










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.11 no.2 Bogotá June 2006
REPLICACIÓN DEL HERPESVIRUS EQUINO Y SU ASOCIACIÓN CON LA PATOGÉNESIS MOLECULAR
Equine Herpesvirus Replication and It’s Association with Molecular Pathogenesis
JULIÁN RUIZ SÁENZ1,2, SILVIO URCUQUIINCHIMA1
1Grupo Inmunovirología – BIOGÉNESIS.2Corporación Ciencias Básicas Biomédicas, Universidad de Antioquia, Medellín, Colombia.
Presentado 2 de septiembre de 2005, aceptado 23 de enero de 2006, correcciones 27 de enero de 2006.
RESUMENEl herpesvirus equino (EHV) es uno de los patógenos virales de mayor importancia en la industria equina mundial, debido a las grandes pérdidas económicas que acarrea. La enfermedad comúnmente asociada con el EHV se denomina rinoneumonitis equina y se caracteriza por ser una infección primaria del tracto respiratorio superior, que progresa a través de la mucosa; puede causar aborto en los últimos meses de gestación, muerte perinatal de potros, mortinatos y mieloencefalitis. La infección productiva es seguida por un estado de latencia viral, etapa en la cual el animal no presenta ningún signo clínico de enfermedad y no hay replicación viral. Bajo una situación de estrés, el virus puede reactivarse y caballos infectados infectar a otros caballos sanos. En esta revisión se presenta de manera sintetizada, los principales hallazgos relacionados con la replicación viral y patogénesis molecular del EHV, relacionando además las proteínas implicadas en la regulación de la replicación del genoma, todas las glicoproteínas estructurales que han sido estudiadas hasta el momento y que son el eje central de investigación de distintos grupos en el mundo. Se discute además, la verdadera importancia de la dispersión directa célulacélula del virus, la formación de placas, el crecimiento in vitro y en algunos casos, la asociación con la patogénesis, bien sea en un modelo animal o en el hospedero natural.
Palabras clave: EHV, genoma, glicoproteína, ORF, replicación.
ABSTRACTEquine herpesvirus (EHV) is one of the most important viral pathogens in worldwide equine industry, due to the dramatic economic looses incurred by these microorganisms. The clinical disease, commonly called equine rhinopneumonitis, is characterized by initial infection of the upper respiratory tract that progresses through the mucosa; it can cause abortion in the last months of gestation, stillbirth, perinatal death of foals and myeloencephalitis. Productive infection is followed by a state of viral latency during which the animal presents no clinical symptoms and there is no viral replication. During a stress situation, the virus in the infected horses is reactivated and can infect healthy horses. This review presents in a synthetic manner, the main findings related with the study of viral replication and molecular pathogenesis of EHV; it also presents the main findings related to proteins involved in the regulation of viral genome replication, all the structural glycoproteins that have been studied to date and that constitute the central axis of different research centres around the world. In addition, it discusses the true importance of direct celltocell dispersion of the virus, the formation of plaques, in vitro growth, and in some cases association with the pathogenesis either in an animal model or in the natural host.
Key words: EHV, genome, glycoprotein, ORF, replication.
INTRODUCCIÓNLA ENFERMEDAD
La rinoneumonitis equina o aborto viral equino, es una de las enfermedades de distribución mundial más graves que pueden padecer los equinos; la enfermedad es causada por los herpesvirus equinos tipos 1 y 4 (EHV1 y EHV4), los cuales son potenciales factores de riesgo para caballos de todas las edades y categorías, especialmente para los potros (Crabb y Studdert, 1996). En Colombia en el año 2001, Ramírez et al. de la Universidad Nacional de Colombia trabajando con muestras tomadas de un feto abortado proveniente de un criadero de equinos, con historia de importación de animales, lograron por primera vez el aislamiento de un EHV (Ramirez et al., 2001). La infección por EHV1 y EHV4 se caracteriza por una infección primaria del tracto respiratorio, la cual puede variar de moderada a severa según el estado inmunológico del animal infectado. La infección por EHV1 particularmente, puede progresar a través de la mucosa respiratoria, dispersarse a otros sistemas orgánicos y causar aborto en los últimos meses de gestación, muerte perinatal de potros, e incluso sintomatología nerviosa diagnosticada como encefalomielitis. En la mayoría de los casos, la infección primaria por EHV1 y EHV4 está seguida por un estado de latencia viral en el ganglio trigémino, estado en el cual los caballos se encuentran aparentemente sanos. Luego de una situación de estrés (transporte, cambio de medio, preñez, etc.) puede ocurrir una reactivación y liberación del virus infectando a otros caballos (Allen, 2002).
Para el diagnóstico de los animales infectados existe una gran cantidad de técnicas; lo más apropiado es el aislamiento viral a partir de muestras clínicas, seguido de la confirmación serológica de la entidad. El virus se puede aislar de secreciones nasales, durante la infección productiva del tracto respiratorio, de muestras de hígado, pulmón, bazo o timo de los fetos abortados y de muestras de sangre tomadas al animal en la fase aguda de la enfermedad para aislamiento del virus en células blancas (Allen, 2004). Las estrategias de prevención existentes a escala mundial, se enfocan en especial al establecimiento y mantenimiento de programas de vacunación, que van desde la inmunización con antígenos virales hasta el uso de las vacunas de DNA, al mantenimiento de los caballos en grupos aislados y a impedir la entrada o diseminación del virus en los criaderos. Cuando las medidas de prevención no son suficientes, se hace necesario tomar otras medidas de control, tales como el diagnóstico oportuno, las medidas de aislamiento, cuarentena y desinfección, y el tratamiento terapéutico de los casos individuales (Ruiz, 2005).
ESTRUCTURA GENÓMICA DEL VIRUS
Se han descrito cinco herpesvirus distintos con capacidad de infectar caballos: EHV1, EHV4 y EHV3 (exantema coital equino), miembros de la subfamilia Alfaherpesvirinae, y EHV2 y EHV5 miembros de la Gammaherpesvirinae, ambos pertenecientes al género Varicelovirus. Existen algunos homólogos de EHV1, EHV2 y EHV3, denominados herpesvirus asino3 (AHV3), AHV2 y AHV1, los cuales han sido aislados de asnos (Crabb y Studdert, 1996; Wyrick, 2006). La partícula madura consta de una nucleocápside que contiene el genoma viral y las proteínas asociadas (core), el tegumento que rodea la nucleocápside y la envoltura viral la cual es derivada de la membrana de la célula del hospedero y en la cual están incorporadas las glicoproteínas de membrana codificadas por el virus (Roizman y Knipe, 2001; Fig. 1).

Figura 1. Microfotografía electrónica de un virión de EHV, indicando algunos de los componenteesenciales del virus (con permiso de Linda M. Stannard, University of Cape Town, http://web.uct.ac.za/depts/mmi/stannard/emimages.html).
El genoma de EHV1 y EHV4 está constituido por una molécula de DNA de cadena doble entremezclada (dsDNA), posee 150,2 y 145,6 Kb respectivamente (Telford et al., 1992; Telford et al., 1998) y la diferencia entre los dos virus se determinó mediante enzimas de restricción (Crabb y Studdert, 1996; Allen, 2002). Los genomas de EHV1 y 4 poseen 80 y 79 genes respectivamente. Ambos poseen 76 fases abiertas de lectura (ORFs) que dan origen a 76 proteínas. En los dos virus, algunos ORFs (64, 65 y 66 para EHV4 y 64, 65, 66 y 67 para EHV1) son expresados en doble copia, pero se desconoce cual es su función (Telford et al., 1992; Telford et al., 1998). El genoma está compuesto de una región única larga (UL) y una región corta (US); US está flanqueada por dos secuencias repetidas indirectas, una interna (IRS) y una terminal (TRS; Fig. 2). La presencia de secuencias repetidas en los dos extremos de la región US, permite que la región pueda invertirse, y dar origen a dos isómeros de DNA en cantidades equimolares (Crabb y Studdert, 1996).
