










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.13 no.1 Bogotá Jan./Apr. 2008
LATENCIA DEL HERPESVIRUS BOVINO-1: EL PAPEL DE LOS TRANSCRITOS RELACIONADOS CON LATENCIA (RL)
Bovine Herpesvirus-1: The Role of Latency-Related Genes
JULIÁN RUIZ1,2, M.Sc.; JAIRO JAIME2, Ph. D.; VÍCTOR VERA2, Ph. D.
1Estudiante de Doctorado, Facultad de Medicina Veterinaria y de Zootecnia. Universidad Nacional de Colombia, Sede Bogotá.
2Grupo de investigación en Microbiología y Epidemiología Universidad Nacional de Colombia, Sede Bogotá, Carrera 30 No. 45-08 julianruizsaenz@gmail.com
Presentado: 26 de julio de 2007, aceptado 11 de diciembre de 2007, correcciones 9 de febrero de 2008.
RESUMEN
El herpesvirus bovino-1 es un virus de distribución mundial causante de graves pérdidas económicas debidas principalmente a la disminución de la eficiencia y en los indicadores de salud y productividad de cualquier hato ganadero infectado. Luego de la infección inicial del tracto respiratorio de los animales, el virus establece un estado de latencia viral en las neuronas sensoriales del ganglio trigémino y en los centros germinales de las tonsilas faríngeas. Periódicamente, el virus es reactivado y excretado en secreciones a través de las cuales puede infectar a otros animales susceptibles. Durante dicho estado de latencia hay disminución dramática de la expresión de genes virales, llevando solo a la expresión de dos transcritos: El RNA codificado por el gen relacionado con latencia (RL) y el ORF-E viral. Múltiples estudios demuestran como el RL y el ORF-E están involucrados en la regulación del complejo ciclo de latencia y reactivación de la infección. La presente revisión de literatura se enfocará en describir y analizar los distintos estudios que han llevado a dilucidar el papel jugado por el gen RL y el ORF-E, sus transcritos y sus productos proteicos en el establecimiento, mantenimiento y reactivación de la latencia del HVB-1.
Palabras clave: Herpesvirus, latencia, transcritos, ORF.
ABSTRACT
Bovine herpesvirus-1 is a world wide spread virus that causes significant economic losses due mainly to a decrease in the efficiency and in the health and productivity indicators in all the infected herds. After a primary infection of the respiratory tract of the animals, the virus establishes viral latency state in sensory neurons of trigeminal ganglia and germinal centers of pharyngeal tonsils. Periodically, the virus reactivates from latency, is shed through secretions, and can infect other susceptible animals. During latency there is a dramatic reduction of viral gen expression; only two transcripts are abundantly expressed: the latency related (LR) RNA and the viral ORF-E. Multiple studies have shown LR and ORF-E role in the regulation of BHV-1 latencyreactivation cycle. This review focuses on the description and analysis of the litherature that had lead to incriminate LR gene and viral ORF-E, their transcripts and protein products in the establishment, maintenance and reactivation of BHV-1 latency state.
Key words: Herpesvirus, Latency, transcripts, ORF.
