











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkAmyotrophic lateral sclerosis is a neurodegenerative disease characterized by the progressive death of motor neurons, producing muscular paralysis 1,2. Its clinical presentation is variable and depends on the anatomical site of onset and the type of motor neuron affected 3-5. There is no cure for amyotrophic lateral sclerosis and most of the patients die of respiratory failure 3 to 5 years after onset of symptoms 1,6. Amyotrophic lateral sclerosis etiology is not completely understood but, like other neurodegenerative diseases, it is thought to be the result of a complex interaction between multiple cellular mechanisms and environmental and genetic factors 7-9.
The genes with the strongest association to the development of both familiar and sporadic amyotrophic lateral sclerosis are SOD1 and C9orf72 (1,4,10). SOD1 encodes the enzyme superoxide dismutase 1, responsible for the degradation of superoxide ions via reactions with zinc and copper ions 1,7,11, while C9orf72 encodes a protein involved in both nuclear membrane transport and as a guanine nucleotide exchange factor that activates GTPases 5,12,13.
G93A and D90A mutations in SOD1 cause oxidative stress and subsequent neuronal death 1,11,14. However, the most common genetic anomaly found in amyotrophic lateral sclerosis patients is a hexanucleotide sequence repetition or short tandem repeat (STR) in C9orf72 gene, which explains 23.5 % of familial cases worldwide and up to 60 % in Spain 1,3,15. This C9orf72 expansion causes a loss of the functional protein, progressively generating cellular death 5,12,13. Healthy individuals generally have a maximum of two repetitions 16, while a pathological form of the gene in amyotrophic lateral sclerosis patients typically has 20 or more hexanucleotide repetitions; however, the number of pathogenic repetitions varies depending on the population 17.
Amyotrophic lateral sclerosis incidence in Europe has been estimated at 2.8 cases per 100,000 people per year. While there is scarce data for Latin America, reports from some countries in the region suggest a range between 0.2 and 3.17 cases per 100,000 people per year. In Colombia, national reports propose its prevalence at 0.5 cases per 100,000 people 18, but in Antioquia (a state within Colombia), this prevalence is much higher, being reported as 4.9 cases per 100,000 people 19,20. Despite this, there are no published studies on genetics of amyotrophic lateral sclerosis in the country.
The population of Antioquia (Colombia) -due to historical processes of its foundation- may have up to 70% of an ancestral component of European origin; more precisely, from the Iberian peninsula 3,21,22. Additionally, Antioquia is considered a genetic isolate due to population bottlenecks and endogamy 23. Hence, this population and others with similar characteristics are recommended for the genetic analysis of complex diseases such as amyotrophic lateral sclerosis, since they tend to show lower etiologic heterogeneity 6,21. All of this could explain the relative high prevalence of this and other neurodegenerative diseases in Antioquia 19.
Knowing amyotrophic lateral sclerosis genetic traits provides advantages for the development of diagnostic and prognostic biomarkers with utility both in the clinical practice and in the development of therapeutic trials for this disease, so far incurable. To date there is no published genetic study of amyotrophic lateral sclerosis in Colombia. The purpose of this research was to evaluate the presence of D90A, G93A mutations in SOD1 gene, as well as the hexanucleotide repetition in C9orf72, in amyotrophic lateral sclerosis patients in Antioquia (Colombia).
Materials and methods
Population sample
Patients found in the Neuromuscular Disorders Service at Neuro Clínica IPS, Medellín, Antioquia, who fulfilled the Escorial and Awaji-Shima diagnostic criteria 24, were included in the study. Only patients from Antioquia, diagnosed in 2017 and whose ancestors had been born in Antioquia, were included. Informed consent was obtained from all participants. This study was approved by the medical ethics committee of the Universidad CES.
DNA extraction and genotyping
DNA was extracted from blood samples using Wizard Genomic DNA Purification Kit™ (Promega, USA), following manufacturer’s instructions.
For the genotyping of SOD1 variants, D90A and G93A (SNP tags rs80265967; rs121912438), positive and negative controls were used besides the samples to be evaluated. With the purpose of confirming the reproducibility of the genotypes obtained through the KASP-PCR methodology (performed by LGC Genomics Inc., USA), 10% of the data were repeated for blind evaluation. Conventional PCR was performed to genotype the C9orf72 STR, using the set of primers (FAM 5’-CAAGGAGGGAAACAACCGCAGCC-3’ and R 5’-GCAGGCACCGCAACCGCAG-3’) and the methodology described by DeJesús-Hernández, et al., (2011) 25. DNA amplification was confirmed through electrophoresis in 1% agarose gel stained with ethidium bromide.