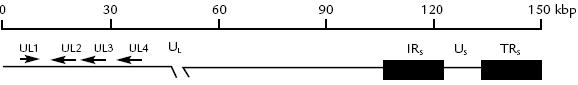
Figura 2. Organización del genoma de EHV1. Nótese la ubicación de las regiones única larga (UL) y una región corta (US), y secuencias repetidas indirectas interna (IRS) y terminal (TRS; adaptado de Zhao et al., 1995).
En los últimos años, las investigaciones se han enfocado a entender la función de los 76 genes o sus productos. Para el herpesvirus simplex (HSV), uno de los virus más estudiados de esta familia (Roizman y Pellet, 2001), se sabe que los genes son transcritos en una cascada ordenada, coordinada, regulada y secuencial; y dependiendo del momento en que son expresados durante la replicación son clasificados en tres clases: Inmediatos tempranos (IT), tempranos y tardíos. Los genes IT son sintetizados en primer lugar, utilizando la RNA polimerasa II de origen celular; y sus productos son requeridos para iniciar la trascripción de los mRNA tempranos. Subsecuentemente, las proteínas tempranas actúan regulando negativamente la trascripción de mRNAs IT, y están involucradas en la síntesis de DNA viral, usualmente como enzimas o como proteínas de unión a DNA. Las proteínas tardías son generalmente sintetizadas en las últimas fases del ciclo de replicación viral; muchas de ellas son componentes estructurales del virión (Crabb y Studdert, 1996; Roizman y Knipe, 2001).
Para EHV se han reportado cuatro proteínas IT de alto peso molecular, codificadas a partir de un solo trascripto de mRNA. Al igual que para otros alfaherpesvirus, se asume que la producción de proteínas IT es necesaria para la expresión de otros genes virales. Purewal et al. (1994), reportaron que el genoma de EHV1 y 4 codifica por una proteína homóloga a la proteína de tegumento VP16 de HSV, la cual está bien caracterizada como fuerte inductora de la expresión de genes IT; se ha descrito que la VP16 de EHV, posee la misma o similar función a la VP16 de HSV (Purewal et al., 1994; Thomas et al., 1999). Se han identificado 33 proteínas en el HSV, incluyendo siete de cápside, al menos ocho glicoproteínas de envoltura y numerosas proteínas del tegumento (Roizman y Knipe, 2001). La composición del genoma de EHV se estima similar al de HSV (Crabb y Studdert, 1996).
PATOGÉNESIS MOLECULAR DEL EHVProductos de EHV asociados con la patogénesis
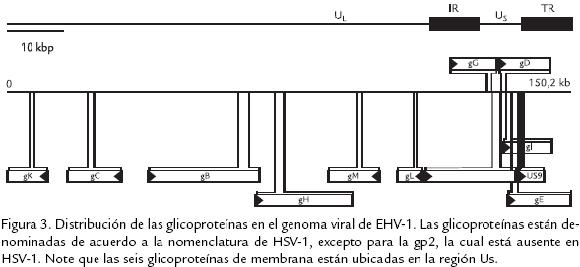
Las glicoproteínas de herpesvirus juegan un papel importante en el proceso de infección, mediando tanto la entrada del virión en la célula como la dispersión célulacélula. Gracias a la localización en la envoltura viral y en la superficie de células infectadas, son los principales blancos para la respuesta inmune del hospedero. Para EHV se han identificado y estudiado 10 glicoproteínas (g) homólogas a 10 de las 11 existentes en HSV. Estas son: gB, gC, gD, gE, gG, gH, gI, gK, gL y gM. La gJ de HSV tiene una contraparte en la región US, correspondiente al gen 71 de EHV. Además en EHV se han identificado otras glicoproteínas denominadas gp2 y gp21/22ª, las cuales no se han descrito en HSV, y gp10 la cual es el homólogo de una de las proteínas de tegumento, VP13/14 de HSV en el UL47 (Whittaker et al., 1991). Por lo tanto se considera que EHV posee por lo menos 13 glicoproteínas diferentes (Crabb y Studdert, 1996; Fig 3 ).
Smith et al. (1992) demostraron que el gen IT de EHV codifica un factor regulador con funciones comunes a las descritas para otros productos génicos en alfaherpesvirus; tales como la autorregulación negativa, la capacidad para activar ciertos promotores en ausencia de productos virales auxiliares, y la capacidad de activar en unión con otros productos virales auxiliares, un subconjunto de promotores virales. Kim et al. (2001) demostraron que la proteína IT (producto del gen IT) interactúa con EAP una proteína ribosomonucleolar, que la interacción no desplaza la unión de la proteína IT a su sitio afín en el DNA, y que durante las fases tardías de la infección, ambas proteínas colocalizan en el citoplasma de las células infectadas, como agregados firmes y densos.
Regiones no codificantes
El genoma de EHV contiene cinco regiones importantes las cuales al parecer no codifican por proteínas, y su función se ha especulado basándose en la secuencia de nucleótidos. La primera es de aproximadamente 1 kb de tamaño, está ubicada al extremo 5’ del genoma viral y se ha sugerido que está implicada en la replicación del DNA viral (Telford et al., 1992). La segunda de aproximadamente el mismo tamaño, está localizada entre el ORF 39 y el ORF 40, y contiene el origen de replicación (ORIL); la tercera es de aproximadamente 1,5 kb y está ubicada entre el ORF 62 y el ORF 63; la cuarta es de 2 kb y se encuentra entre los ORF 63 y 64 en la unión UL/IRS, y la quinta región no codificante tiene 2,5 kb y es la ubicada entre los ORF 64 y ORF 65 en IRS y TRS, y contiene un origen de replicación ORIS (Ibrahim et al., 2004).
Csellner et al. (1998) reportaron que la inserción del gen lacZ en la región entre el ORF 62 y el ORF 63 de la cepa HVS25A, sin ninguna delección en el genoma, no interfería con la expresión de los genes contiguos y el mutante resultante exhibía propiedades de crecimiento y replicación similares a las del tipo silvestre in vitro; y en ratones BALB/c infectados causó enfermedad respiratoria con sintomatología, histopatología y títulos virales similares a los de la causada por el tipo silvestre. En contraste, Ibrahim et al. (2004) al insertar en la misma región de la cepa Ab4p el gen de la proteína verde fluorescente, encontraron que el mutante resultante presentaba menor fenotipo de placa y tenía menor virulencia en hámsters y ratones. Estos resultados sugieren que la región no codificante entre los ORFs 62 y 63, juega un papel importante en el crecimiento vial de esta cepa. Purewal et al. (1998) demostraron que los productos del ORF63, el cual es expresado como un producto tardío, es transactivado por la proteína IT del ORF 64, y delecciones en este gen 63 indican que no es esencial para un eficiente crecimiento del EHV en cultivos celulares.