INTRODUCCIÓN
El Herpesvirus Bovino-1 (HVB-1) es un virus de genoma DNA perteneciente a la familia Herpesviridae, subfamilia Alfaherpesvirinae (Wyler et al., 1989); este virus posee una amplia diversidad de cepas, las cuales son serológicamente indistinguibles. Sin embargo, con base en el análisis del genoma con endonucleasas de restricción y sus respectivos patrones de migración en gel, se han clasificado en tres tipos y en cinco subtipos: HVB-1.1, HVB-1.2a, HVB-1.2b, HVB-1.3a y HVB-1.3b (Wyler et al., 1995). Posterior-mente, los subtipos HVB-1.3a y HVB-1.3b fueron reclasificados como HVB-5, el cual presenta un alto potencial neuropatogénico (Büchen-Osmond, 1998). El HVB-1 afecta naturalmente al bovino, especie en la que provoca un amplio espectro de manifestaciones clínicas: rinotraqueitis, vulvovaginitis pustular infecciosa, balanopostitis pustular infecciosa, conjuntivitis, aborto, enteritis y encefalitis (Fig. 1)
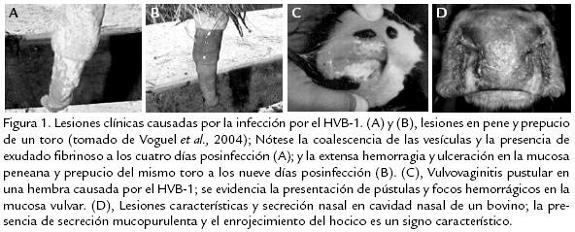
La distribución de la infección es mundial y la prevalencia de reactores serológicos indica que prácticamente todos los bovinos mayores de tres años han estado en contacto con el virus. Se han registrado brotes de la forma respiratoria en EE.UU. y Canadá; la vulvovaginitis pustular infecciosa es común en Europa y se han notificado brotes importantes de meningoencefalitis en Argentina, Australia, EE.UU., Canadá, Siria, Hungría, Uruguay, Italia y Brasil (Pidone et al., 1999). El HVB-1 se transmite fácilmente en forma directa de un animal a otro, pues gran cantidad de virus es eliminado principalmente en las secreciones respiratorias, oculares y reproductivas de animales infectados; sin embargo, la transmisión también puede hacerse en forma indirecta, a través de personas o equipos. El periodo de incubación varía entre dos y seis días, dicha variabilidad depende de la cantidad del inóculo, ruta de inoculación y otros factores inherentes del hospedero (Wyler et al., 1995). Aunque se acepta que, para el caso de la rinotraqueitis, el virus se excreta en las secreciones nasales por aproximadamente 12-14 días posinfección, se ha recuperado el virus en forma intermitente por un periodo de hasta 578 días (Jubb, 1993). Otras fuentes importantes de diseminación son el semen y la transferencia embrionaria (Bielanski et al.,1994). Se ha podido comprobar que toros serológicamente positivos sin sintomatología son portadores del virus, el cual se ha logrado aislar de muestras de semen congelado, incluso 12 meses después de su almacenamiento a -196 ºC (Bielanski et al., 1988; van Oirschot et al., 1993). El riesgo de transmisión de HVB-1 en hembras fertilizadas con semen de toros seropositivos proveniente de centros de inseminación artificial no se considera muy alto; sin embargo el tratamiento del semen infectado con gammaglobulinas de suero hiperinmune o con una solución de tripsina (Bielanski et al., 1988) puede reducir el riesgo de transmisión de HVB-1 en la inseminación artificial. El bovino es el principal reservorio del HVB-1; sin embargo, muchas especies de rumiantes tales como: caprinos, ovinos, etc., e incluso el cerdo, son susceptibles a la infección por este virus (Wyler et al., 1989). En Colombia, se han realizado diferentes estudios seroepidemiológicos, los cuales además de demostrar el alto impacto económico y epidemiológico ocasionado por la infección, han permitido el aislamiento de algunas cepas nativas, las primeras a comienzos de los años 70 (CIAT, 1972; CIAT, 1975); otra a mitad de los años 90 a partir de muestras de tomadas en los Llanos orientales (Molano et al., 1996); y más recientemente una aislada en 2001 en la Sabana de Bogotá (Piedrahita et al., 2005). Estas dos últimas han sido sujeto de caracterización molecular por el Grupo de Microbiología y Epidemiología de la Universidad Nacional, clasificando los aislamientos como HVB-1.1 a la cepa de los Llanos orientales y HVB-1.2a para la cepa de la sabana de Bogotá (Piedrahita et al., 2005).
