










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Biológica Colombiana
versión impresa ISSN 0120-548X
Acta biol.Colomb. v.12 n.2 Bogotá jul./dic. 2007
THREE-DIMENSIONAL STRUCTURE OF Bacillus thuringiensis TOXINS: A REVIEW
Estructura tridimensional de las toxinas de Bacillus thuringiensis: revisión
LÓPEZ PAZOS SA1,M.Sc.; CERÓN SALAMANCA JA1, Ph. D. 1Instituto de Biotecnología, Universidad Nacional de Colombia, Sede Bogotá, Edificio Manuel Ancízar, ciudad universitaria, Carrera 30 No. 45-03. AA. 14490. Bogotá, Colombia. jacerons@unal.edu.co
Presentado 4 de octubre de 2006, aceptado 28 de marzo 2007, correcciones 30 de marzo de 2007.
SUMMARY
Structure-based protein engineering of Bacillus thuringiensis d-endotoxins may direct the search for variants with broader susceptible species spectra, optimal potency, and stability properties. Here, we revised the more important characteristics of the Cry and Cyt proteins three-dimensional structure; it is possible to conclude that an obvious general model exists with specific properties according to its function and target organism.
Key words: Bacillus thuringiensis, Cry toxins, Cyt toxins, Parasporin.
RESUMEN
La ingeniería de proteínas de las d-endotoxinas de Bacillus thuringiensis puede orientar la búsqueda de variantes con un espectro mayor de especies susceptibles, potencia optimizada, y estabilidad apropiada. Aquí, nosotros revisamos las características más importantes de la estructura tridimensional de las proteínas Cry y Cyt. Es posible concluir que existe un modelo general obvio con propiedades específicas de acuerdo a su función y organismo susceptible.
Palabras clave:Bacillus thuringiensis, delta-endotoxina.
INTRODUCTION
The bacterium Bacillus thuringiensis produces crystalline protein inclusions (d-endotoxins) that have a natural insecticidal effect on pest insects of the Lepidoptera, Coleoptera, Diptera, Hymenoptera, Homoptera, Orthoptera and Mallophaga orders, and against mites, platyhelminthes and nematodes; they have also shown activity on cancer cells and on protozoa of medical importance such as Plasmodium berghei (Schnepf et al., 1998; Mizuki et al., 1999; Xu et al., 2004). These proteins, denominated d-endotoxins, are a useful alternative to synthetic chemical pesticides and this led to the development of bioinsecticides and transgenic expression to provide pest resistance in plants (Schnepf et al., 1998); they have been divided in Cry and Cyt proteins (Crickmore et al., 1998).
For the Cry proteins to become active, a target organism must eat them, then these endotoxins are solubilized and processed by proteases in the insect midgut to become activated toxins; the next steps involve binding to specific receptors, insertion of the toxin into the apical membrane to create ion channels or pores that disrupt the membrane potential resulting in cell lysis (Schnepf et al., 1998; de Maagd et al., 2001). The Cyt protein assembles into cation-selective channels in the membrane, resulting in colloid-osmotic lysis and cell death, or alternatively the protein adsorbs onto the membrane surface and the resulting aggregates cause defects in lipid packing through which the cytoplasm can leak into the extracellular space; these two models are not mutually exclusive, where each could operate at different toxin concentrations or on different time scales (Li et al., 1996; Schnepf et al., 1998; Butko, 2003; Manceva et al., 2004).
Cry toxins possess three domains. Domain I, consists of a bundle of antiparallel a- helices in which helix 5 is encircled by the remaining helices and might be responsible for the formation of lytic pores in the insect midgut. Domain II, consists of three antiparallel b-sheets joined in a typical Greek key topology, arranged in a so-called b- prism fold; this domain contains the surface-exposed loops and because they show similarities to immunoglobin antigen-binding sites, they are candidates for involvement in receptor binding. Domain III, consists of two twisted, antiparallel b-sheets forming a b-sandwich with a jelly roll topology; this domain could play a number of key roles in the biochemistry, structural integrity, receptor binding, membrane penetration, and ion channel function (Schnepf et al., 1998). Cyt protein consists of a single domain in which two outer layers of a-helix wrap around a mixed b-sheet; the predominantly b- sheet structure suggests a pore based on a b-barrel, three of the strands are sufficiently long to span the hydrophobic core of the membrane, and the sheet formed by them shows an amphiphilic or hydrophobic character; this molecule self-assembles within the membrane and two a-helices (A and C) appeared to be involved in both membrane interaction and intermolecular assembly, Cyt protein exerts its effect via a general, detergent-like perturbation of the membrane (Butko 2003; Promdonkoy et al., 2004).
