA), and inserted 17-19 mm from the ECA to the ICA to occlude the MCA and its origin. Then, we verified that the monofilament was not inside the pterygopalatine artery. The incision was sutured leaving a bulge of 1 cm. After 60 minutes, the nylon was removed allowing reperfusion and the wound was closed.
Sham animals were subjected to the same procedures, but the filament was not introduced. Half of the animals were euthanized 15 days after reperfusion and the other half, 30 days after for which all animals were anesthetized with xylazine 10 mg/kg + ketamine 90 mg/kg and immediately perfused with 4% paraformaldehyde (0.1 M phosphate buffer [PBS], pH 7.4). Brains were carefully removed and post-fixed with 4% paraformaldehyde at 4°C for 48 hours, then preserved by a sucrose gradient (7%, 25%, 30%) at 4°C; they were then placed in a cryopreservation solution at -20°C. Coronal sections were sectioned at 50 μm with a vibrating blade microtome (VT1000S™, Leica Microsystems, Nussloch, Germany).
Short hairpin RNAmiR delivery
During the t-MCA occlusion, at 30 minutes, we performed a right hippocampal stereotaxic injection with anteroposterior (AP) coordinates of -2.56 mm, mediolateral (ML) 0.8 mm and dorsoventral (DV) 4.1 mm. We used a 10 μL Hamilton syringe for injection at a rate of 0.2 μL/min of a solution containing adeno-associated viral (AAV) particles obtained from the Davidson Laboratory (University of Iowa Viral Vector Core) with the RNAi sequences for silencing CDK5 (short hairpin CDK5miR) and a control scrambled RNA sequence (shSCRmiR) 12,15-18 for treating ischemic and control animals, respectively. Five minutes elapsed after the infusion before withdrawing the syringe. We injected a volume of 2.5 μl of AAV2.5.shSCRmiR.GFP or AAV2.5.shCDK5miR.GFP as previously described 12,18. After the injection, we proceeded to suture the animals keeping them in observation until their awakening (figure 1 A,B).
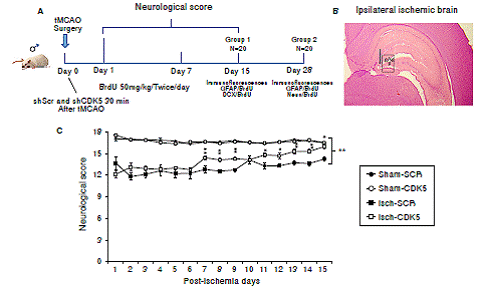
Figure 1 Neurological recovery induced by CDK5 silencing in ischemic rats. A) Experimental design to evaluate the effect of CDK5 RNAi on neural population and neurological performance after an ischemic insult. B) Ipsilateral injection site at stereotaxic coordinates AP -2.56, ML 0.8, and DV 4.1. and evaluated hippocampal regions (g: granular; sg: subgranular; p: polimorphic zone. C) Neurological analysis of CDK5 silencing in ischemic rats. The error bars indicate the SEM. All data are presented as mean ± SEM. Sham-SCR: Control group treated with shSCRmiR; Sham-CDK5: Control group treated with shCDK5miR; Isch-SCR: Ischemic group treated with SCRmiR; Isch- CDK5: Ischemic group treated with shCDK5miR. n=10, p<0.05 = *; p<0.001 = **.
Recovery and neurological evaluation
Animals were evaluated from day 1 to 15 after the t-MCAO (N=10). Neurological evaluation was scored on an 18-point scale. The evaluation was divided into six independent tests scored from 0 to 3 to determine the following parameters:
The highest possible score in the tests was 18, which indicated there were no neurological deficits, while the lowest was 3 for the most severe impairment. Neurological evaluation was performed every day and in the same order in all animals. To validate and reduce bias, a double-blind procedure was performed recording videos for every animal during the test.
Insertion of 5-Bromo-2-deuxyuridine
For the BrdU (Catalog Number B5002, Sigma- Aldrich) insertion, we injected rats via IP twice a day during seven days after ischemia at 50 mg/kg (0.1 M phosphate buffer [PBS], pH 7.4).