Alleles were resolved through capillary electrophoresis using an ABI PRISM 3130 Genetic Analyzer™ (IdentiGEN Laboratory, Colombia).
Statistical analysis
Correlation between the amyotrophic lateral sclerosis status and allele variants was performed descriptively and mathematically using Fisher’s exact test 26. Population genetic parameters, such as allele and genotype frequencies as well as Hardy-Weinberg equilibrium, were also calculated using Genepop, version 4.2 (GENEPOP, RRID:SCR_009194) (27,28). The endogamy model was evaluated through a c2 test and the selection model through calculations for different selection coefficients. All of this was done considering that by having chosen individuals already diagnosed with amyotrophic lateral sclerosis, an artificial selection was exerted over the study sample.
Results
Out of a total of 63 cases with confirmed diagnosis of amyotrophic lateral sclerosis during that year, 34 patients, evaluated and followed during 2017, were included in the study. The sample size of this study represents 54% of amyotrophic lateral sclerosis cases diagnosed in 2017 in Antioquia, Colombia.
Clinical characteristics
Approximately 32% of the patients presented with established disease of more than 5 years of duration. No patient had other family members with amyotrophic lateral sclerosis, although 26% reported a family history of other neurodegenerative diseases. In 19% of the cases, the disease began before 35 years of age.
Table 1 shows the characteristics of the patients classified by age ranges of onset of symptoms. Earlier ages of onset of symptoms correlated with longer duration of the disease (coeficiency correlation=-0.37) (p=0.03). Categorization by age group was done to show the difference between the age at onset of amyotrophic lateral sclerosis in the population of Antioquia and the usual age described in other populations (figure 1).
Table 1 Clinical characteristics of 32 amyotrophic lateral sclerosis patients grouped by age
| Age of onset (years) | Number of patients | Spinal onset | Family history of NDD | Tobacco use | Exposure to substances* | Average disease duration (months) |
|---|---|---|---|---|---|---|
| 20-35 | 6 | 4 | 1 | 0 | 3 | 78 |
| 36-50 | 3 | 3 | 1 | 0 | 1 | 77 |
| 51-65 | 17 | 13 | 5 | 13 | 6 | 52 |
| 66-80 | 6 | 2 | 2 | 2 | 2 | 34 |
NDD: Neurodegenerative diseases
* Alcohol, cocaine derivatives, solvents and detergents
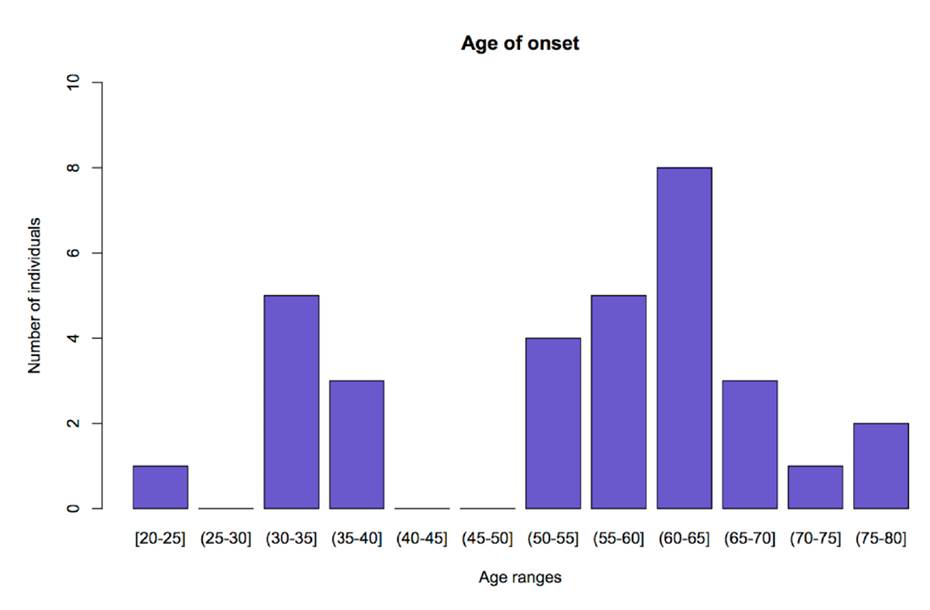
Figure 1 Age of onset and number of patients. The graph shows the distribution of patients according to the age at onset of ALS, presented as age ranges. The y-axis represents the number of patients and the x-axis represents age ranges. Figure done using R version 3.3.2 (R Project for Statistical Computing, RRID:SCR_001905).