The three-dimensional (3D) structure of B. thuringiensis d-endotoxins will allow understanding its folding to achieve a better interaction with the receptor and to develop its activity (Grochulski et al.,1995; Morse et al., 2001). The present paper reviews the structures of B. thuringiensis d-endotoxins experimentally determined, which are in the Protein Data Bank (PDB; http://www.rcsb.org/pdb/), and theoretical models
CRY1AA (PDB: 1CIY)
This protein has specificity towards Lepidoptera order; domain I (residues 33-253) forms a bundle of eight anti-parallel helices with helix a5 in the center; the outer helices, especially the long ones, display an amphipathic character; this is also the case for the central helix a5, although the charged side-chains (Arg173 and Asp174) are located only at the C-terminal end while the polar residues in the remaining helix are uncharged (Ser159, Gln163, Asn166, His168 and Ser170) and concentrated on one side (except for His168), buried underneath helices a1 and a7; the outer faces of the other helices are sprinkled more or less evenly with charged side-chains. Helix a4 is an exception and contains charged side-chains (Arg127, Glu128, Glu129, Arg131 and Asp136) only in the N-terminal; together with the central helix a5 and the loop that connects them, they form a hairpin that contains the least polar segment of domain I. This domain has a hydrophobic helical hairpin, a4-a5, and is likely to undergo a conformational rearrangement of tertiary structure in order to form ion channels in the membrane, by pH or receptor interaction. Domains I and II are connected by a linker, extending from the end of helix a7 (domain I) to the first b- strand of sheet 3 (domain II) and most of the interactions occur at the N-terminal end where all the interdomain salt bridges are located. They likely play an important role either in freeing domain I from the rest of the protein or in keeping the toxin in a compact globular form during solubilization and activation of the protoxin (Grochulski et al., 1995). Domain II (residues 265-461) consists of three antiparallel b-sheets and two short a-helices; the sheets are related by 3-fold symmetry around an axis parallel to the direction of the strands. The first two sheets are each formed by four strands b2 to b5 and b6 to b9, respectively; the strands in both sheets are connected according to the typical Greek-key topology. The third sheet contains three strands and is formed by two separate fragments: two central strands, b10 and b11 (C-terminal), and the outer strand b1b with helix a8 (N-terminal); the packing of the sheets, provides a core filled with many hidrophobic, with aromatic and hydrophobic residues. The two middle strands of each of the three b-sheets form long b-hairpin extensions; the apex of this domain is formed by the loops at the tips of these hairpins (residues 310 to 313, 367 to 379 and 438 to 446 from sheets 1, 2 and 3, respectively). Domain II contacts domain I through sheet 3 and the external faces of sheets 1 and 2 are exposed to the solvent (Grochulski et al.1995). The specificitydetermining regions have been mapped primarily to domain II; residues 365 to 371 are essential for binding to the membrane of the midgut cells which form a loop at the bottom of sheet 2 and its intrinsic flexibility does play an important role in receptor recognition (Grochulski et al.1995). The C-terminal domain (residues 463 to 609) is a b-sandwich of two anti-parallel, highly twisted b-sheets and comprises with an additional, with an outer strand (residues 254 to 264) in one of the b-sheets; the outer sheet (facing the solvent) comprises five strands: b13b, b16, b22, b18 and b19. The short strand b12 runs parallel to b16 near its C-terminal end, back to back with strand b13b; the inner sheet, facing the other two domains, contains five strands: b20, b17 and b23, are contiguous, the fourth strand contains an insertion in the middle (residues 475 to 497) and is made of two sections (b13 and b15), the fifth strand is made of b14 and b15, strand b1a is adjacent and parallel to b13. Two loops extend from the main body of domain III; one of the loops (residues 489-497) connects strands b14 and b15 (half-strands 4 and 5), and the second loop (residues 554-565) joins strands b19 (outer sheet) and b20 (inner sheet). These two loops provide the interface for interactions with domain I; domain III plays a important role in protein stability due to the network of interactions provided by the arginine residues (Grochulski et al.1995). There are numerous van der Waals, hydrogen bond and electrostatic interactions between the domains; most of the contacts are of the van der Waals type and are formed between domains I and II and domains I and III (Grochulski et al.1995). The electrostatic effects may play an important role in the appropriate orientation of toxin molecules towards the cell membrane during the initial binding with the receptor; most of the surface has a positive potential, one side of domain I (helices a2a, a3 and the N-terminal part of a4) has a significant negative potential (Grochulski et al., 1995). All but two of Cry1Aa basic residues are arginine, maybe as a consequence of the high pH environment found in the insect midgut where the toxin displays its activity; the high pKa of arginine (12.4) ensures that most of these side-chains will still be charged which is important either for protein stability or for its activity (Grochulski et al., 1995).
CRY2AA (PDB: 1I5P)
Cry2Aa protein exhibits high specific activity against two insect orders, Lepidoptera and Diptera (Schnepf et al., 1998). Cry2Aa contains an N-terminal 49-amino acid peptide that is cleaved upon activation; the structures of the three domains are surprisingly similar in the overall topology to those of the activated toxins Cry, in spite of its little sequence identity, suggesting that removal of the activation peptide serves to expose regions of the toxin rather than to alter its conformation. In the mature toxin, domain I (residues 1-272) is a pore-forming seven-helical bundle; domain II (residues 273-473) is a receptor binding b prism (a three-fold symmetric arrangement of b sheets, each with a Greek key fold); domain III (residues 474-633) is implicated in determining both larval receptor binding and pore function and is a C-terminal b sandwich. The dipteranspecific residues are 278-340, while lepidopteran specificity is conferred by residues 341-412; this defines a continuous hydrophobic 106 amino acid block (307-412) and 800 Å2 of specificity-distinguishing residues, within or about domain II/III and surrounding residues from the b5-b6, b7-b8, and b4-b5 loops. The toxin-receptor binding surface is comprised of a distribution of hydrophobic residues (Ile474-Ala477 from b12a, Val365-Leu369 from the b5-b6 loop, and Leu402-Leu404 from the b7-b8 loop) across the solvent-exposed surface of the middle and C-terminal domains. Proteolytic activation of the toxin involves the removal of the 49 N-terminal amino acids and exposes residues comprising this putative toxin-receptor binding surface without affecting the structure of the seven-helical membrane insertion domain; the structure of Cry2Aa suggests that the N-terminal residues should sterically hinder access to the putative binding epitope b5-b6 and b7-b8 loops and the exposed parts of domain III closest to domain II; the occlusion of the hydrophobic patch of the putative binding epitope prevents nonspecific aggregation of the toxin with itself or other host proteins, or/and forms environmentally stable crystalline inclusions (Morse et al., 2001).