Immunohistochemistry
Brain sections were treated with methanol and hydrogen peroxide (H2O2) in 0.1M PBS (pH 7.4) to inhibit endogenous peroxidase activity for 20 minutes while free-floating with moderate shaking. Subsequently, we washed them three times with 0.3% (v/v) Triton X-100™ (Sigma-Aldrich) in 0.1 M PBS to increase tissue permeability and we then incubated for 30 minutes with Triton™ 0.5% (v / v) X-100 in 0.1 M PBS. Then, we backwashed with 0.3% (v/v) Triton X-100™ in 0.1 M PBS for three times.
For BrdU, we denaturalized brain DNA sections with HCl 2N for 20 minutes at 37°C, we washed three times with 0.3% (v/v) Triton X-100™ in 0.1 M PBS, then incubated with citrate buffer (Sodium citrate 10Mm, 0.05% tween 20, pH 6.0) for 10 minutes at room temperature and, finally, we washed three times with 0.3% (v/v) TritonX-100™ in 0.1 M PBS. Nonspecific binding sites were blocked by preincubation with BSA (1%) and 0.3% (v/v) Triton X-100™ in 0.1 M PBS for 1 hour.
Sections were incubated 24 hours while freefloating with moderate shaking and with the primary anti-body diluted in incubation solution (0.1 M PBS with 0.3% (v/v) Triton X-100™ and 0.3% BSA) at 4°C.
Anti-BrdU (1:100 mouse monoclonal B8434, Sigma- Aldrich) was used to detect bromodeoxyuridine (BrdU) specifically incorporated into the DNA. Next day, sections were washed 5 minutes with 0.1 M PBS three times and incubated with the secondary antibody (1:250 biotinylated mouse, Pierce) for 1 hour at room temperature and then washed 3 times with 0.1 M PBS, incubated with Avidin-Biotin complex (Pierce complex™; 1: 250 reagents A and B), and washed again three more times and developed with a chromogenic reaction diaminobenzidine (DBA) in 0.01% H2O2. For negative controls, we followed the same protocol for each experimental group without incubation with the respective primary antibody.
Sections were mounted on glass slides, dehydrated with increasing concentration of alcohol (70%, 96%, 100%) and cleared with xylene. Finally, the slides were sealed using Vecta-shield mounting medium (Vector Laboratories, Burlingame, CA, USA), and then analyzed through a light microscope (Nikon Eclipse E200™). Images were captured using a Nikon digital camera (Sight DS-L1™). Quantification of immunoreactivity in the hippocampus was determined using a 10X objective. Photomicrographs were analyzed using the ImageJ software program (version 1.45, US National Institutes of Health, Bethesda, MD, USA). To quantify the relative intensity of immunostaining, we modified the images to a binary system and obtained integrated densities (relative units) substracting automatically the background for each image.
Immunofluorescence
Brain slices were initially heated (80°C) in 10mM Tris pH 6.0 to increase exposure of the antigen and then treated with ammonium chloride (NH4Cl) for 20 minutes to avoid autofluorescence. Three washes with 0.3% v/v Triton X-100™ in 0.1M PBS were made to increase tissue permeability. For BrdU, we denaturalized DNA brain sections with HCl 2N for 20 minutes at 37°C, washed them three times with 0.3% (v/v) Triton X-100™ in 0.1 M PBS, and then incubated them with citrate buffer (sodium citrate 10 Mm, 0.05% tween 20, pH 6.0) for 10 minutes at room temperature to finally wash them three times with 0.3% (v/v) Triton X-100™ in 0.1 M PBS. Nonspecific binding sites were blocked by preincubation with BSA (3%) and 0.5% (v/v) Triton X-100™ in 0.1 M PBS for 1 hour.
Sections were incubated overnight while freefloating with moderate shaking at 4°C with the respective primary anti-BrdU (mouse monoclonal 1: 100, Sigma) for the new cells produced, anti-DCX (polyclonal rabbit 1: 250, Abcam) for immature neurons, anti-GFAP (rabbit polyclonal 1: 500, Sigma) for astrocytic cells and anti-NeuN (polyclonal rabbit 1: 500, Millipore) for mature neuronal cells. After three washes, tissues were incubated with secondary antibodies: Rabbit Alexa Fluor 594™, and mouse Alexa Fluor 350™ (molecular probes, 1:1,000) for 90 minutes at room temperature. Finally, we washed with PBS and slides were coverslipped using Gel Mount™ (BioMeda, Hatfield, PA, USA) in dark conditions. For negative controls, we followed the same protocol for each experimental group without incubation with the respective primary antibodies. Tissues were observed with a motorized spinning disk confocal microscope (Disk Scan Unit) Olympus IX 81™ (Olympus, Latin America, Inc., Miami, FL, USA). Images were photographed at 10X and the analysis was performed by evaluating the fluorescence intensity (FI) using the Image Pro Plus (Media Cybernetics) software.
