











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkFabry disease is a rare X-linked disorder caused by alpha-galactosidase deficiency which leads to a progressive lysosomal glycosphingolipids accumulation, mainly globotriaosylceramide (Gb3) 1 in endothelial and smooth muscle cells of several organs vessels including the eye 1-4.
Clinical manifestations begin during infancy and adolescence and include intermittent acroparesthesias 5, limbs chronic pain 3,6, “Fabry crises” 1,5, angiokeratomas 5,6, hypo-anhidrosis 5, gastrointestinal and cardiac abnormalities 1,5,7,8, cerebrovascular attacks, and ocular abnormalities 2,9,10.
The disease occurs primarily in the cornea, conjunctiva, lens, and retina. Ophthalmological features of Fabry disease have been described not only in the anterior segment but also in the posterior pole of the eye. Anterior segment manifestations are aneurysmal vessels in the bulbar conjunctiva, cornea verticillata, and opacification of the lens. In the posterior segment, the most common features are vessel tortuosity and retinal vascular occlusions 1.
Early diagnosis is important to start a prompt enzyme replacement therapy 11,12 and to prevent new ocular complications and irreversible systemic organ damage, although enzyme replacement therapy does not reverse the established ocular manifestations of Fabry disease 2.
The purpose of this case series is to present the ocular findings of Fabry disease in a Colombian series of cases to improve diagnosis, remission, and early treatment of these patients, avoiding long-term complications that can increase morbidity and mortality.
Case series
Four male patients diagnosed with Fabry disease and a carrier woman, all from Bogotá, Colombia, were referred to a reference ophthalmologic center for evaluating their ophthalmological features. The diagnosis was previously made by geneticists considering the clinical findings by testing for deficient α-galactosidase A (α-GAL) enzyme activity and confirmed by DNA analysis of the α-GAL A gene. Four patients belonged to the same family: Three brothers and the mother identified as patients 1, 2, 3, and 4 (table 1).
Table 1 Demographic, systemic and ophthalmological findings in five Colombian patients with Fabry disease
| Case | Gender | Age | Family history of Fabry disease | Neuropathy | Organs affected besides eye at the moment of the ophthalmic evaluation | Ametropy | Ambliopy | Cornea verticillata | Lens opacity | Conjuctival vascular tortuosity | Retinal tortuous vessels | Lacrimal abnormality |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 14 | Yes | No | No | Mixed astigmatism | Yes | Yes | Yes | Yes | Yes | No |
| 2 | Male | 19 | Yes | Yes | No | Simple myopic astigmatism | Yes | Yes | Yes | Yes | Yes | No |
| 3 | Female | 38 | Yes | No | No | Simple myopic astigmatism | No | Yes | No | No | No | No |
| 4 | Male | 17 | Yes | Yes | No | Mixed astigmatism | Yes | Yes | Yes | Yes | Yes | No |
| 5 | Male | 43 | No | No | Kidney | Simple myopic astigmatism/ mixed astigmatism | Yes | Yes | Yes | Yes | Yes | Yes |
All of the patients (or relatives/guardians) signed the assent and/or informed consent. A complete clinical record with family and personal history, optometric-ophthalmological examination and ocular photographic register of anterior and posterior segments of the eye were completed (table 1).
Refractive defects were found in all patients as composed myopic astigmatism, three with mixed astigmatism and amblyopia. They all had punctate brown-cream corneal deposits with a central corneal radiated pattern, which in advanced cases generated a verticillata pattern (figure 1). Four patients had a wedge shape brown-cream deposit on the anterior internal lens capsule periphery, predominantly inferior (figure 2). All of them except for the carrier female presented with conjunctival and retinal vessels tortuosity (table 1).
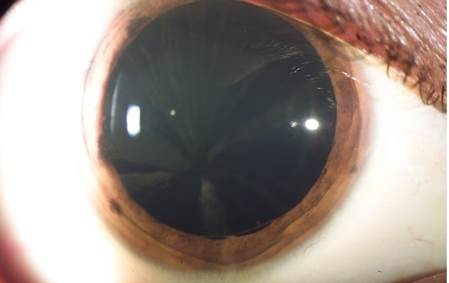
Figure 1 Cornea verticillata pattern characterized by brown-cream color deposits in a radiated pattern observed in patient 4

Figure 2 Lens opacity described as brown-cream deposits in the periphery of the anterior internal capsule of the lens observed in patient 2
Four of the five patients presented the ‘classic variant’ of Fabry disease and only one presented the ‘kidney variant’ with severe renal compromise requiring kidney transplantation (table 1). The starting time of enzyme replacement therapy was not specified in the clinical charts of the patients.
Discussion
Ophthalmological findings are common in Fabry disease compromising various ocular structures 1,2,8,9,13. In our case series, one of the most typical ocular findings was cornea verticillata pattern found in all of the patients; four out of five presented with lens opacity and with tortuosity and dilatation of retinal and conjunctival vessels. Although four of five presented with tortuous vessels, none of them developed retinal venous-arterial occlusions and none presented with optic disc edema.