CRY3AA (PDB: 1DLC) Y CRY3BB (PDB: 1JI6)
The cleaved Coleoptera specific Cry3Aa has a wedge-shaped molecule with three domains. Domain I (residues 1-290), is a seven-helix bundle in which a central helix is completely surrounded by six outer helices; this central helix (a5) is oriented with its Cterminal towards the bulky end of the molecule; the outer helices are arranged anticlockwise in the order: a1, a2, a3, a4, a6, a7, with the helices a1 and a7 adjacent to the b-sheet domains. The helices are long, especially a3 to a7, which contain respectively 8, 7, 6, 9 and 7 complete helical turns and hence would be long enough to span the 30 Å thick hydrophobic region of the membrane bilayer; the six outer helices bear a strip of hydrophobic residues down their entire length on the side-facing helix a5, so they are amphipathic. All polar groups in domain I are hydrogen-bonded or in salt bridges; the concentric arrangement of the seven-helix bundle may be a soluble form of packaging for the hydrophobic and amphiphilic helices that will form pores in the membrane after a large change in conformation (Li et al., 1991). The middle domain (residues 291-500), contains three antiparallel b sheets packed around a hydrophobic core; the chain connections b4, b3, b2, b5 and b8, b7, b6, b9 respectively, follow the order +3, -1, -1, +3, which is typical of the Greek-key topology; the pseudo-symmetry between these sheets is very approximate; the three stranded-sheet 3 is formed by two separate polypeptide segments and is in contact with helix a7 of domain I; the b ribbons from all three sheets terminate in loops in a small region on the molecular apex, in a manner reminiscent of the complementarity-determining region of immunoglobins which correlates with receptor recognition (Li et al., 1991). Domain III (residues 501- 644), is a sandwich of two antiparallel sheets and with domain I make up the bulky end of the protein; this domain has the jelly-roll topology because it can be generated by folding an antiparallel b ribbon which starts with b13 (N-terminal) and b23 (Cterminal) on the inner sheet and end in the loop between b18 and b19 on the outer sheet, b14 forms the fifth antiparallel strand of the outer sheet. In addition, small parallel sheets are formed at the edge of the b-sandwich through hydrogen bonding of strand b12 to b16 at the edge of the outer sheet, and b1 to b13 at the edge of the inner sheet; the inner sheet plays a critical role in the structural integrity and stability of the protein through interaction with the helical bundle (Li et al., 1991).
Similar to Cry3A the first 60 N-terminal residues of Cry3Bb1 were also not observed in this crystal structure (Galitsky et al., 2001).There are seven additional single amino acids (Ala104, Lys416, Gln453, Lys554, Leu557, Lys624 and Glu626) in the sequence of Cry3Bb1 compared with that of Cry3A, the majority of these insertions are located in domain III (Galitsky et al., 2001). The seven-helical bundle of domain I is long and twisted around the central helix a5 surrounded by six outer helices, which creates greater structural stability for the bundle; helix a2 is interrupted by a non-helical segment between residues 100-105 and includes an additional residue, Ala104, compared with the sequence of Cry3A, there is greater surface exposure of residues Ser102 and Asp103 in Cry3Bb1 as a result of this insertion. The major changes in domain I of these two toxins involve the conformation of loop regions (residues 81-90, 100- 105, 155-159 and 216-222), and three more N-terminal residues were observed in the tertiary structure of Cry3A than in Cry3Bb1 (Galitsky et al., 2001). Domain II (residues 295-501) contains three antiparallel b-sheets, sheet 1, composed of strands b5, b2, b3 and b4 and sheet 2, composed of strands b8, b7, b6 and b9, form the distinctive Greek key motif, similar to that observed in Cry3A. The outer surface of sheet 3 (b1, b11 and b10) makes contact with helix a7 of domain I (Galitsky et al., 2001); interactions between domains I and II are strengthened by a hydrogen bond between Gln316of domain II and Asp261 of domain I, this interaction is not observed in the structure of Cry3A owing to changes in the local conformation as a consequence of the differences in the sequence of these two proteins (Galitsky et al., 2001). There are two additional residues in domain II of Cry3Bb1 not present in the sequence of Cry3A: Lys416 and Gln453. The possible effect of the insertion of Lys416 in domain II is to stabilize the structure by formation of a hydrogen bond to Asp486, Gln453 participates in hydrogen bonds with the backbone of Asp307 and a water molecule, which in turn hydrogen bonds to the hydroxyl group of Thr403 (Galitsky et al., 2001). The most significant differences between the tertiary structure of Cry3Bb1 and that reported for Cry3A are in domain III (residues 503-652) which has a jelly-roll b-barrel topology with a hydrophobic core; there are four single-residue insertions in domain III of Cry3Bb1 (Lys554, Leu557, Lys624 and Glu626) not present in the sequence of Cry3A, these residues all lie on one surface of domain III between strands b16-b17 and b22- b23 (Galitsky et al., 2001). The differences in domain III between the structure of Cry3Bb1 and Cry3A may be important in the regulation of channel function at the dimer interface, as well as imparting specificity of action (Galitsky et al., 2001).