Genes inmediatos tempranos
Los genes IT solos, al ser los primeros genes trascritos durante la primera fase de la infección, codifican proteínas reguladoras claves de EHV. La trascripción de los IT ocurre en ausencia de la síntesis de proteínas virales y es inducida por ETIF (una proteína transinductora de EHV1). Las proteínas producto de los genes IT activan la expresión de promotores tempranos, autorregulan la expresión de sus propios promotores y son esenciales para la replicación, pues una delección de ambas copias de estos genes lleva a la producción de un virus incapaz de replicarse en líneas celulares complementarias (GarkoBuczynski et al., 1998). Actualmente, se conocen cinco proteínas reguladoras de EHV con capacidad para controlar la trascripción específica de genes: EICP22, EICP27, EICP0, IE y ETIF (Smith et al., 1995).
Genes tempranos y tardíos
El segundo grupo de genes que es trascrito durante la infección son los genes tempranos, los cuales codifican proteínas reguladoras adicionales (EICP0, EICP22, EICP27), al igual que proteínas implicadas en la replicación del genoma viral. La transcripción de genes tempranos requiere la presencia de proteínas IT y ocurre antes de la iniciación de la síntesis de DNA. Los genes tardíos, como se mencionó anteriormente, son los últimos trascritos y la mayoría codifican para proteínas estructurales. Estos genes tardíos pueden dividirse en dos subclases: genes permeables, cuya expresión comienza antes de la replicación del DNA viral pero que no alcanza su máxima expresión hasta después del inicio de la replicación del DNA; y genes verdaderos tardíos, cuya síntesis es totalmente independiente de la replicación viral. La proteína EICP0 es la única proteína reguladora que transactiva independientemente todas las clases de promotores virales (IT, tempranos y tardíos; Bowles et al., 1997).
Estudios realizados por Bowles et al. (2000) muestran que IT y EICP0 no funcionan en sinergismo, por el contrario, tienen una función antagónica, forman heterodímeros directamente, se unen al factor de trascripción celular TFIIB (Jang et al., 2001; Albrecht et al., 2003), y a la caja TATA celular. La interacción proteínaproteína y la posible competencia de ambas por los factores de transcripción pueden explicar, por lo menos en parte, la relación antagonista entre ellas (Kim et al., 2003). EICP27 (UL3) tiene la capacidad de aumentar la transactivación inducida por EICP0 de genes tempranos y tardíos, vía interacción proteínaproteína con las proteínas de la caja TATA celular (Zhao et al., 1995; Albrecht et al., 2004). Análisis mutacionales de EICP0 de EHV indican que distintas regiones son importantes para su función transactivadora (Bowles et al.,
2000). Sin embargo, no puede sobreregular la expresión de su propio promotor, debido a la presencia de un elemento regulador negativo (NER) dentro de su promotor (Osterrieder et al., 2001); su actividad está regulada por la unión de proteínas nucleares específicas a NER (Kim et al., 2004). Las proteínas IT se unen al sitio de iniciación de la trascripción de la secuencia promotora de la gK reprimiendo así la trascripción de este gen tardío verdadero (Kim et al., 1999). EICP22, la cual forma homodímeros de alto orden de complejidad durante la infección y se une con la proteína IT (Derbigny et al., 2000), para regular la expresión de genes de EHV (Kim et al., 1997).
La fosfoproteína de tegumento ETIF, la cual es el producto del ORF12, solo transactiva los promotores de genes IT, no así los de los genes tempranos o tardíos y estudios publicados por Kim y O’Callaghan (2001) indican que ETIF interactúa con factores de trascripción tales como TFIIB y dTAFl40. Esas interacciones son importantes en la formación de un complejo de preiniciación el cual lleva a la trascripción de genes IT. La interacción de EICP27 con IT permite el reclutamiento de promotores virales; Albrecht et al. (2005) concluyen que EICP27 se une al complejo de preiniciación, estimulando la iniciación de la trascripción y/o la elongación por la RNA polimerasa II celular.
El shutoff o supresión temprana de la síntesis de proteínas del hospedero es un comportamiento característico de la infección con distintas cepas de HSV; este fenómeno es secundario a la desestabilización y degradación de los mRNA del hospedero. Como ocurre en ausencia de la expresión de genes virales, se ha sugerido que el shutoff es causado por la entrada del virus o de una proteína de él (Feng et al., 1996; Roizman y Knipe, 2001). Además, se ha asociado con una proteína del tegumento codificada por el UL41 de HSV1 (Feng et al., 1996). En contraste, se ha observado que las infecciones por EHV no inducen una rápida reducción de la síntesis de proteínas; de hecho, no hay un shutoff abrupto temprano, sino una disminución gradual de la síntesis a través de la cual persiste alguna síntesis proteica. Feng et al. (1996) en cultivos celulares infectados, encontraron que tanto los aislados patogénicos como las cepas de laboratorio de EHV tienen deficiencias en la presentación del fenómeno de apagado de la síntesis de proteínas del hospedero a causa del virus. Los autores postulan que posiblemente, la proteína del ORF19, homóloga del ORF41 de HSV, no está presente en los viriones o está presente como un componente viral pero intrínsicamente inactiva. Sin embargo, los análisis muestran que el gen del ORF19 es transcrito y traducido en células infectadas, la proteína es empaquetada dentro de las partículas virales en cantidades detectables y su función puede ser abolida in vitro por mutaciones específicas que introducen sustituciones de aminoácidos en una región altamente conservada.
Partículas interferentes defectuosas (Dips)
EHV1 (cepa KyA) ha servido como modelo experimental para el estudio de los aspectos moleculares de las infecciones persistentes por herpesvirus. Se ha reportado que la presencia de DIPs de EHV1, media el coestablecimiento de la infección persistente y la transformación oncogénica en líneas celulares. Se han generado numerosas líneas celulares infectadas persistentemente con EHV1, capaces de liberar continuamente partículas de EHV1 normales y DIPs (Harty et al., 1993). Dichas partículas también han sido aisladas a partir de hámsters infectados y estudios de microscopía electrónica mostraron una producción alterada de proteínas de cápside en cultivos infectados con EHVDIPs comparado con cultivos infectados con virus silvestres. La infección de células de ovario de hámster con EHVDIPs, resulta en infección persistente y transformación. Se ha observado que líneas celulares infectadas persistentemente, exhiben propiedades biológicas inherentes a su transformación y continúan liberando virus infecciosos y DIPs en cultivo durante varios años (Chen et al., 1996).
La digestión con endonucleasas de restricción, análisis de Southern blot y análisis de secuencias del genoma de DIPs, permitieron determinar la presencia de secuencias conservadas del DNA, derivadas de tres regiones del genoma de virus silvestre: a) El extremo 5’ de la región UL que constituye una porción de la secuencia CPS (señal de clivaje y empaquetamiento), los genes UL1 y UL2 y parte de la secuencia de ICP27; b) la región media del IR, la cual constituye una secuencia Oris y una parte de la secuencia ICP22 y c) la región terminal del IR, que constituye una parte de la secuencia CPS. Estas tres regiones son ensambladas juntas y repetidas en tandem, y se empaquetan como DIPs DNA con un tamaño similar al del virus silvestre. Chen et al. (1996) demostraron la presencia de un ORF híbrido IR4/UL3, el cual se genera en cultivos infectados con DIPsEHV; el ORF IR4 codifica para la proteína IT ICP22, la cual se expresa en abundancia y se localiza predominantemente en el núcleo. La proteína UL3 es el homólogo del IT ICP27, el cual tiene un papel esencial en el crecimiento del virus pero no es necesario en la síntesis de DNA. Teniendo en cuenta que ICP22/ICP27 puede modificar la regulación de genes virales (IT, tempranos y tardíos), los investigadores postulan que esas alteraciones pueden afectar las funciones citolíticas del virus, retardando la expresión de promotores de EHV (Chen et al., 1996). Además, la proteína híbrida ICP22/ICP27 regula negativamente de manera moderada los promotores de proteínas IT y de ICP22, sobreregula los promotores de genes tardíos como el IR5 y altera la función reguladora de proteínas IT y ICP22 en células cotrasfectadas. Estos resultados demuestran que las DIPs alteran la expresión de genes por medio de la expresión de un gen híbrido en su genoma (Chen et al., 1999).
