











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkLas protocaderinas, proteínas transmembrana de adhesión cuya estabilidad mecánica depende del calcio, constituyen un subgrupo de la superfamilia de las caderinas. Se han reconocido más de 70 genes de protocaderinas 1,2. En el sistema nervioso central están ampliamente involucradas en el proceso de neurulación anterior y en la morfogénesis.
Son numerosas las enfermedades cerebrales de etiología metabólica y genética que producen encefalopatía progresiva y pueden estar asociadas con la epilepsia. La encefalopatía ligada al cromosoma X por mutación del gen PCDH19 que codifica para la protocadherina 19, se caracteriza por epilepsia resistente, trastorno de espectro autista, retardo del desarrollo, síndrome hipotónico, microcefalia y convulsiones recurrentes. Las crisis epilépticas pueden desencadenarse por diversos estímulos, como fiebre, vacunación, cirugías o infecciones adquiridas.
El PCDH19 se localiza en el cromosoma Xq22.3 y su función se expresa en el desarrollo cerebral, específicamente en la zona subventricular intermedia, subplato, capas II,IV,V y VI de la corteza cerebral, en el hipocampo y el subículo 3,4. La proteína protocadherina 19 podría regular la formación de conexiones neuronales durante el desarrollo cerebral y remodelar las conexiones sinápticas durante la etapa posnatal temprana 5,6.
Reporte del caso
Se trata de una niña nacida de un segundo embarazo, con adaptación y antropometría adecuadas para la edad de gestación, pero con síndrome hipotónico. Su madre había tenido un aborto cuya causa no pudo establecerse. A los 10 meses de edad, aparecieron las primeras convulsiones de diferente tipo: focales, discognitivas, autonómicas y tónico-clónicas, algunas de las cuales fueron desencadenadas por la fiebre debida a una infección respiratoria aguda. La condición evolucionó a epilepsia resistente, con regresión del neurodesarrollo y del habla, agnosia visual y auditiva, y conversión autista. La evaluación neuropsicológica corroboró estos hallazgos y y se obtuvo un puntaje de 59, el cual indica moderada afectación, en la escala Bayley de desarrollo infantil.
La resonancia magnética (RM) cerebral simple practicada a los 10 meses, mostraba leucoencefalopatía parieto-occipital, e hiperintensidad del tracto tegmental central y de los núcleos delgado y cuneiforme (figura 1). En el electroencefalograma (EEG) se evidenció actividad epileptiforme focal témporo-parietal izquierda. Con los estudios metabólicos se descartó aminoacidopatía, aciduria orgánica, leucodistrofia metacromática y enfermedad del peroxisoma.
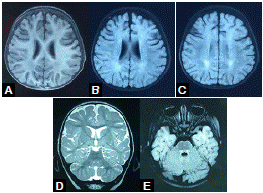
Figura 1 Resonancia magnética cerebral a los 10 meses de vida de la paciente. Se observa leucoencefalopatía parietooccipital e hiperintensidad del tracto tegmental central. A. Secuencia T1. B-C-E. Secuencia FLAIR. D. Secuencia T2
Dada la sospecha de síndrome epiléptico genético, se hizo una secuenciación completa del exoma y se confirmó la mutación del PCDH19 (c.142G>T/p.Glu48X). Para el control de las convulsiones, fue necesario utilizar hasta cinco medicamentos anticonvulsivos.
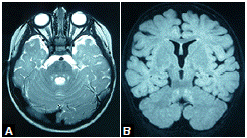
A partir de los 18 meses de vida, la niña no volvió a tener convulsiones, pero en la RM cerebral persistía la hiperintensidad del tracto tegmental central, aunque habían desaparecido las lesiones de la sustancia blanca parieto-occipital (figura 2). A los dos años de vida, recibía un solo medicamento anticonvulsivo y presentaba rasgos autistas, en tanto que en el electroencefalograma y la RM cerebral ya no aparecían las anomalías previamente descritas (no se muestran las imágenes).
Discusión
Las mutaciones en el gen PCDH19 causan epilepsia desde la primera infancia únicamente en el sexo femenino. Hasta hoy, se han encontrado alrededor de 100 mutaciones distintas en este gen, la mayoría de las cuales son esporádicas y, el 50 %, sin sentido (nonsense). Después del gen del canal de sodio alfa 1 (SCN1), el PCDH19 es el más relevante en la encefalopatía epiléptica 7,8. El gen SCN1A se relaciona con convulsiones febriles simples, epilepsia genética, convulsiones febriles plus y epilepsia mioclónica temprana de la infancia (síndrome de Dravet) 9,10. Hasta un 16 % de los pacientes que no presentan mutaciones en el SCN1A, las tienen en el PCDH1911,12.
En los casos reportados, las crisis ocurren principalmente al final del primer año de vida, con un rango de aparición de 6 a 36 meses de vida 13,14. Es usual encontrar diferentes tipos de crisis, siendo las más comunes las tónicoclónicas, las focales y aquellas en racimo. Con menor frecuencia, se pueden presentar otros tipos de crisis, como las mioclónicas, las parciales complejas y las ausencias atípicas 15,16. El estatus epiléptico se ha descrito en 30 % de los pacientes. En el electroencefalograma son usuales las anormalidades frontotemporales. Las crisis se acompañan de fiebre en 54 a 100 % de los casos y en un porcentaje considerable de las pacientes dejan de presentarse al llegar a la adolescencia 17,18.
El fenotipo de las pacientes afectadas por mutaciones en el gen PCDH19 es heterogéneo. Puede incluir desde la epilepsia con escasas crisis, hasta la encefalopatía epiléptica con deterioro cognitivo y rasgos autistas persistentes. El deterioro cognitivo ocurre en el 70 % de los casos, en el 42 % hay discapacidad intelectual leve, en el 54 %, discapacidad intelectual moderada, y en el 4 %, discapacidad intelectual grave. Hay rasgos autistas en 28 % de las pacientes 19,20. En este caso, se presentaban muchas de las características del fenotipo descrito por mutación del gen PCDH19: hipotonía, inicio de la epilepsia antes del año, retardo del desarrollo, deterioro del lenguaje y conversión autista, aunque no desarrolló encefalopatía epiléptica. Además, la frecuencia, la duración y la gravedad de las crisis convulsivas, comenzaron a disminuir con la administración de los medicamentos anticonvulsivos, de los cuales solo recibe uno actualmente.
Dado que en la RM cerebral se demostró leucoencefalopatía a los 10 meses de vida, se descartaron leucodistrofia metacromática, adrenoleucodistrofia ligada al cromosoma X (dada su inactivación), enfermedad de la orina con olor a jarabe de arce, hiperglucinemia no cetósica y aciduria orgánica.
Este es el primer reporte de caso de una paciente afectada por la mutación del gen PCDH19 asociada con leucoencefalopatía y tractopatía posterior reversible. Mediante las RM cerebrales se evidenció que las lesiones involucionaron progresivamente y, a los 24 meses de vida, las imágenes eran normales.
Conclusión
Este reporte de caso aporta información adicional para estudiar la mutación del gen PCDH19 en pacientes de sexo femenino con leucoencefalopatía de causa no metabólica. En este sentido, sería recomendable ofrecer la posibilidad de secuenciación del gen PCDH19 a las pacientes con epilepsia, retardo del desarrollo y lesión de los cuadrantes posteriores, una vez se descarte un error innato del metabolismo.