CRY4
The models of the activated Cry4A (residues 69-678) and Cry4B (residues 40-634) were determinated by homology modeling, and its secondary structures by Circular dichroism spectroscopy (Angsuthanasombat et al., 2004). The N-terminal domain (Cry4A, residues 69-317; Cry4B, residues 40-271) formed a seven antiparallel helix bundle in which the central helix 5 was entirely encircled by six outer helices; the middle domain (Cry4A, residues 318-527; Cry4B, residues 272-469) was a three-fold symmetric assembly that primarily consisted of b-sheets with sheets 1 and 2 being antiparallel. The C-terminal domain (Cry4A, residues 528-678; Cry4B, residues 470- 634) consisted of two twisted, anti-parallel b-sheets that formed a face-to-face sandwich (Angsuthanasombat et al., 2004). The domain I topology is equipped for membrane insertion and pore formation, the toxin-induced pore/channel is initiated by the insertion of the a4-a5 hairpin into the lipid membranes with subsequent association with other molecules to form an oligomeric helical bundle pore; this critical loop is comprised of one highly conserved tyrosine residue (Cry4A: Tyr202; Cry4B: Tyr170) whose aromatic structure plays a crucial role in mosquito-larvicidal activity, conceivably being involved in an interaction with the phospholipid head groups for stabilizing the oligomeric pore structure. The orientation of the hydrophobic faces of the amphipathic helices of the pore-forming domain is the reverse of that needed to form an aqueous pore/channel in the membranes, this indicates that perhaps substantial conformational changes may be occurring in the membrane-insertion and pore-formation stages, probably due to the proteolysis of one or more of the solvent-exposed loops connecting the helices in the pore-forming bundle which impart greater flexibility to the toxin molecule and might allow protrusion or release of the helical hairpin (Angsuthanasombat et al., 2004).
Later, the Cry4Ba structure (residues 84-641; PDB: 1W99; Boonserm et al., 2005) was experimentally determined; its N-terminal domain (residues 84-282) is an a-helical bundle of five helices (helices a3-a7) all longer than 30 Å and amphipathic in character; helix a3 contains 39 amino acid residues, making it the longest helix in Cry4Ba, the central helix a5 (residues 170-199) is the least amphipathic character and is relatively hydrophobic; the middle domain is a b-prism of three antiparallel b- sheets, sheet 1 (strands b5, b2, b3 and b4) and sheet 2 (strands b8, b7, b6 and b9) show the Greek-key motif, while in sheet 3 (strands b1, b11 and b10) together with the a8 helix form a Greek-key-like motif; there are three cis-proline residues (proline preceded by cis peptide bonds): Pro299, Pro389 and Pro414, located at the bottom of domain II, in loops b6-b7 and b8-b9 of sheet 2, and b1-a8 of sheet 3 (Boonserm et al., 2005). C-terminal domain III (residues 467-641) is a b-sandwich of two antiparallel b-sheets; the inner sheet, facing the other two domains, has five strands across its width; sheets b20 and b21 together make up its top edge, followed by b17, b23, then b13 and b15 together filling the next strand, and finally b14 at the bottom edge; the outer sheet, facing the solvent, also has five strands from top to bottom: b19, b18, b22, b16, and b12 plus b13b together forming the edge strand. The C-terminal residues 634-640 form a small amphipathic helix, a9, which follows the last strand b23; this helix may be a vestige of the C-terminal fragment of the protoxin that was cleaved in the proteolytic activation (Boonserm et al., 2005). A major conformational change is required to transform the initially soluble Cry toxin into a structure able to insert into the membrane, the conformational change was envisaged to expose a relatively non-polar helix hairpin, probably a4 and a5, from domain I to initiate membrane penetration, the exposure of hydrophobic surfaces in the N-terminal domain of Cry4Ba is expected to promote toxin interaction with the membrane, directly and through oligomerisation, thereby facilitating pore formation (Boonserm et al., 2005). The domain II loop region, particularly the loops 1 and 2, are important in the specificity determination for Aedes and Anopheles; the domain II apical loops are shorter in Cry4Ba than in other Cry toxin structures, possible interactions with the receptor in these loops are more likely to perturb the structure of the core b-sheets of domain II, and may impinge on the domain I-II contacts; Cry2Aa, which exhibits lepidopteran /dipteran dual specificity might be suspected to have structural features in common with the Cry4Ba toxin that correlate with their overlapping dipteran specificity; but this appears unlikely, because the Cry4Ba and Cry2Aa structures diverge strongly within each domain and in the interactions between domains (Boonserm et al., 2005).