Cepas atenuadas
En estudios previos relacionados con los factores de virulencia de EHV, se comparó la arquitectura genómica de la cepa HH1 (Fig. 4) con la de su descendiente, la cepa atenuada BK343, obtenida a partir de pasajes seriales en células de riñón bovino (BK). La digestión con la endonucleasa BamHI mostró que BK343 implicaba nueve fragmentos variables: Bam HI A, B, D, E, N, P, QR, S y T. Entre los genes localizados en dichos fragmentos, se encontró un cambio en tres genes (gen 1 en el fragmento T; gen 24 en el fragmento A y gen 71 en el fragmento D) después de su pasaje seriado en células BK (Kirisawa et al., 2003). La función del producto del gen 1 permanece desconocida; el gen 24 codifica para una gran proteína de tegumento, que es homóloga del producto del gen UL36 de HSV y el gen 71 codifica para una glicoproteína polimórfica de membrana, gp2, la cual es Oglicosilada (Wellington et al., 1996; Huang et al., 2002). Los genes 1 y 71 de EHV se encuentran ausentes en su contraparte HSV, y ninguno de ellos es necesario para la replicación viral in vitro (Sun et al., 1996). Sin embargo, un estudio publicado en 1997 reportó que la remoción del gen 71 del genoma de EHV resulta en una maduración viral y encapsidación deficientes, lo cual deteriora la capacidad del virus mutante para dispersarse en las células; además, el mismo estudio demostró que in vivo, esta remoción conlleva a una disminución de la patogenicidad en un modelo de enfermedad pulmonar en ratones (Marshall et al., 1997). Estos resultados demuestran el importante papel que tienen estos tres fragmentos en la patogenicidad de la cepa HH1 de EHV (Kirisawa et al., 2003).
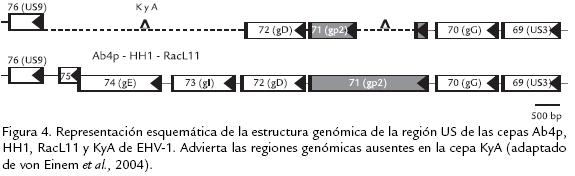
Para evaluar la relación existente entre las mutaciones inducidas en los genes 1 y 71 por pasajes seriados y la patogenicidad de la cepa HH1, Kirisawa et al. (2003) desarrollaron virus mutantes, en los que se inactivaron dichos genes por la inserción del gen lacZ. Sus resultados mostraron que los genes 1 y 71 no están implicados en la reducción del tamaño y el fenotipo de placa visto en la cepa, y el hecho que el fenómeno, solo se presente en células equinas (EDerm) y no en MDBK o RK13, sugiere que un(os) factor(es) celular(es) contribuye(n) en la dispersión célulacélula del virus. Se concluyó que la mutación en los genes 1 y 71 inducida por los pasajes seriados en células, no confería la naturaleza atenuada de la cepa; más aún, es necesario estudiar el producto del gen 24 y su relación con la patogenicidad atenuada de la cepa BK343. Recientemente von Einem et al. (2004) demostraron que delecciones en el ORF71 presentes en la cepa KyA, codifican para una forma truncada de gp2 ( Fig. 4 ), la cual no es funcionalmente equivalente a la gp2 del virus silvestre y exhibe diferentes características de crecimiento en cultivo. En ratones BALB/c conlleva a una disminución de la patogenicidad y a una reducción en tres veces de los títulos virales en pulmón de ratones infectados comparado con el tipo silvestre; además de la pérdida de peso corporal (Kirisawa et al., 2003). De igual forma, Smith et al. (2005), estudiando la patogenicidad de la cepa KyA en ratones CBA, confirmaron que la delección del gp2 lleva a una disminución de la patogenicidad en un modelo de infección respiratoria. Ellos reportaron menor pérdida de peso corporal y menor consolidación pulmonar de linfocitos B y T en ratones infectados con la cepa KyA comparados con animales infectados con una cepa mutante a la cual se le había reparado la gp2.
Como se mencionó anteriormente, EHV posee en su genoma una región UL y una región Us, flanqueadas por dos regiones repetidas (IRS y TRS). La comparación de las secuencias de nucleótidos de la región UsIRs de cepas Ab4p, KyA y RacL11 (Fig. 4) con la cepa vacunal atenuada RacH revelan algunas variaciones genéticas y una delección que afecta los genes 67 y 68 de RacH (IR6 y US2 respectivamente). La proteína US2 varía de tamaño entre estas cepas, predominantemente localiza en las membranas de las células infectadas, encontrándose presente en la envoltura de viriones purificados; no es necesaria para el crecimiento del virus en cultivo celular, pero contribuye a la penetración viral y a una dispersión célulacélula eficiente. Además, desempeña un papel importante para mantener la replicación in vivo (Meindl y Osterrieder, 1999). Por otra parte, la proteína única IR6 forma estructuras filamentosas en las células infectadas y la presencia de estas estructuras se correlaciona con la virulencia de la cepa RacH de EHV; esta proteína es trasportada de célula a célula independientemente de la infección viral, se colocaliza con la lámina nuclear en fases tardías de la infección y codifica una función que facilita el egreso de la nucleocápside viral (Osterrieder et al., 1998).
En un estudio de factores de virulencia de EHV, Schimmer y Neubauer (2003), reportaron que el UL11, una proteína tempranatardía, tiene fuerte influencia en la virulencia; similar a su homólogo UL11 de HSV, el cual está bien caracterizado. UL11 se encuentra en abundante cantidad en los viriones extracelulares; se localiza en las membranas celulares, especialmente en la región transGolgi de las células infectadas, y aunque esta no es esencial para la replicación viral en cultivos celulares, su delección tiene efectos negativos en la formación de placas en células RK13 y en la unión de la cepa RacH a estas mismas células.
Las glicoproteínas
Yao et al. (2003), usando un mutante de la cepa KyA de EHV, deficiente de los genes que codifican para EICP0 y para la glicoproteína gp2, mostraron que esos genes no son necesarios para la replicación viral en cultivo celular. Sin embargo, comparados con la cepa silvestre, los mutantes presentan menor expresión de genes IT, tempranos y tardíos, lo cual se explica por la pérdida de la actividad transactivadora de EICP0. Además, también presentaron menor título viral extracelular y reducido fenotipo de placa. Estos defectos del crecimiento están relacionados con la reducción de las proteínas virales, especialmente de glicoproteínas, las cuales juegan un papel importante en la adhesión, penetración, encapsidación, maduración, egreso y liberación de virión. Estudios paralelos sugieren que la gp2 está involucrada tanto en la adhesión viral como en el egreso, y en el caso de la cepa KyA, en la dispersión directa célulacélula de virus (Rudolph y Osterrieder, 2002). Sin embargo, el posible papel de EICP0 en la patogénesis de EHV permanece poco claro. Actualmente se está evaluando su implicación con mutantes deletéreos de EICP0 de la cepa patogénica Racl11 en un modelo murino (Yao et al., 2003).