CICLO DE REPLICACIÓN VIRAL
El HVB-1 entra a las células por fusión de su envoltura con la membrana plasmática celular, en un proceso independiente de pH y dependiente de la unión de glicoproteínas virales presentes en la envoltura del virus con los receptores en la membrana celular (Wittels y Spear, 1990). Diversos estudios han demostrado que las diferentes glicoproteínas virales (gB, gC, gD, gE, gH, gK y gL) son requeridas en el proceso de entrada del virus a la célula (Li et al.,1995; Schroder y Keil, 1999; Dasika y Letchworth, 1999; Hanon et al., 1999; Dasika y Letchworth, 2000). Los HVB-1.1 y HVB-1.2 difieren en algunos epítopes de la gC; dichos cambios pueden alterar la adhesión viral y pueden ocasionar diferencias en la virulencia del subtipo viral (Rijsewijk et al., 1999). Aunque la totalidad de las moléculas involucradas en la entrada del HVB-1 a la célula no están completamente dilucidadas, está claramente establecido que el virus se une a las moléculas de heparán sulfato presentes en la membrana celular (Hanon et al., 1998; Tyler y Nathanson, 2001) a través de las glicoproteínas virales gB y gC (Li et al., 1996). Luego de esta unión inicial, las gB y gD se unen con alta afinidad a sus receptores putativos en la membrana celular (Li et al., 1995). Estudios más recientes para identificar estos receptores de alta afinidad, han mostrado que la gD de HVB-1 puede unirse débilmente con el HveC (receptor 1 de nectina) o con el receptor humano para el poliovirus (PVR) expresados en células de humano o de hámster (Geraghty et al.,1998; Connolly et al., 2001). Esta unión débil, indica que la entrada del HVB-1 en el hospedero bovino puede estar mediada por la interacción de la gD con receptores diferentes a los homólogos bovinos del HveC o PVR (Geraghty et al., 1998; Connolly et al., 2001).
Después de la entrada a las células del hospedero, el HVB-1 es transportado a través de los microtúbulos hasta el núcleo; donde de manera ordenada y secuencial replica su ge-noma usando proteínas virales y celulares; dependiendo del momento en que los genes virales son expresados durante la replicación son clasificados en tres clases: inmediatos tempranos, tempranos y tardíos (Roizman y Pellet, 2001). Luego del ensamblaje, los viriones abandonan el núcleo y se dirigen hacia la membrana citoplasmática externa por procesos de gemación a partir de membranas intracitoplasmáticas, adquiriendo su envoltura y tegumento. Luego son transportados dentro de vesículas intracelulares a la membrana citoplasmática para su posterior liberación de la célula (Hunter, 2001). La replicación del genoma del HVB-1 comienza dentro de las dos horas siguientes a la infección inicial del animal (Meurens et al., 2004) con la expresión de antígenos en la superficie celular entre las tres a cuatro horas siguientes a la infección y liberación de partículas virales y dispersión a las ocho horas posinfección (Babiuk et al., 1996; Fig. 2).
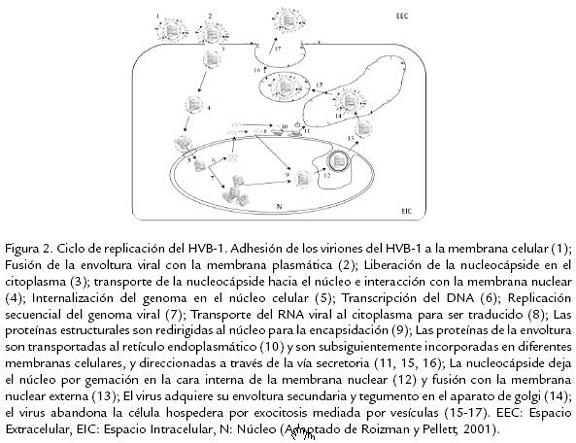
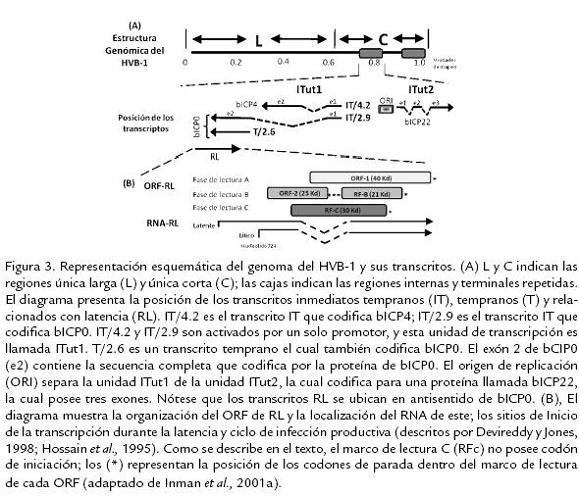