Recently, the crystal structure of the active Cry4Aa single mutant R235Q was reported (PDB: 2c9kA; Boonserm et al., 2006). Domain I (residues 68 to 321), is composed of seven amphipathic helices; the most hydrophobic helix a5 is located centrally and is surrounded by the six remaining helices. Helix a2 is interrupted by a short loop section and thus can be divided into a2a and a2b, both the unique disulfide bridge (Cys192- Cys199) and the proline-rich motif (Pro193-Pro-Asn-Pro196) play essential roles in Cry4Aa toxin activity, conceivably by maintaining the a4-a5 loop structural integrity, which may be required for efficient membrane insertion of the a4-a5 transmembrane hairpin. Domain II, consists of three antiparallel b-sheets packed through formation of a central hydrophobic core; an interesting feature is the presence of a conserved hydrophobic patch on the molecular surface corresponding to a domain I-domain II interface, hydrophobic patches on protein surface are generally determinants of proteinprotein or protein-ligand interactions. It also possesses a cluster of aromatic amino acids, Tyr341, Tyr343, Tyr344, and Tyr-348 in the b1-a8 loop and Tyr513 in loop 3; these five aromatic amino acids form a potential binding site with dimensions that could accommodate a short oligosaccharide. The C-terminal domain III (residues 525 to 679), contains two antiparallel b-sheets with Cry4Ba close structural similarity except for some loops exposed to the solvent, in Cry4Aa, the loops connecting b15 to b16 and b19 to b20 are shorter than the loops in Cry4Ba, and the loop connecting b17 to b18 is longer (Boonserm et al., 2006). Mutagenesis experiments suggests that there is direct participation of loop 2 in receptor binding, interestingly, a single substitution within loop 2, replacement of Asn435 by a tyrosine residue, preserved most of the toxicity indicating that the length of the loop rather than its precise sequence is an important determinant of specificity, likewise, replacement of Lys-514 in loop 3 by an Asn residue, as found in Cry4Ba, resulted in an active toxin. Interestingly, loops 2 and 3 are clustered in the vicinity of the b1-a8 loop, close to the interface between domain II and domain I, this region appears to be crucial for receptor binding by Cry4Aa, an event which could trigger conformational changes that lead to disruption of the interface between domains I and II and prime domain I for insertion into the host membrane (Boonserm et al., 2006).
CRY11BB
A Cry11Bb model (residues 15-620) was proposed based on the hypotheses of structural similarity with Cry1Aa and Cry3Aa toxins which contains all the general features of the Cry toxins (an a+b structure with three domains; Gutiérrez et al., 2001). Domain I (residues 15-256), consists of 9 a-helices and two small b-strands. The identified helices and strands are: a1 (Leu19-Leu32); a2a (Ala39-Gln53), a2b (Ile62- Lys73), a3 (Gln79-Phe108), a4 (Phe117-Pro138), a5 (Ser151-Ile168), a6 (Pro178- Arg209), a7a (Leu215-Phe230), a7b (Glu234-Tyr241), b0 (Ala34-Ala36) and b1a (Thr249-Leu252). The most exposed helices are a1, a2a, a2b, a3 and a6; a4 and a5 insert into the membrane in an antiparallel manner as an helical hairpin with their polar sides exposed to the solvent (Gutiérrez et al., 2001). Domain II (residues 257- 478), is formed of three Greek key b-sheets arranged in a b prism topology distributed this way: one helix (a8, Ala279-Ala285) and 11 b-strands (b2, Ser292-Asn305; b3, Pro319-Ser332; b4, Ile341-Lys343; b5, Thr365-Iso369; b6, Val374-Phe381; b7, Trp389-Leu396; b8, Asn401-Arg407; b9, Ile418-420; b10, Pro437-Thr450; b11, Tyr458-Val468 and b12, Phe470-Lys476); the two loops joining the apical b-strands (b2-b3 and b4-b5) located between L307-Y313 and I348-N358 are implicated in receptor binding and specificity; the first insertion between strands b2 and b3, the Nterminal part of this loop (Ile306-Thr311) is mostly hydrophobic, while the C-terminal half (Thr312-Thr318) is polar and have one positively charged residue (Glu316); this loop probably interacts with the receptor through both hydrophobic and electrostatic interactions. Gly315 probably helps in receptor binding by providing more mobility to Glu316 that may interact through salt bridges with the receptor, loop b4-b5 is mostly hydrophilic and the charged residues located at the tip of the loop (Lys353, Asp355 and His356) are probably important determinants of insect specificity (Gutiérrez et al., 2001). Domain III, is characterized by conservation of residues and the only important modification is a 3-residue deletion between b16 and b17; the b-strands in this domain are b13a (Tyr485-Asn490), b3b (Ile495-Ala497), b14 (Ala501-Val503), b15 (Pro513-Ala516), b16 (Ser520-Gly529), b17 (Lys533-Asn543), b18 (Thr546- Arg553), b19 (Lys555-Ala562), b20 (Gly579-Glu583), b21 (Ile592-Leu601) and b22 (Thr608-Val619); the sequence AKYSIRLNTGF is homolog to conserved block 4; in the conserved block 5 there is only one identity and two conserved residues (Ile614, Phe611 and Asp615, respectively), this possibly reveals an alternative mechanism of membrane permeabilization (Gutiérrez et al., 2001).