Statistical analysis
For the neurological analysis, we used ten (10) animals per group and for the histological analysis, five 5 per group. The data obtained from the neurological and tissue immunoreactivity evaluations were analyzed using a homogeneity of variance test, followed by ANOVA of repeated measurement and one-way ANOVA tests, and a Tukey’s post hoc test to define differences between groups. We used the SPSS Statistics™ software (20.0 version) and a p ≤0.05 significance. All experimental groups were processed in parallel for reducing variability. The data are presented as mean ± SEM.
Results
CDK5 reduction promoted neurological recovery in ischemic rats
The neurological assessment showed that the Isch-SCR animals had a lower score (12.8 and 14.3 points) at days 7 and 15 post-ischemia, respectively, compared with the sham groups with and without CDK5 RNAi treatment (16.5 points). Interestingly, the CDK5 RNAi-treated ischemic group presented a significantly higher score of 14.5 on day 7 and of 16 points on day 15, recovering neurological skills similar to those of the control animals (figure 1C; p<0.05 = *, p<0.001 = **, repeated-measures in ANOVA).
BrdU incorporation in the hippocampus was promoted by CDK5 RNAi treatment in ischemic rats
We found a significantly increased number of BrdU+ cells in the Isch-SCR group compared with the sham-SCR and sham-CDK5 controls (figure 2A a; p<0.05 = *, p<0.001 = **, repeatedmeasures in ANOVA and Tukey’s test). However, the CDK5 RNAi treatment induced a significantly greater incorporation of BrdU in ischemic animals compared with the Isch-SCR group without treatment (figure 2A a). Although there was no significant incorporation of BrdU in the polymorphic area of the Isch-SCR animals compared to controls (figure 2A b; p<0.05 = *, p<0.001 = **, repeated-measures in ANOVA and Tukey’s test), CDK5 silencing significantly increased BrdU+ cells compared to Isch-SCR and controls in the cerebral zone (figure 2A b). This effect of CDK5 RNAi on ischemic rats remained for a month in the granular/ subgranular and polymorphic zones (figure 2 B b). The Isch-group had a slight tendency or significantly increased BrdU+ cells in comparison with the control groups (figure 2 B a, b).
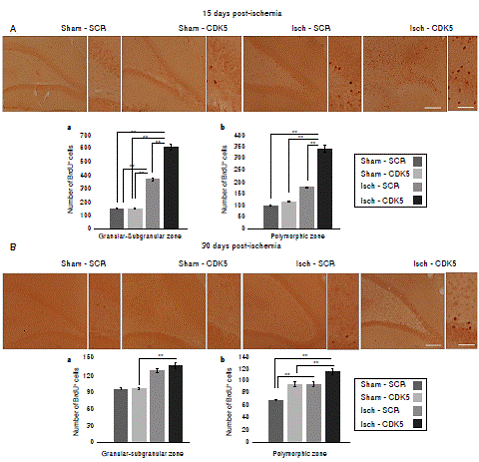
Figure 2 BrdU incorporation in the hippocampus of CDK5 RNAi-treated ischemic rats. A) Representative images at bregma -2.56 mm of BrdU immunoreactivity in the granular-subgranular (a) and polymorphic (b) zones at 15 days and B) 28 days post-ischemia in treated and untreated rats. The error bars indicate the SEM. n= 5, p<0.05 = *; p<0.001 = **. All data are presented as mean ± SEM. 10X. Scale bar=100 µm.