La infección inicial del HVB-1 en líneas celulares permisivas (Células MDBK) conduce primordialmente a una rápida muerte celular ocasionada por un estado apoptótico dado por la activación de diferentes caspasas celulares (principalmente caspasa 3) y de la proteína P53 (Devireddy y Jones, 1999; Henderson et al., 2004b). Resultados similares han sido reportados ante la infección in vivo de terneros con la cepa Cooper de HVB-1, llevando a un aumento de los niveles de apoptosis en linfocitos T CD4+ infectados (Winkler et al., 1999). Durante la expresión génica en la infección primaria, los genes son transcritos en una cascada coordinada, regulada y secuencial, la cual es común a todos los miembros de esta familia viral (Wirth et al., 1989; Roizman y Pellet, 2001); y dependiendo del momento en que los genes son expresados durante la replicación son clasificados en tres clases: Inmediatos tempranos (IT), tempranos y tardíos. La expresión de los genes IT es regulada por un componente del virión, α-TIF, el cual es un transactivador viral de los genes α o IT (Misra et al., 1995). Adicionalmente, se ha reportado la existencia de dos unidades de transcripción de genes IT; la primera (ITut1) codifica dos homólogos funcionales de los genes IT del Herpesvirus Simplex tipo 1 (HSV-1), denominados ICP0 e ICP4. Por otra parte, la segunda unidad de transcripción (ITut2) codifica una proteína homologa a la IT ICP22 de HSV-1 (Wirth et al., 1991). Subsecuentemente, las proteínas IT activan la expresión de los genes tempranos, asegurando así la replicación del ADN viral. Las proteínas tardías son generalmente sintetizadas en las últimas fases del ciclo de replicación viral; se conoce que bICP0, el principal gen IT, está involucrado en la activación de genes tardíos, llevando al ensamblaje y liberación de los nuevos viriones. bICP0 es importante para la infección, ya que se ha demostrado que es expresado en altos niveles durante la fase productiva y que tiene la capacidad de activar transcripcionalmente todos los demás promotores virales (Fraefel et al., 1994; Wirth et al., 1992); además, aunque su expresión, no es requerida para la infección en cultivos celulares, estudios realizados usando un mutante negativo de bICP0 demuestran que éste es necesario para una eficiente replicación y formación de placas (Geiser et al., 2005; Fig. 3).
LATENCIA Y REACTIVACIÓN
El herpesvirus bovino, al igual que la gran mayoría de miembros de la subfamilia Alfaherpesvirinae, establece latencia durante toda la vida del hospedero en neuronas ganglionares del sistema nervioso periférico. Luego de la replicación inicial en el epitelio mucoso, la nucleocápside del virus es direccionada por transporte axonal retrógrado hasta el cuerpo de la neurona sensorial dentro del ganglio trigémino, allí el ADN es insertado en el núcleo de la neurona; entrando en un estado de transcripción restringida, el cual le permite evadir la respuesta inmune del hospedero (Fig. 4). Tiempo después, durante eventos de estrés, inmunosupresión o uso prolongado de corticoides, el virus puede reactivarse, produciendo partículas virales, las cuales migran en forma centrífuga usando el transporte axonal anterógrado de la misma neurona por la cual ascendieron, alcanzando el sitio inicial de replicación, para poder así dispersarse a otros animales susceptibles (Sheffy y Davies, 1972; Rock et al., 1992). El sitio primario de establecimiento de latencia son las neuronas sensoriales del ganglio trigémino; sin embargo, existe evidencia que demuestra la persistencia y reactivación viral también dentro de los centros germinales de las tonsilas faríngeas, teniendo este sitio un papel importante, tanto en la patogénesis como en la transmisión del virus a nuevos hospederos (Winkler et al., 2000a). Así mismo, se ha evidenciado infección latente en mononucleares de sangre periférica (Fuchs et al., 1999), en nódulos linfoides y tejidos esplénicos, aun en ausencia de infección productiva (Mweene et al., 1996).