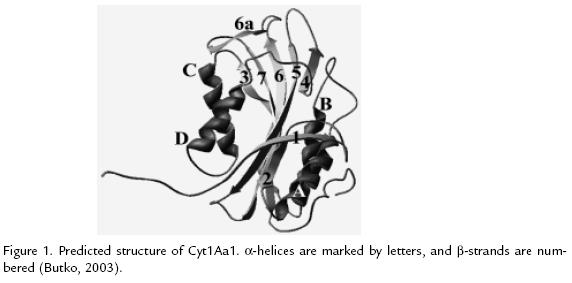
CYT1AA1
This structure was determined by homology modeling (Figure 1 ; Butko, 2003). The toxin consist of two outer a-helix hairpins (helices A-B and C-D) flanking a core of mixed b-sheet (strands 1 to 7); the amino acid sequences of six Cyt proteins from different subspecies of B. thuringiensis were aligned, four conserved blocks: a) helix A (consensus sequence YILQAIQLANAFQGALDP), b) the loop after helix D plus strand 4 (TFTN LNTQKDEAWIFW), c) strands 5 and 6 (TNYYYNVLFAIQNEDTGGVMACVPI GFE), d) strand 6a and the following loop (LFFTIKDSARY; Butko, 2003). The most important part of the protein, responsible for toxicity and lipid binding, comprises the loops at the top of the molecule, charged residues exposed on the surface of proteins are important for electrostatic interactions with polar head groups of lipids; Trp132, Trp154, and Trp157 are essential for folding and activity of this protein (Butko 2003; Promdonkoy et al., 2004)
CYT2AA1 (PDB: 1CBY)
Cyt2Aa1 (before CytB; 259 residues) consists of a single domain protein of a/b architecture, with two outer layers of a-helices and a b-sheet in between; its helical layers consist of hairpins, which can be moved relative to the b-sheet without unravelling the sheet (Li et al., 1996). The N-terminal arm consists of a b-strand and its extensions, which are involved in dimerisation; starting at strand b2 the polypeptide chain forms the three-layered core of Cyt2Aa1; b2 forms one half of an edge strand in the b-sheet, and it is followed by the helix hairpin A-B lying on one face of the sheet, this is connected to the opposite edge strand, b3, and followed by the helix hairpin C-D on the other face of the sheet, after a loop containing a 310 helix, strand _4 completes the edge of sheet which is half formed by b2, but b4 runs in the opposite direction from b2, from then on the polypeptide chain forms a b-meander of b5, b6 and b7 to fill in the middle of the sheet, except a short helix aE inserted in the loop between b6 and b7; then the chain reemerges onto the molecular surface, forming an extended segment and then a small helix aF, and finally a C-terminal tail which crosses helix A on the outside (Li et al., 1996). The six a-helices in the Cyt2Aa1 monomer involve a total of 55 residues (24%) of the polypeptide; there are three 310 helices (N and C termini of aA, and in the loop connecting aD and strand b4). The b-sheet involves 75 residues (33%) of the polypeptide; its helices are not long enough to span the width of the hydrophobic zone in a cell membrane, in contrast the lengths of strands b5, b6, and b7, estimated on the basis of an average rise of 3.3 Å per residue in twisted parallel and antiparallel sheets, are sufficient to span this width; the helices aA, aB, aC and aD all have an amphiphilic character, with hydrophobic residues packed against the b-sheet, and polar and charged residues on the molecular surface, the long strands in the b-sheet also show an amphiphilic character; on the long strands b5, b6 and b7, the residues on the helix AB face are predominantly hydrophilic, while those on the helix C-D face are predominantly hydrophobic (Li et al., 1996). Cyt2Aa1 monomer is in contact with ten neighbours, their extended N-terminal segments stretch to another 2-fold axis where pairs of Cys19 from different dimers are within the distance to form a disulphide link, also it must be linked by hydrogen bonds between interleaved b-strands in the sheet and van der Waals contacts observed between the cysteine residues (Li et al., 1996). Cyt2Aa1 shows no larvicidal or cytolytic activities until processed to the 23 kDa form, extending from Thr34 to Phe237, by insect gut extract or proteinase K (Li et al., 1996). The following events are present during membrane pore formation by Cyt2Aa1: firstly, the proteolytically activated toxin molecules approach the membrane and bind as monomers; secondly, if the segments forming the edge of the b-sheet adjoining the helix pair C-D are to remain external to the membrane, whereas a region near the other end of the sheet is to form the site of initial contact with the membrane, then the toxin would enter the membrane oriented towards the intracellular compartment; thirdly, the amphiphilic helices are unlikely to participate in membrane binding and pore formation, the helices C-D are lifted off the sheet to lie on the surface of the membrane; this would expose the underlying hydrophobic face of the sheet and cause some part of it to partition into the membrane core; lastly, it was thought that the membrane bound toxin molecules oligomerize to form the trans-membrane pore (Li et al., 1996). The higher proportion of unsaturated phospholipids in Dipteran insects can be expected to lead to a greater affinity of the Cyt d-endotoxins for their cell membranes.