En los últimos años se ha logrado un gran entendimiento de las funciones y propiedades de las glicoproteínas de EHV. Para todos los herpesvirus, las glicoproteínas de envoltura gI y gE no son indispensables para la entrada viral y la replicación, pero juegan un importante papel en la virulencia (Damiani et al., 1998). Estudios realizados por Matsumura et al. (1998) reportaron que la delección de gI y gE reduce la virulencia en potros infectados; los autores usaron la cepa KyA de EHV, adaptada a cultivos celulares, la cual posee deficiencia por lo menos de seis genes, incluyendo los que codifican por la gI y gE y es avirulenta en potros (Matsumura et al., 1996). Encontraron que cepas KyA mutantes con rearreglos en los genes codificantes por las gI y la gE exhibían características de crecimiento y fenotipos de placa mayores que el de KyA tipo silvetre, sugiriendo que las gI y/o gE de EHV están involucradas en la dispersión viral célulacélula e in vivo tiene fuerte influencia en la virulencia de la cepa (Matsumura et al., 1998). Además, se postula que gI/gE se encuentran en EHV formando un complejo estable, el cual tiene un importante papel en la virulencia (Damiani et al., 2000). De forma similar, la glicoproteína B (gB) de EHV se ha visto implicada de manera esencial en el crecimiento viral en cultivos celulares. Estudios con mutantes deletéreos para esta glicoproteína muestran infección individual de células sin formación de placas, lo que sugiere que está implicada en la dispersión directa célulacélula; además, gB es altamente necesaria para la penetración del virus en las células (Neubauer et al., 1997). Un estudio realizado en el mismo año, demostró que ratones infectados con la cepa RacL11 mutante de EHV, la cual no poseía los ORF para las gB o gM, no presentaban ningún signo de infección clínica, indicando que la delección de éstas glicoproteínas neutralizaba la virulencia de la cepa viral (Neubauer et al., 1997).
Osterrieder (1999) demostró que la glicoproteína C (gC) es necesaria para la interacción del virión con glicosaminoglicanos (GAG) de la superficie celular en líneas celulares RK13 y en la línea celular de células dérmicas equinas (Edmin337); es decir, es esencial para la unión a las células blanco. Además, en el mismo estudio, usando un mutante negativo para gC se encontró un crecimiento deficiente y disminución de más de 100 veces del título viral extracelular, demostrando la implicación de la gC tanto en penetración como en el egreso viral. La infección de ratones con el mutante defectuoso en la gC no causo ningún signo de enfermedad compatible con la infección por EHV, mientras que ratones infectados con la cepa parental (cepa RacL11) mostraron pérdida de peso masiva, altos títulos virales en pulmón y viremia, lo cual implica que la gC está fuertemente relacionada con la virulencia de EHV (Osterrieder, 1999).
La glicoproteína M (gM) es una glicoproteína no esencial, altamente conservada en todas las subfamilias herpesvirales. La forma madura de gM se encuentra formando dímeros a nivel de la red transGolgi en las células infectadas y en los viriones extracelulares (Osterrieder et al., 1996); y requiere de la presencia del UL49.5 (gN) para su completa maduración a la forma completamente glicosilada y funcional (Rudolph et al., 2002). La delección de la gM en la cepa RacL11 lleva a una reducción del 50% en el tamaño de la placa, comparado con la cepa silvestre, y a 50 a 100 veces menos producción de virus extracelulares (Osterrieder et al., 1996). Estudios realizados por Seybold et al. (2000), usando la cepa KyA, la cual es deficiente de gI y gE, demostraron que la delección de la gM en esta cepa, lleva a un crecimiento débil del virus en cultivo celular. Además la delección de los dominios de transmembrana de la gM de EHV llevan a un procesamiento inadecuado de la glicoproteína y a la retención de moléculas en el retículo endoplásmico, las cuales en consecuencia no son incorporadas en los viriones extracelulares; esto demuestra que la delección simultánea de las gM y gI/gE, lleva a una disminución en el crecimiento, dispersión y liberación de las células infectadas. Rudolph y Osterrieder (2002) establecieron que la delección simultánea de las glicoproteínas gp2 y gM de la cepa RacH de EHV, llevan a una reducción en 200 veces de la producción de virus extracelular, pero tiene un efecto moderado en la dispersión del virus célulacélula, demostrando que la patogenicidad de la cepa requiere un efecto aditivo de éstas glicoproteínas. De igual manera, estudios in vivo usando ratones BALB/c muestran que la delección del gen que codifica para la gM de la cepa RacH de EHV, lleva a una ausencia de signos clínicos y bajos títulos virales en los pulmones, disminuyendo así la patogenicidad en ratones, confiriendo un alto potencial inmunogénico (Neubauer et al., 1997; Osterrieder et al., 2001).
La glicoproteína G (gG) se encuentra presente en la envoltura de los viriones y juega un papel importante en la morfogénesis, adhesión a células y tropismo viral. Es secretada de la célula infectada como un homodímero y existe en dos configuraciones diferentes: puede entrar asociada al virión formando homodímeros unidos por puentes disulfuro; su clivaje proteolítico no requiere de otras proteínas virales y puede existir en forma libre (Drummer et al., 1998). Bryant et al. (2003) reportaron que la gG de EHV tiene la capacidad de unirse a quimoquinas con alta afinidad, inhibiendo su actividad biológica in vitro. La gG bloquea la interacción de las quimioquinas con los receptores celulares y con GAG, inhibiendo las señales de traducción y migración. La actividad de unión a quimioquinas, sugiere que esta actividad puede mediar la adhesión del virus a la superficie de la célula que expresa quimioquinas, jugando un papel importante en la determinación del tropismo celular y tisular in vivo.
La glicoproteína K (gK) es un trascripto bicistrónico tardío, el cual es codificado por el gen ICP27 (Telford et al., 1992). Neubauer y Osterrieder (2004) estudiaron la importancia de la gK en la patogénesis de la cepa KyA. Usando un virus mutante deficiente de esta glicoproteína, demostraron que ella es importante para la replicación in vitro, puesto que este mutante disminuyó significativamente sus títulos virales en células RK13. Está críticamente implicada en la dispersión viral directa célulacélula y en la encapsidación y salida del virus, ya que estas preparaciones mostraron disminución en la producción de virus extracelulares. Además, lograron demostrar que la gK facilita la penetración del virus y es parcialmente responsable de la formación de sincitios en fases tardías de la infección. En conclusión gK está involucrada ya sea en etapas tempranas del ensamblaje viral, antes de la división de las rutas para dispersión célulacélula y liberación al ambiente extracelular, o bien se encuentra involucrada en el proceso de fusión de membranas. También en el mismo estudio, evaluando la expresión de proteínas tardías representativas como la gM, gB, UL34 o UL45, se comprobó que la delección de gK no tiene influencia detectable sobre la producción de otras proteínas tempranas o tardías (Neubauer y Osterrieder 2004).