CDK5 RNAi exerted neuroprotection associated with the induction of immature neurons and down-regulation of the astroglial population in ischemic rats
Our findings showed that CDK5 RNAi treatment induced proliferation of immature neurons in the granular/subgranular zones. This is supported by the significant increase of BrdU+ and DCX+ cells in comparison with ischemic and control groups at 15 days post-ischemia (figure 3A, C; p<0.05 = *, p<0.001 = **, repeated-measures in ANOVA and Tukey’s test). Although the sham-CDK5 and Isch- SCR groups presented a significant increase of BrdU in the granular/subgranular zones, this was not confirmed by the variations in the amount of DCX+ or GFAP+ cells (figure 3A, C). On the other side, the polymorphic zone presented variability between groups and treatments (figure 3B, c; p < 0.05 = *, p<0.001 = **, repeated-measures in ANOVA and Tukey’s test), however, there was a significant increase of BrdU+ and GFAP+ cells in the ischemic-SCR group 15 days post-ischemia, which was significantly down-regulated by the CDK5 RNAi treatment in ischemic rats (figure 3B, C). The green fluorescent protein (GFP) was used to confirm the expression of AAV2.5.shSCRmiR. GFP and AAV2.5.shCDK5miR.GFP in each group, respectively.
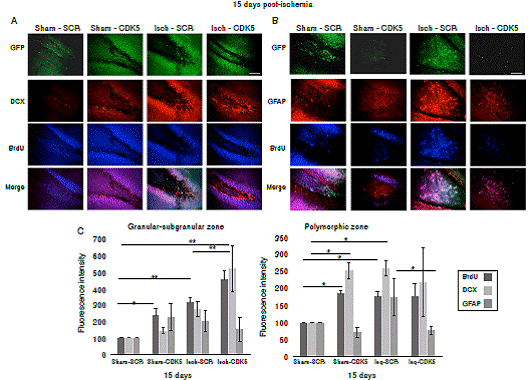
Figure 3 CDK5 RNAi induced immature neurons and down-regulation of astroglial population in ischemic rats. A) BrdU and DCX immunoreactivities in the granular-subgranular and polymorphic zones at 15 days post-ischemia in treated and untreated rats (A, C). BrdU and GFAP in the granular-subgranular and polymorphic zones (B, C) at 15 days post-ischemia in treated and untreated rats. The error bars indicate the SEM. n=5, p<0.05 = *; p<0.001 = **. All data are presented as mean ± SEM. Representative images at bregma -2.56 mm. 10X. Scale bar = 100 µm.
CDK5 RNAi preserved the down-regulation of astrogliosis at one month post-ischemia
Interestingly, when we evaluated the neural population at 30 days post-ischemia, we did not find changes in the mature neuron immunoreactivity neither in the granular/subgranular nor in the polymorphic zones in the untreated and treated rats, because there was a significant increase in the BrdU+ cells but without changes in the NeuN immunoreactivity (figure 4A, B; p<0.05 = *, p<0.001 = **, repeated measures in ANOVA and Tukey’s test). The GFAP+ population did not present changes in the granular/subgranular zone either (figure 4A, B), but stayed increased in the polymorphic zone at one-month post-ischemia in untreated rats, which was reversed by the CDK5 RNAi treatment as in the control groups (figure 4A, C, p<0.05 = *, p<0.001 = **, repeated measures in ANOVA and Tukey’s test).
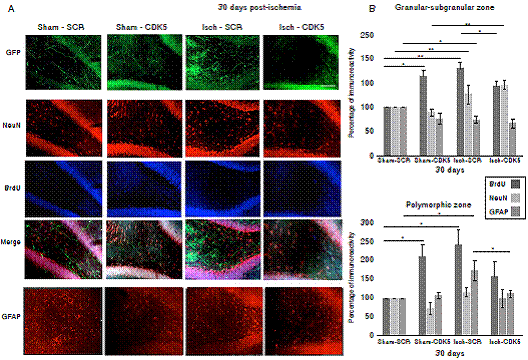
Figure 4 CDK5 RNAi prevents astrogliosis at one-month post-ischemia. A) NeuN, BrdU and GFAP immunoreactivities in the granular-subgranular (A, B) and polymorphic (A, C) zones at 30 days post-ischemia in treated and untreated rats. The error bars indicate the SEM. All data are presented as mean+/-SEM. Representative images at bregma -2.56 mm. n=5, p<0.05 = *; p<0.001 =**. 10X. Scale bar = 100 µm
Discussion
Our data suggest that CDK5 RNAi exerts part of its neuroprotection effect by controlling immature neuron proliferation and reducing astroglial cells, which reflects in the recovery of the neurological function after an ischemic stroke.
CDK5 is an effective form of protection against an ischemic event 7, preventing the excitotoxic spreading of glutamate by the reduction of calpain and CDK5 activation 12,20.