Corneal epithelial-subepithelial deposits are the most important ocular manifestation 7 presenting in more than 95% of affected males and 88% of carrier females. The injury starts during infancy as a diffuse cloud, which progresses to cornea verticillata, characterized by radial, linear, symmetric, and bilateral cream to brown-gold color deposits 14.
Cornea verticillata is the most distinctive finding in females. All males in our case series presented cornea verticillata, as well as other ocular findings such as conjunctiva, lens, and retinal alterations. In contrast, the only woman among our patients presented cornea verticillata without the aforementioned findings. Cornea verticillata is rare in subjects without Fabry disease and, therefore, this finding has high diagnostic sensitivity and specificity, often related to disease severity 15.
Other causes of cornea verticillata are amiodarone prolonged therapy, aminoquinoline, atovaquone, subconjunctival gentamicin, gold salts, ibuprofen, indomethacin, naproxen, phenothiazine, tamoxifen, multiple myeloma, and environmental exposure to silica dust16. All pathologies and/ or drugs that can produce similar findings were discarded in the patients.
Vascular conjunctival abnormalities include vessel dilatation, tortuosity, and aneurysms, which are more common in the inferior bulbar conjunctiva 1,14. Vascular tortuosity is related with disease severity 17. Similarly, except for the female, all of our patients presented with conjunctival abnormalities.
In the literature, lens opacities are present in 70% of males and 35% of carrier females. Posterior opacities are the most common in both genders. Linear white opacities reveal the Gb3 epithelium deposits through the posterior suture lines known as Fabry cataracts, the only ocular diagnostic criteria 1. Anterior cataract, seen in 31% of males and 5% of females, is usually bilateral and wedge-shaped, with a variable density. Helix form, granular cream color deposits are found in the anterior capsule 4,14. According to this, lens opacities were observed in all four males of our case series. Figure 2 shows the linear-white opacities compatible with Fabry cataract.
The most common retinal finding is vascular tortuosity 1, found in 77% of males and 19% of carrier females. A narrowing of small vessels can cause a vascular-occlusive phenomenon secondary to Gb3 accumulation. Therefore, Fabry disease should be considered when a young patient presents a retinal or peripapillary vascular-occlusive event. Several patients have visual loss caused by retinal central artery-vein occlusion, ischemic optic neuropathy, optic atrophy, retinal ischemia or choroid perfusion defects 1. Although retinal vascular tortuosity was observed in four of our male patients, there were no vascular-occlusive events.
Increased blind-spot has been reported without an afferent disc defect or dyschromatopsia. Other findings include myopia, nerve fibers myelination, and disc edema 14,18. None of our patients had optic nerve alterations. All of them presented with refractive errors consistent with astigmatism and three of them with myopic astigmatism.
Because most ocular findings do not compromise visual acuity, patients rarely consult ophthalmologists. Nevertheless, when a characteristic Fabry sign is observed, it should be suspected and referred for a complete screening, especially when the patient is young, as in such cases ophthalmologists may identify an early-stage disease before complications appear. Ocular signs can act as markers of the disease with diagnostic and prognostic implications. As the eye is an external organ easily investigated with minimally invasive technologies, it may be useful for monitoring the natural history of Fabry disease and its response to enzyme replacement therapy 17.
All the patients in this series were referred to our clinic with the diagnosis of Fabry disease for ophthalmological examination and ocular findings were present in all of them.
The clinical systemic presentation is heterogeneous and the signs and symptoms change with the age of the patient due to the slow progressiveness of the disease 17,19. Three of our patients were male siblings under 20 years of age who presented with acroparesthesia, neuropathic pain, joint and abdominal pain crisis, and dyshidrosis. They did not report cardiac, respiratory symptoms or stroke, probably because such symptoms often appear at a later age 19. Patient 5, the oldest in the case series (43 yearsold), had a renal variant, several of the symptoms already mentioned, such as pain attacks, and was under enzymatic treatment. Additionally, he was taking immunosuppressant medications for post-transplant management.
An exhaustive ophthalmological assessment offers an important opportunity for early diagnosis. Any patient with corneal opacity, cornea verticillata or Fabry cataract in association with retinal vessels tortuosity, conjunctival telangiectasia or lens opacities should have a detailed evaluation and appraisal of family history. If there is a high risk of Fabry disease, the patient should be referred to a geneticist for evaluation and genetic counseling. If it is confirmed, the patient can be treated and multidisciplinary management should be started before irreversible complications appear 19. The treatment does not depend only on the diagnosis; rather, since each patient is different, the treatment pathway should be adjusted and managed according to the individual.
Greater awareness of Fabry disease among optometrists and ophthalmologists could reduce diagnostic delays and decrease early mortality of these patients.