Allele frequencies
Regarding SOD1 gene mutations G93A and D90A, all the evaluated patients presented the ancestral genotypes (GG and AA, respectively); thus, being monomorphic loci.
For the C9orf72 gene, 6 different alleles were found in 33 analyzed samples (table 2). The allele of 8 repeats of the hexanucleotide expansion GGGGCC was found in 84.85% (28 of 33 people) of the samples from patients with amyotrophic lateral sclerosis (figure 2).
Table 2 C9orf72 locus genotypes and number of repeats
| Genotypes | Observed | Expected |
|---|---|---|
| (CAGCAG)1/(CAGCAG)8 | 12 | 8.12 |
| (CAGCAG)8/(CAGCAG)8 | 16 | 14.55 |
| (CAGCAG)11/(CAGCAG)12 | 1 | 0.02 |
| (CAGCAG)13/(CAGCAG)13 | 3 | 0.32 |
| (CAGCAG)13/(CAGCAG)14 | 1 | 0.11 |

Figure 2 C9orf72 alleles. Capillary electrophoresis results, using an ABI PRISM genetic analyzer. Alleles are represented in the x-axis with relative fluorescence units (RFUs) shown in the y-axis. Alleles are identified with number of base pairs: Allele 1 (82. 41pb); Allele 2 (127.77pb).
The C9orf72 STR was not found to be in Hardy-Weinberg disequilibrium (p<0.01). SOD1 variants could not be evaluated with this parameters due to the finding of ancestral (no mutated) genotype in all the patients. Upon performing the calculations to evaluate the selection model against the genotypes not carrying allele of 8 repeats with different values of selection coefficient, it was found that when selection coefficient was 0.9 the genotype frequencies are similar to those observed in the data from the patients (table 3).
Table 3 Observed frequencies versus calculated model frequencies for each genotype
| Genotype | Observed frequency | Expected frequency with S=0.9 and a dominant model for allele 2 |
|---|---|---|
| (CAGCAG)1/(CAGCAG)8 | 0.3636 | 0.3195 |
| (CAGCAG)8/(CAGCAG)8 | 0.4848 | 0.5858 |
| (CAGCAG)11/(CAGCAG)12 | 0.0303 | 0.0001 |
| (CAGCAG)13/(CAGCAG)13 | 0.0909 | 0.0015 |
| (CAGCAG)13/(CAGCAG)14 | 0.0303 | 0.0004 |
Correlations between variables and the predominant genotype
Upon performing Fisher’s exact test, it was found: i) that there is a greater frequency of genotypes with 8 repeats allele in patients below 65 years old (X 2=6.72; p<0.01); ii) this genotype is more frequent in patients with spinal onset (X 2=8.90; p<0.01); and iii) that there is no difference between its distribution and the disease duration (X 2=0.22; p=0.64).
Discussion
This study evaluated the association between variants in SOD1 and C9orf72 and the occurrence of amyotrophic lateral sclerosis, finding the existence of an 8 repeat allele in the C9orf72 locus with a high frequency amongst amyotrophic lateral sclerosis cases in Antioquia, Colombia. All cases included in this study were sporadic amyotrophic lateral sclerosis diagnosed at young ages (< 65 years old) compared to the average of 65 years of age reported in other populations. None of the patients had a history of amyotrophic lateral sclerosis in their family. This is of remarkable importance since it is very likely that the genetic bases differ between familial and sporadic amyotrophic lateral sclerosis 29.
In this investigation we had 78.13% of patients whose age of disease onset was between 20 and 65 years of age (figure 1) and an average of 54 ± 15.23 years which differs from what has been reported in the literature (65 years in other populations) 30. The duration of the disease in the study group was higher than that reported in global literature, which can be explained by a larger proportion of young patients. As it has been described, an earlier age at onset of the symptoms relates to an increased life expectancy of the patients 31,32; this phenomenon was observed in this research.
Although environmental factors that explain the particularities of the group of patients must always be taken into account, it is possible that genetics has a crucial role. First, Antioquia is a genetic isolate that has experienced population bottlenecks and endogamy, which produce a reduced effect of etiologic heterogeneity and leads to an increase in the prevalence of hereditary or rare diseases 21,33. Additionally, the action of a phenomenon called genetic anticipation, which produces increased severity of a phenotype and/or reduces the onset time, can also be involved 34.