La glicoproteína D (gD) de EHV es una proteína de 55 kDa, presente en viriones purificados y en extractos de células infectadas, compuesta por dos polipéptidos (Flowers y O’Callaghan, 1992). Un estudio realizado por Wellington et al. (1996), usando un virus con gD mutante, el cual tenía la gD clivada en su extremo Nterminal, mostró que, in vitro, esta glicoproteína aumentaba los niveles de fusión célulacélula, indicando el papel de esta en la transmisión directa del virus. Csellner et al. (2000), usando mutagénesis insercional, construyeron un virus mutante en el cual la fase abierta de lectura para la gD había sido cambiada, confirmaron que esta glicoproteína es importante para la entrada del virus, y como el virus mutante no fue capaz de dispersarse para formar sincitios y placas, se concluyó que también es importante para la fusión célulacélula. Además in vivo estos virus con la gD delecionada, poseen baja patogenicidad en un modelo murino de enfermedad respiratoria, en el cual los ratones presentaron muy bajo título viral en pulmones, sugiriendo que la progenie de una ronda de replicación viral puede dispersarse más allá del sitio de la infección debido a una deficiencia de gD en la envoltura del virión (Csellner et al., 2000; Ruitenberg et al., 2001). Se ha observado que esta delección está asociada con baja virulencia en equinos adultos, yeguas preñadas y potros, e induce una respuesta inmune similar a la inducida por la infección natural (Ruitenberg et al., 2000; Foote et al., 2005).
CONCLUSIONESEn la actualidad la infección por el EHV continúa siendo de gran importancia mundial (Allen, 2004); por lo tanto, el estudio del virus y su conjunto (replicación viral, regulación de la expresión génica, respuesta inmune etc.) son fundamentales para entender la patogénesis. Estos estudios permitirán entender no solo las manifestaciones clínicas, sino también, servirán como una alternativa para el desarrollo de técnicas que se puedan aplicar y permitan controlar la infección, y por ende, la patogenia. La utilización de cepas virales con patogenicidad disminuida para inmunizar equinos susceptibles a la enfermedad es una de las mayores alternativas (Neubauer et al., 1997; Osterrieder et al., 2001; Kirisawa et al., 2003). Igualmente el área de desarrollo de vacunas para estos virus continúa abierto a la investigación, ya que los resultados obtenidos con las vacunas comerciales con virus modificado (como Rhinomune®, Pneumabort K® etc.), no son convincentes o son cuestionables (Ruiz, 2005). Por tanto, para el diseño de nuevos modelos de vacunas modificadas, las cuales confieran alta protección y sean completamente apatogénicas, así como para evaluar la virulencia de distintas cepas de EHV, es necesario entender los mecanismos por los cuales el virus puede salir de una célula infectada y ser transportado a grandes distancias, o infectar a una célula vecina, dispersarse localmente dentro de un tejido o de un órgano infectado.
Consecuentemente, la comprensión de la patogénesis de EHV, puede conducir a un mejor entendimiento de las características de crecimiento de las diferentes cepas de virus en cultivos celulares y su presentación en los animales. Estos conocimientos son fundamentales para combatir una de las enfermedades virales más importantes de los equinos, tema en el cual pretendemos encabezar una lucha pionera en el país, tratando de abarcar desde los aspectos epidemiológicos hasta los aspectos moleculares, en un campo poco explorado en la investigación en el país.
AGRADECIMIENTOS
Los autores desean agradecer al Comité para el Desarrollo de la Investigación de la Universidad de Antioquia (CODI) por el apoyo financiero, proyecto código E01069, y al Grupo de InmunovirologíaBiogénesis por el apoyo constante y espíritu formador.
BIBLIOGRAFÍAALBRECHT RA, JANG HK, KIM SK, O’CALLAGHAN DJ. Direct Interaction of TFIIB and the IE Protein of Equine Herpesvirus 1 is Required for Maximal TransActivation Function. Virology. 2003;316:302–312.
ALBRECHT RA, KIM SK, ZHANG Y, ZHAO Y, O’CALLAGHAN DJ. The Equine Herpesvirus 1 EICP27 Protein Enhances Gene Expression Via an Interaction with TATA BoxBinding Protein. Virology. 2004;324:311326.
ALBRECHT RA, KIM SK, O’CALLAGHAN DJ. The EICP27 Protein of Equine Herpesvirus 1 is Recruited to Viral Promoters by Its Interaction with the ImmediateEarly Protein. Virology. 2005;333:7487
ALLEN GP. Respiratory Infections by Equine Herpesvirus Types 1 and 4. In: Equine Respiratory Diseases, Lekeux P. (Eds) International Veterinary Information Service, Ithaca NY, 2002; (Citado Ene 2006). Disponible en: :http://www.ivis.org/special_books/Lekeux/allen/IVIS.pdf.
ALLEN GP. Equine Rhinoneumonitis en: Truszczynski M, Pearson JE, Edwards S and Schmitt B, editors. OIE Manual of standars for diagnostic test and vaccines, 5th edn. Paris: OIE press; 2004. p. 707716.
[ Links ]BOWLES DE, HOLDEN VR, ZHAO Y, O’CALLAGHAN DJ. The ICP0 Protein of Equine Herpesvirus 1 is An Early Protein that Independently Transactivates Expression of All Classes of Viral Promoters. J Virol. 1997;71:4904–4914.
BOWLES DE, KIM SK, O’CALLAGHAN DJ. Characterization of the TransActivation Properties of Equine Herpesvirus 1 EICP0 Protein. J Virol. 2000;74:12001208.
BRYANT NA, DAVISPOINTER N, VANDERPLASSCHEN A, ALCAMI A. Glycoprotein G Isoforms From Some Alphaherpesviruses Function as BroadSpectrum Chemokine Binding Proteins. EMBO J. 2003;22:833846.
[ Links ]CHEN M, HARTY R, ZHAO Y, HOLDEN RV, O’CALLAGHAN DJ. Expression of an Equine Herpesvirus 1 ICP22/ICP27 Hybrid Protein Encoded by Defective Interfering Particles Associated with Persistent Infection. J Vriol. 1996;70:313320.
CHEN M, GARKOBUCZYNSKI KA, ZHANG Y, O’CALLAGHAN DJ. The Defective Interfering Particles of Equine Herpesvirus 1 Encode an ICP22/ICP27 Hybrid Protein that Alters Viral Gene Regulation. Virus Res. 1999;59:149164.
CRABB BS, STUDDERT MJ. Equine Rhinoneumonitis (Equine Herpesvirus 4) and Equine Abortion (Equine Herpesvirus 1). In: Studdert MJ (eds) Virus Infections of Equines. Holanda: Elsevier; 1996.
[ Links ]CSELLNER H, WALKER C, LOVE DN, WALLEY JM. An Equine Herpesvirus 1 Mutant with a lacZ Insertion Between Open Reading Frames 62 and 63 is Replication Competent and Causes Diseases in the Murine Respiratory Model. Arch Virol. 1998;143:22152231.
[ Links ]CSELLNER H, WALKER C, WELLINGTON JE, MCLURE LE, LOVE DN, WHALLEY JM. EHV1 Glycoprotein D (EHV1 gD) is Required for Virus Entry and CellCell Fusion, and An EHV1 gD Deletion Mutant Induces a Protective Immune Response in Mice. Arch Virol. 2000;145:23712385.
[ Links ]DAMIANI AM, MATSAMURA T, YOKOYAMA N, MAEDA K, MIYAZAWA T, et al. Nucleotide Sequence of Glycoprotein I and E Genes of Equine Herpesvirus Type 4. J Vet Med Sci. 1998;60:219225.
[ Links ]DAMIANI AM, MATSAMURA T, JANG HK, IZUMIYA Y, MIKAMI T. Identification of the Products of the Equine Herpesvirus type 4 gI and gE Genes. Arch Virol. 2000;145:14891496.