Complementarily, the present study suggests that the silencing of CDK5 in the hippocampus has positive consequences on the behaviour associated with the motor cortex (M1 and M2) and the sensory area (S1) 21-24 as the exploratory behaviour integrate the interaction of those areas in the spatial activity performed by the hippocampus 24-26, whisker movement through the motor cortex by M1 stimulation, which strongly innervates reticular nuclei containing cholinergic premotor neurons of the vibrissae and, in parallel, the sensory cortex S1, which innervates the trigeminal interpolar nuclei of brainstem and plays a role in the retraction of the whiskers and, in turn, in the sensitivity of the paretic side of the animals 25,26.
In addition, our recent observations support the influence of hippocampal CDK5 down-regulation on the neurological tasks and the performance of learning and memory functions, where the CDK5 RNAi avoids cell death on cingulate cortex and thalamus at one and four months post-ischemia (unpublished data); these cerebral areas are involved in motivation and motor control behaviours 27,28. Its mode of action also involves the BDNF/ TRKB pathway and the restoration of long-term potentiation (LTP) dysfunction after stroke in the hippocampus at one-month post-ischemia 12,18.
On the other hand, BrdU has been widely used for labelling new-born cells and proliferation of different cell types. It has been shown that in cerebral ischemia the inclusion of BrdU is a marker of proliferation or neurogenesis in brain niches 29, which was confirmed in our observations. It is well known that CDK5 plays an important role in post-mitotic neurons, where its inhibition prevents immature neuron migration and progression to a mature stage in physiological conditions 30. But the cellular and molecular fate depend on the contexts and homeostatic balance between normal and pathological conditions 12,18.
In our findings, CDK5 silencing in ischemia increased the BrdU/DCX+ cells, maybe because its reduction could regulate the re-entry in the cell cycle for a correct balance between Cdk6 and Cdk4 and the E2F1/CDK5 nuclear complex 31-36, or by the prevention of stem cell differentiation as CDK5 activity promotes neural differentiation through phosphorylation of the p27 protein in its 187-threonine residue 37. Moreover, CDK5 may phosphorylate to DCX, an immature neurons marker, in the 297-serine residue during migration 38. Therefore, our data would suggest that CDK5 RNAi induces DCX in an early stage, but it does not facilitate its migration and maturation since NeuN labelling was unmodified.
It has also been described that CDK5 RNAi induces the production of BDNF 14,18, a neurotrophic factor highly involved in neurogenesis, neuronal plasticity, and protection preventing proinflammatory response 39,40. The neuroprotective action of CDK5 RNAi increased activation of a typical plasticity pathway, the BDNF/ERK/CREB, and its action is dependent on the TRKB receptor activity 18,39,40,41. Also, ERK signalling has been widely described as a mediator of neuronal plasticity and survival in physiological and neurodegenerative contexts 41.
Those sequential cascades of CDK5 RNAi-induced survival and plasticity events could also be associated with the control of astrocytes reactivity. GFAP is a marker of astrogliosis on brain disease, and its proliferation is related to the adverse progression of brain impairment, pro-inflammatory response, lymphocytic infiltration, cytokines release, and neurovascular uncoupling 1,3,6. Interestingly, CDK5 knock-down could prevent the astrogliosis in the short and long time post-injury, thus supporting the potential translation of CDK5 RNAibased therapy 7,12,18,42,43.
On the other hand, when CDK5 levels are decreased in astroglial cells, BDNF is released from those cells protecting neurons 42 and endothelial cells in co-cultures (unpublished data), and recovering motor and neurological functions in ischemic rats 43.
Moreover, our data suggest that CDK5 RNAi induces proliferation of other types of cells besides neurons, which were not detected by our current approach. However, in agreement with our previous studies we have found that silencing of CDK5 induces an increase of endothelial cell marker PECAM143 and the adhesion cell protein p120ctn in the membrane of endothelial cells (unpublished data), suggesting that endothelial cells may be the other potential cell population involved in the CDK5 RNAi-induced neuroprotection. This is supported by the recovery of the neurovascular unit as we have suggested previously 6, and is reflected in the physiological recovery of the neurological performance 14,18,42,43.
In conclusion, our data suggest CDK5 reduction induces proliferation of immature neurons and blocks astrogliosis in the long-term after ischemic injury improving neurological function, which, together with previous scientific evidence, supports the potential translation of CDK5 RNAi-based gene therapy in acute and chronic neurodegeneration.