26 KDA PROTEIN LIKE PARASPORIN-4 (CRY45AA1; ID PDB 2D42)
Recently, it has also been noted that non-insecticidal B. thuringiensis strains produces novel crystal proteins with cytotoxicity against particular human carcinoma cells; this group has been named parasporins (Akiba et al., 2006; Mizuki et al., 1999; Mizuki et al., 2000). A 26-kDa protein structure, for which no toxicity has been detected but shares a considerable sequence identity with parasporin-4 (Cry45Aa1, cytotoxic against human leukemic T cells), was identified (Akiba et al., 2006). The 26-kDa protein is an unusually elongated molecule dominated by b-strands (67%, S1-12), most of which are remarkably extensive, running all or most of the longer axis of the molecule; the longest strand (S9), comprising 36 residues, runs the whole length of the molecule; three others (S5, S8, S12) span two-thirds of the molecules length (Akiba et al., 2006). The molecule can be divided into three domains: domain I (Ala2-Pro35, Gly176-Pro210), domain II (Ile36-Pro53, Ala81-Asp138, Glu160-Ser175, Gly211-Gln236), and domain III (Val54- Asn80, Phe139-Leu159, Thr237-Ala250). Domain I, is composed of a short N-terminal b-strand (S1), an a/b structure (H1 and S2), a b-hairpin (S9 and S10), and an a-helix (H2); the two a-helices, H1 and H2, are close together and short occupying only 8.9% of the molecule. Domains II and III, are both b-sandwiches: the former is made of a twostranded b-hairpin (S6 and S7) and a curled anti-parallel five-stranded b-sheet including S3, S9, S12, S5, and S8; and the latter is of antiparallel three-stranded and two-stranded b-sheets (S4, S9, and S12; S5 and S8; Akiba et al., 2006). Association of two molecules in the asymmetric unit contact each other at the side of domain III, forming a V shape is mediated mainly by hydrogen bonds between main-chain carbonyl and amide groups in two S5 b-strands, the shape may imply the presence of local twofold symmetry (Akiba et al., 2006). The structure of the 26-kDa protein is distinct from the available structures of B. thuringiensis crystal proteins; unexpectedly, the structure of the 26-kDa protein is remarkably similar to that of e-toxin from Clostridium perfringens, in spite of the rather low sequence identity (22.7%), secondary structures are organized virtually in the same way in the two proteins. e-Toxin is very potent and is responsible for fatal enterotoxemia in livestock, it is considered as a b-pore-forming toxin (b-PFT); the 26-kDa protein is an inactive variant of a certain b-PFT (Akiba et al., 2006). In contrast, there are remarkable differences in the surface distributions of amino acids between the 26-kDa protein and e-toxin: the C. perfringens toxin has a large distinct patch of hydroxylated amino acids (Ser and Thr) on the opposite side of the b-hairpin in domain 2, in addition, the removal of the C-terminal inactivation peptide from the protoxin model exposes a small hydrophobic patch on the surface of domain 3; both of these characteristic patches are supposed to be responsible for the oligomerization of e-toxin within the membrane, which is necessary for pore formation; the 26-kDa protein lacks both features and this could be a reason for the loss of toxicity in the protein (Akiba et al., 2006). A substantial sequence identity (38%) between the 26-kDa protein and a cancer-cell-targeting toxin, parasporin- 4, suggests the structural similarity of the two proteins; hence, parasporin-4 probably has a structure similar to that of e-toxin, the cytopathic effects of parasporin- 4 on leukemic T cells including nuclear decondensation and cell-ballooning are also similar to those observed for e-toxin, thus, parasporin-4 and e-toxin would share a structure and a mode of action as active b-PFTs (Akiba et al., 2006).
CONCLUSION
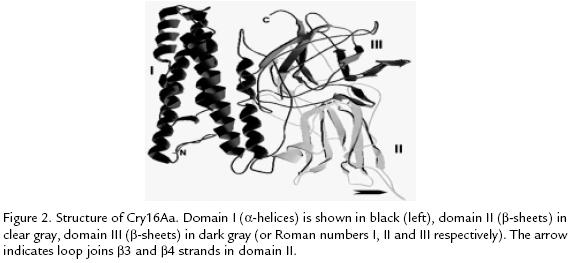
B. thuringiensis d-endotoxins can be considered in three groups, according to their 3D structure: Cry proteins, Cyt proteins and parasporins. A d-endotoxin general model exists for each group with several particular characteristics which are important for specificity and mode of action. The 3D structures are necessary to both understand their functionality and proteins engineering experiments. Here, a Cry16Aa theoretical 3D model (Figure 2), isolated from Clostridium bifermentans subsp. malaysia (Diptera activity; Barloy et al., 1996) was built. Sequence alignment between Cry16Aa, Cry3Aa (28% identity) and Cry4Ba (27% identity) was generated using the structural alignment tool of the program Deep Viewer (http://www.expasy.org/spdbv/; Guex and Peitsch, 1997) and corrected manually until a satisfactory placement of conserved blocks and amino acid identities was obtained. This alignment was submitted to Swiss-Model in the Expasy server (http://www.expasy.ch/spdbv/) and a preliminary model for Cry16Aa was retrieved. The model was optimized with Deep Viewer program and validated with WHAT IF program (http://swift.cmbi.kun.nl/whatif/; Vriend, 1990). Structural comparison of the Cry3Aa and Cry4Ba toxins with the theoretical model of the Cry16Aa protein indicates correspondence to the general model for a Cry protein. N-terminal domain (residues 57-274) is formed by five helix bundle in which the central helix 5 was encircled by four outer helices (a3-a7); the a4-a5 loop may be required for efficient membrane insertion. Domain II, have three antiparallel b-sheets; the C-terminal domain (residues 452-613) is a b-sandwich of two anti-parallel b-sheets; the overlapping with well-known Cry structures showed high similarity with Cry4 proteins maybe by Diptera activity, this suggests a conserved structure according to its specific biological activity. The main difference is in the longest loop joins b3 and b4 strands in the middle domain (Figure 2), this could be a specificity determinant. The conserved blocks I (domain I), II (domains I and II), III and IV (domains II and III) are in the Cry16Aa primary sequence, it corresponds structural and functionally with another Cry proteins (Barloy et al., 1996; Schnepf et al., 1998).