[ Links ]DERBIGNY WA, KIM SK, CAUGHMAN GB, O’CALLAGHAN DJ. The EICP22 Protein of Equine Herpesvirus 1 Physically Interacts with the ImmediateEarly Protein and with Itself To Form Dimers and HigherOrder Complexes. J Virol. 2000;74:14251435.
DRUMMER HE, STUDDERT MJ, CRABB BS. Equine Herpesvirus4 Glycoprotein G is Secreted as a DisulphideLinked Homodimer and is Present as Two Homodimeric Species in the Virion. J Gen Virol. 1998;79:12051213.
[ Links ]FENG X, THOMPSON YG, LEWIS JB, CAUGHMAN GB. Expression and Function of the Equine Herpesvirus 1 VirionAssociated Host Shutoff Homolog. J Virol. 1996;70:87108718.
[ Links ]FLOWERS CC, O’CALLAGHAN DJ. Equine Herpesvirus 1 Glycoprotein D: Mapping of the Transcript and a Neutralization Epitope. J Virol. 1992;66:64516460.
FOOTE CE, LOVE DN, GILKERSON JR, ROTA J, TREVORJONES P, et al. Serum Antibody Responses to Equine Herpesvirus 1 Glycoprotein D in Horses, Pregnant Mares and Young Foals. Vet Immunol Immunopathol. 2005;105:4757.
[ Links ]GARKOBUCZYNSKI KA, SMITH RH, KIM SK, O’CALLAGHAN DJ. Complementation of a ReplicationDefective Mutant of Equine Herpesvirus Type 1 by a Cell Line Expressing the ImmediateEarly Protein. Virology. 1998;248:8394.
HARTY R, HOLDEN VR, O’CALLAGHAN DJ. Transcriptional and Translational Analyses of the UL2 Gene of Equine Herpesvirus 1: a Homolog of UL55 of Herpes Simplex Virus Type 1 That Is Maintained in the Genome of Defective Interfering Particles. J Virol. 1993;67:22552265.
HUANG JA, FICORILLI N, HARTLEY CA, ALLEN GP, STUDDERT MJ. Polymorphism of Open Reading Frame 71 of Equine Herpesvirus4 (EHV4) and EHV1. J Gen Virol. 2002;83:525531.
[ Links ]IBRAHIM ESM, PAGMAJAV O, YAMAGUCHI T, MATSAMURA T, FUKUSHI H. Grown and Virulence Alterations of Equine Herpesvirus 1 by a Insertion of a Green Fluorescent Protein Gene in the Intergenic Region Between ORFs 62 and 63. Microbiol Immunol. 2004;11:831842.
[ Links ]JANG HK, ALBRECHT RA, BUCZYNSKI KA, KIM SK, DERBIGNY WA, O’CALLAGHAN DJ. Mapping the Sequences That Mediate Interaction of the Equine Herpesvirus 1 ImmediateEarly Protein and Human TFIIB. J Virol. 2001;75:1021910230.
KIM SK, HOLDEN R, O’CALLAGHAN DJ. The ICP22 Protein of Equine Herpesvirus 1 Cooperates with the IE Protein To Regulate Viral Gene Expression. J Virol. 1997;71:10041012.
KIM SK, BOWLES DE, O’CALLAGHAN DJ. The g2 Late Glycoprotein K Promoter is Differentially Regulated by the IE and EICP0 Proteins. Virology. 1999;256:173179.
KIM SK, BUCZYNSKI KA, CAUGHMAN GB, O’CALLAGHAN DJ. The Equine Herpesvirus 1 ImmediateEarly Protein Interacts with EAP, a NucleolarRibosomal Protein. Virology. 2001;279:173184.
KIM SK, O’CALLAGHAN DJ. Molecular Characterizations of the Equine Herpesvirus 1 ETIF Promoter Region and Translation Initiation Site. Virology. 2001;286:237247.
KIM SK, JANG HK, ALBRECHT RA, DERBIGNY WA, ZHANG Y, O’CALLAGHAN DJ. Interaction of the Equine Herpesvirus 1 EICP0 Protein with the ImmediateEarly (IE) Protein, TFIIB, and TBP May Mediate the Antagonism Between the IE and EICP0 Proteins. J Virol. 2003;77:26752685.
KIM SK, ALBRECHT RA, O’CALLAGHAN DJ. A Negative Regulatory Element (Base Pairs 204 to 177) of the EICP0 Promoter of Equine Herpesvirus 1 Abolishes the EICP0 Protein’s TransActivation of its Own Promoter. J Virol. 2004;78:1169611706.
KIRISAWA R, KOBAYASHI T, UEMATSU R, IKEDA A, KUROIWA R, et al. Growth of Recombinant Equine Herpesvirus 1 (EHV1) Replaced with PassageInduced Mutant Gene 1 and Gene 71 Derived From an Attenuated EHV1 in Cell Cultures and in the Lungs of Mice. Vet Microbiol. 2003;95:159174.
[ Links ]MARSHALL KR, SUN Y, BROWN SM, FIELD HJ. An Equine Herpesvirus1 Gene 71 Deletant Is Attenuated and Elicits a Protective Immune Response in Mice. Virology. 1997;231:2027.
[ Links ]MATSUMURA T, O’CALLAGHAN DJ, KONDO T, KAMADA M. Lack of Virulence of the Murine Fibroblast Adapted Strain, Kentucky A (KyA), of Equine Herpesvirus type 1 (EHV1) in Young Horses. Vet Microbiol. 1996;48:35365.
MATSUMURA T, KONDO T, SUGITA S, DAMIANI AM, O’CALLAGHAN DJ, IMAGAWA H. An Equine Herpesvirus Type 1 Recombinant with a Deletion in the gE and gI Genes is Avirulent in Young Horses. Virology. 1998;242:6879.
MEINDL A, OSTERRIEDER N. The Equine Herpesvirus 1 US2 Homolog Encodes a Nonessential MembraneAssociated Virion Component. J Virol. 1999;73:34303437.
[ Links ]NEUBAUER A, BEER M, BRANDMULLER C, KAADEN OR, OSTERRIEDER N. Equine Herpesvirus 1 Mutants Devoid of gGlycoprotein B or M are Apathogenic for Mice but Induce Protection Against Challenge Infection. Virology. 1997;239:3645.
[ Links ]NEUBAUER A, BRAUN B, BRANDMULLER C, KAADEN OR, OSTERRIEDER N. Analysis of the Contributions of the Equine Herpesvirus 1 Glycoprotein gB Homolog to Virus Entry and Direct CellToCell Spread. Virology. 1997;227:28194.
[ Links ]NEUBAUER A, OSTERRIEDER N. EHV1 Glycoprotein K is Required for Efficient CellToCell Spread and Virus Egress. Virology. 2004;329:1832. OSTERRIEDER N. Construction and Characterization of an Equine Herpesvirus 1 Glycoprotein C Negative Mutant. Virus Res. 1999;59:165177.
[ Links ]OSTERRIEDER N, NEUBAUER A, BRANDMULLER C, BRAUN B, KAADEN OR, et al. The Equine Herpesvirus 1 Glycoprotein gp21/22a, the Herpes Simplex Virus Type 1 gM Homolog, Is Involved in Virus Penetration and CelltoCell Spread of Virions. J Virol. 1996;70:41104115.
[ Links ]OSTERRIEDER N, NEUBAUER A, BRANDMULLER C, KAADEN OR, O’CALLAGHAN DJ. Equine Herpesvirus 1 IR6 Protein that Colocalizes with Nuclear Lamins is Involved in Nucleocapsid Egress and Migrates from Cell To Cell Independently of Virus Infection. J Virol. 1998;72:9806–9817.