In general, this occurs when there is an expansion in DNA triplet repeats, as in the case of C9orf72 marker 17. Also, hexanucleotide repeat elongation with variants in other amyotrophic lateral sclerosis causative genes are associated with an earlier age of onset, suggesting that both mutations affect this feature 35. Allele with 8 CAGCAG repeats present in either homozygous or heterozygous condition, was found in 88.46% of the patients, suggesting a dominant allele effect on the amyotrophic lateral sclerosis phenotype. This same gene has been previously associated with amyotrophic lateral sclerosis. It has been proposed that the C9orf72 hexanucleotide repeat could be considered the cause of amyotrophic lateral sclerosis at around 20-25 repeats 25,36; however, in recent research studies it has been reported that the number of repeats considered pathologic varies according to the population 17,19,37.
Given the unusual reported genetic characteristics of the population of Antioquia, we do not rule out a phenotypic association between amyotrophic lateral sclerosis and a lower number of repeats than the one reported for the C9orf72 gene, or the possibility of this allele to be a risk factor for the disease. For this reason, it is necessary to complement the current study with a control population. This will allow to determine the distribution of alleles in the general population and confirm that the registered observations are not due to chance alone.
Despite that mutations G93A and D90A of the SOD1 gene were expected to be found, they were not present in any of the patients of the studied sample. Possible hypotheses that could explain this phenomenon are: i) that migration of these mutations to this population is scarce or non-existent; or ii) that the mutations are expressing a different phenotype due to additional genetic and environmental factors 3,4,22.
Given that the C9orf72 locus was, of the three studied in the diagnosed patients, the polymorphism related to amyotrophic lateral sclerosis in Antioquia, corresponding analyses were performed. It was found that the data do not fit the Hardy-Weinberg equilibrium model (table 3), which could be indicative of a direct association with amyotrophic lateral sclerosis 38,39. It is known that when an allele/genotype is associated to a phenotype this one can be augmented (risk) or diminished (protection) causing a deviation from the model, as shown in our research 40.
However, there are multiple causes for which a population may be out of Hardy-Weinberg equilibrium, such as selection, endogamy, genetic flux or genotyping errors 39,40. However, several of them can can be ruled out by the use of experimental controls and by the differences between observed and expected heterozygotes, which were not significant in our data. With these alleles, 21 possible combinations could have been generated, but only 5 were found in the study population. This suggests a selection effect that was done with the samples (previously diagnosed patients) reinforcing the possibility of an association between this loci and the occurrence of amyotrophic lateral sclerosis.
Furthermore, C9orf7 allele with 8 CAGCAG repeats was most commonly observed in patients below 65 years old than in their older counterparts (>65 years old), supporting the effect of genetic anticipation in this population.
In our sample, patients with this allele were more likely to have an spinal onset. This predominance of spinal over bulbar amyotrophic lateral sclerosis in our cohort can be explained by the small sample size, since in other international reports this variant has been associated with both spinal and bulbar amyotrophic lateral sclerosis 16. The number of hexanucleotide repeats in C9orf72 that has been considered pathologic or as the cause of amyotrophic lateral sclerosis is of 20-25 25,36.
In our study, the allele candidate for being related to the disease has only 8 repeats. While this variant has not been associated previously with amyotrophic lateral sclerosis cases, the relationship of the disease with the number of repeats is highly variable between different populations, lacking a definitive correlation between a specific number and the pathology 37. Analyses of common causal variants in different populations are important in pathophysiology investigation of amyotrophic lateral sclerosis in each population would have valuable implications 41,42.
This, along with the complete absence of G93A and D90A mutations of SOD1 in the patients, is an unexpected and relevant finding about amyotrophic lateral sclerosis genetics, its physiopathology and other neurodegenerative conditions. It is therefore necessary to confirm whether this genotype of 8 repeats in C9orf72 is pathologic in our population and to determine its importance as a diagnostic biomarker or as a risk factor for amyotrophic lateral sclerosis, as it is currently being done in other populations 43-45.
It is also relevant to define the influence of homozygosity or heterozygosity of these repetitions as a disease risk factor. Considering the significance of these findings for amyotrophic lateral sclerosis genetics and their clinical implications, it is necessary to carry out further studies to confirm the hypotheses we have presented.