BIBLIOGRAPHY
ANGSUTHANASOMBAT C, UAWITHYA P, LEETACHEWA S, PORNWIROON W, OUNJAI P, KERDCHAROEN T, KATZENMEIER G, PANYIM S. Bacillus thuringiensis Cry4A and Cry4B Mosquito-larvicidal Proteins: Homology-based 3D Model and Implications for Toxin Activity. J Biochem Mol Biol. 2004;37:304-313. [ Links ]
AKIBA T, HIGUCHI K, MIZUKI E, EKINO K, SHIN T, OHBA M, KANAI R, HARATA K. Nontoxic Crystal Protein from Bacillus thuringiensis Demonstrates a Remarkable Structural Similarity to b-pore-Forming Toxins. Proteins. 2006;63:243-248. [ Links ]
BARLOY F, DELÉCLUSE A, NICOLAS L, LECADET M-M. Cloning and Expression of the First anaerobic Toxin Gene from Clostridium bifermentans subsp. malaysia, Encoding a New Mosquitocidal Protein with Homologies to Bacillus thuringiensis Delta-Endotoxins. J Bacteriol. 1996;178:3099-3105. [ Links ]
BOONSERM P, DAVIS P, ELLAR DJ, LI J. Crystal Structure of the Mosquitolarvicidal Toxin Cry4Ba and Its Biological Implications. J Mol Biol 2005;348:363- 382 [ Links ]
BOONSERM P, MO M, ANGSUTHANASOMBAT C, LESCAR J. Structure of the Functional Form of the Mosquito Larvicidal Cry4Aa Toxin from Bacillus thuringiensis at a 2.8-Angstrom Resolution. J Bacteriol. 2006;188:3391-3401 [ Links ]
BUTKO P. Cytolytic Toxin Cyt1A and its Mechanism of Membrane Damage: Date and Hypotheses. Appl Environ Microbiol. 2003;69:2415-2422. [ Links ]
CRICKMORE N, ZEIGLER DR, FEITELSON J, SCHNEPF E, VAN RIE J, LERECLUS D, BAUM J, DEAN DH. Revision of the Nomenclature for the Bacillus thuringiensis Pesticidal Crystal Proteins. Microbiol Mol Biol Rev. 1998;62:807-813. [ Links ]
DE MAAGD RA, BRAVO A, CRICKMORE N. How Bacillus thuringiensis has Evolved Specific Toxins to Colonize the Insect World. Trends Genet. 2001;17:193-199. [ Links ]
GALITSKY N, CODY V, WOJTCZAK A, GHOSH D, LUFT JR. Structure of the Insecticidal Bacterial Delta-Endotoxin Cry3Bb1 of Bacillus thuringiensis. Act Crystallogr. 2001;D57:1101-1109. [ Links ]
GROCHULSKI P, MASSON L, BORISOVA S, PUSZTAI-CAREY1 M, SCHWARTZ JL, BROUSSEAU R, CYGLER M. Bacillus thuringiensis CryIA(a) Insecticidal Toxin: Crystal Structure and Channel Formation. J Mol Biol. 1995;254:447-464. [ Links ]
GUEX N, PEITSCH MC. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis. 1997;18:2714-2723. [ Links ]
LI J, CARROLL J, ELLAR DJ. Crystal Structure of Insecticidal d-endotoxin from Bacillus thuringiensis at 2.5 Å Resolution. Nature. 1991;353:815-821. [ Links ]
LI J, KONI PA, ELLAR DJ. Structure of the Mosquitocidal d-endotoxin CytB from Bacillus thuringiensis sp. Kyushuensis and Implications for Membrane Pore Formation. J Mol Biol. 1996;257:129-152. [ Links ]
MANCEVA SD, PUSZTAI-CAREY M, BUTKO P. Effect of pH and Ionic Strength on the Cytolytic Toxin Cyt1A: A Fluorescence Spectroscopy Study. Biochim Biophys Acta. 2004;1699:123-130. [ Links ]
MIZUKI E, OHBA M, AKAO T, YAMASHITA S, SAITOH H, PARK YS. Unique Activity Associated with Non-Insecticidal Bacillus thuringiensis Parasporal Inclusions: in vitro Cell-Killing Action on Human Cancer Cells. J Appl Microbiol. 1999;86:477 486. [ Links ]
MIZUKI E, PARK YS, SAYITO H, YAMASHITA S, AKAO T, HIGUCHI K, OHBA M. Parasporin, a Human Leukemic Cell-Recognizing Parasporal Protein of Bacillus thuringiensis. Clin Diagn Lab Immunol. 2000;7:625-634 [ Links ]
MORSE RJ, YAMAMOTO T, STROUD RM. Structure of Cry2Aa Suggests an Unexpected Receptor Binding Epitope. Structure. 2001;9:409-417. [ Links ]
PROMDONKOY B, PATHAICHINDACHOTE W, KRITTANAI C, AUDTHO M, CHEWAWIWAT N, PANYIM S. Trp132, Trp154, and Trp157 are Essential for Folding and Activity of a Cyt Toxin from Bacillus thuringiensis. Biochem Biophys Res Commun. 2004;317:744-748. [ Links ]
SCHNEPF E, CRICKMORE N, VAN RIE J, LERECLUS D, BAUM J, FEITELSON J, ZEIGLER DR, DEAN DH. Bacillus thuringiensis and its Pesticidal Crystal Proteins. Microbiol Mol Biol Rev. 1998;62:775-806. [ Links ]
VRIEND G. What if: A Molecular Modeling and Drug Design Program. J Mol Graph. 1990;8:52-56. [ Links ]
XU Z, YAO B, SUN M, YU Z. Protection of Mice Infected with Plasmodium berghei by Bacillus thuringiensis Crystal Proteins. Parasitol Res. 2004;92:53-57. [ Links ]