OSTERRIEDER N, SEYBOLDT C, ELBERS K. Deletion of Gene 52 Encoding Glycoprotein M of Equine Herpesvirus Type 1 Strain RacH Results in Increased Immunogenicity. Vet Microbiol. 2001;81:219226.
[ Links ]PUREWAL AS, ALLSOPP R, RIGGIO M, TELFORD EA, AZAM S, et al. Equid Herpesvirus 1 and 4 Encode Functional Homologs of Herpes Simplex Virus Type 1 Virion Transactivador Protein, VP16. Virology. 1994;198:385389.
[ Links ]PUREWAL AS, IQBAL J, EDINGTON N. The Equid Herpesvirus1 Gene 63 is Expressed as a Leaky Late (gamma 1) Transcript and is Nonessential for Replication in vitro. Virus Res. 1998;54:18995.
[ Links ]RAMÍREZ GC, CHAPARRO JJ, VERA VJ, VILLAMIL LC, ROMERO JR. Primer aislamiento de herpesvirus equino en Colombia. Rev Col Cienc Pec. 2001;14:71.
[ Links ]ROIZMAN B, KNIPE DM. Herpes Simplex Viruses and Their Replication. In Fields Virology. Fourth edition. Knipe DM and Howley PM, Editors. USA: Lippincott Williams & Wilkins; 2001. p. 23992461.
[ Links ]ROIZMAN B, PELLETT PE. The Family Herpesviridae: A Brief Introduction. In Fields Virology, Fourth edition. Knipe DM and Howley PM, Editors. USA: Lippincott Williams & Wilkins; 2001. p. 23812397
[ Links ]RUDOLPH J, OSTERRIEDER N. Equine Herpesvirus Type 1 Devoid of gM and gp2 is Severely Impaired in Virus Egress but not Direct CellToCell Spread. Virology. 2002;293:356–367.
RUDOLPH J, SEYBOLDT C, GRANZOW H, OSTERRIEDER N. The Gene 10 (UL49.5) Product of Equine Herpesvirus 1 Is Necessary and Sufficient for Functional Processing of Glycoprotein M. J Virol. 2002;76:29522963.
[ Links ]RUITENBERG KM, LOVE DN, GILKERSON JR, WELLINGTON JE, WHALLEY JM. Equine herpesvirus 1 (EHV1) Glycoprotein D DNA Inoculation in Horses with Preexisting EHV1/EHV4 Antibody. Vet Microbiol. 2000;76:117127.
[ Links ]RUITENBERG KM, GILKERSON JR, WELLINGTON JE, LOVE DN, WHALLEY JM. Equine Herpesvirus 1 Glycoprotein D Expressed in Pichia pastoris is Hyperglycosylated and Elicits a Protective Immune Response in the Mouse Model of EHV1 Disease. Virus Res. 2001;79:125135.
[ Links ]RUIZ J. Prevención y control de la rinoneumonitis equina. Rev Col Cienc Pec. 2005;18:6474.
[ Links ]SCHIMMER C, NEUBAUER A. The Equine Herpesvirus 1 UL11 Gene Product Localizes to the TransGolgi Network and is Involved in CellToCell Spread. Virology. 2003;308:2336
[ Links ]SEYBOLDT C, GRANZOW H, OSTERRIEDER N. Equine Herpesvirus 1 (EHV1) Glycoprotein M, Effect of Deletions of Transmembrane Domains. Virology. 2000;278:477489.
[ Links ]SMITH RH, CAUGHMAN GB, O’CALLAGHANL DJ. Characterization of the Regulatory Functions of the Equine Herpesvirus 1 ImmediateEarly Gene Product. J Virol. 1992;66:936945.
SMITH RH, HOLDEN VR, O’CALLAGHAN DJ. Nuclear Localization and Transcriptional Activation Activities of Truncated Versions of the ImmediateEarly Gene Product of Equine Herpesvirus 1. J Virol. 1995;69:38573862.
SMITH PM, KAHAN SM, ROREX CB, VON EINEM J, OSTERRIEDER N, O’CALLAGHAN DJ. Expression of the FullLength Form of gp2 of Equine Herpesvirus 1 (EHV1) Completely Restores Respiratory Virulence to the Attenuated EHV1 Strain KyA in CBA mice. J Virol. 2005;79:51055115.
SUN Y, MACLEAN AR, AITKEN J D, BROWN SM. The Role of the Gene 71 Product in the Life Cycle of Equine Herpesvirus 1. J Gen Virol. 1996;77:493500.
[ Links ]TELFORD EA, WATSON MS, MCBRIDE K, DAVISON AJ. The DNA Sequence of Equine Herpesvirus1. Virology. 1992;189:304316.
[ Links ]TELFORD EA, WATSON MS, PERRY J, CULLINANE AA, DAVISON AJ. The DNA Sequence of Equine Herpesvirus4. J Gen Virol. 1998;79:11971203.
[ Links ]THOMAS SK, LILLEY CE, LATCHMAN DS, COFFIN RS. Equine Herpesvirus 1 Gene 12 Can Substitute for vmw65 in the Growth of Herpes Simplex Virus (HSV) Type 1, Allowing the Generation of Optimized Cell Lines for the Propagation of HSV Vectors with Multiple ImmediateEarly Gene Defects. J Virol. 1999;73:73997409.
[ Links ]VON EINEM J, WELLINGTON J, WHALLEY JM, OSTERRIEDER K, O’CALLAGHAN DJ, OSTERRIEDER N. The Truncated Form of Glycoprotein gp2 of Equine Herpesvirus 1 (EHV1) Vaccine Strain KyA Is Not Functionally Equivalent to FullLength gp2 Encoded by EHV1 WildType Strain RacL11. J Virol. 2004;78:30033013.
WELLINGTON JE, ALLEN GP, GOOLEY AA, LOVE DN, PACKER NH, et al. The Highly Oglycosylated Glycoprotein gp2 of Equine Herpesvirus 1 is Encoded by Gene 71. J Virol. 1996;70:8195–8198.
WELLINGTON JE, LAWRENCE GL, LOVE DN, WHALLEY JM. Expression and Characterization of Equine Herpesvirus 1 Glycoprotein D in Mammalian Cell Lines. Arch Virol. 1996;141:178593.
[ Links ]WHITTAKER GR, RIGGIO MP, HALLIBURTON IW, KILLINGTON RA, ALLEN GP, MEREDITH DM. Antigenic and Protein Sequence Homology between VP13/14, a Herpes Simplex Virus Type 1 Tegument Protein, and gp1O, a Glycoprotein of Equine Herpesvirus 1 and 4. J Virol. 1991;65:23202326.
[ Links ]WYRICK C. Equine Herpesvirus (EHV4) and 1 (EHV1) Equine Viral Rinoneumonitis en: (Citado Ene 2006) Disponible en: URL: http://www.vet.uga.edu/vpp/ia/SRP/ERD/EHV-4and1.html
[ Links ]YAO H, OSTERRIEDER N, O’CALLAGHAN DJ. Generation and Characterization of an EICP0 Null Mutant of Equine Herpesvirus. Virus Res. 2003;98:163172. ZHAO Y, HOLDEN RV, SMITH RH, O’CALLAGHAN DJ. Regulatory Function of the Equine Herpesvirus 1ICP27 Gene Product. J Virol. 1995;69:27862793.