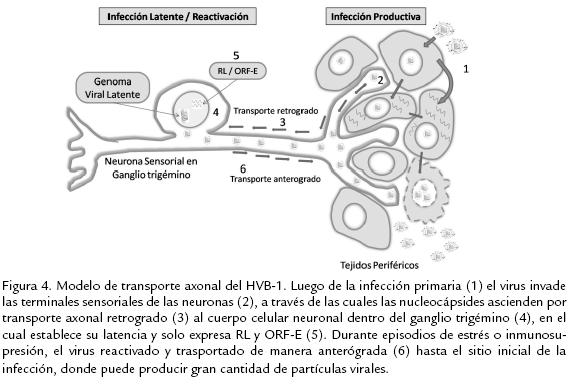
Recientemente, Voguel et al. (2004) demostraron la presencia de infección latente en la raíz dorsal de ganglios nerviosos sacros, en plexo simpático pélvico y en nódulos linfoides locales luego de la inoculación intraprepucial de toros con una cepa de campo del subtipo HVB-1.2 (Voguel et al., 2004). Los pasos del ciclo de latencia y reactivación han sido operacionalmente divididos en tres pasos mayores: establecimiento, mantenimiento y reactivación. El establecimiento del estado de latencia involucra la entrada de la cápside viral en una neurona sensorial, el posterior acceso del genoma al núcleo y la infección aguda; luego, la expresión de genes virales es suspendida o disminuida drásticamente, y en lugar de los 80 genes virales que se expresan durante la infección lítica, en la fase latente solo se da la expresión de dos transcritos virales: un transcrito relacionado con latencia (RL) y el ORF E, que son abundantemente expresados en ganglios trigéminos durante la latencia (Bratanich y Jones, 1992).
El mantenimiento de la latencia es un paso que puede durar toda la vida del hospedero; técnicamente, se define como un periodo durante el cual no es posible detectar virus infecciosos mediante los procedimientos estándar de aislamiento viral, y en general tampoco hay expresión de los genes virales necesarios para el establecimiento de una infección productiva (genes de ciclo lítico). Los mensajeros RL son los únicos transcritos virales que son expresados abundantemente durante esta fase, y la evidencia demuestra que la abundante expresión de RNA de RL en neuronas sensoriales potencialmente inhibe bICP0, bloqueando así la infección productiva, promoviendo el establecimiento y mantenimiento de la latencia (Geiser et al., 2002). La reactivación del estado de latencia es iniciada principalmente por estímulos externos (estrés o inmunosupresión, glucocorticoides, etc.) los cuales favorecen la expresión de genes IT virales; por tanto después de la reactivación viral es posible evidenciar la expresión de genes virales en neuronas sensoriales infectadas y aislar virus infecciosos de ganglio trigémino y secreciones nasales y/o oculares (Jones, 2003). Estudios realizados con el Herpesvirus Simplex tipo 1 (HSV-1), han permitido determinar que la reactivación del estado de latencia inducida por el tratamiento con glucocorticoides (dexametasona) está ocasionada por un mecanismo indirecto de supresión de la función de neutrófilos y linfocitos, llevando a la reexpresión de los genes virales de ciclo lítico (Schwyzer y Ackerman, 1996). Usando la técnica de microarreglos, se ha reportado un aumento de la expresión de genes tales como: el receptor de la prostaglandina E2, la ciclina D2, el factor de transcripción AP1 y la proteína kinasa activada por mitógenos (MAP-K) en ganglios trigéminos de ratones infectados por el HSV-1 y tratados con dexametasona; postulando así, un papel importante de estos genes en la reactivación viral inducida por corticoides (Higaki et al., 2002).
EL GEN RL EN EL CICLO DE LATENCIA Y REACTIVACIÓNEl RNA del RL es expresado abundantemente en neuronas sensoriales durante la infección latente (Kutish et al., 1990) y su secuencia es transcrita en dirección antisentido con respecto a la secuencia de los genes IT y tempranos que codifican el gen bICP0 (Bratanich y Jones, 1992). El gen RL tiene dos marcos abiertos de lectura (ORF1 y ORF2) y dos marcos de lectura (RF-B y RF-C) los cuales carecen del codón de iniciación (metionina). Estudios realizados utilizando RT-PCR del RNA del RL indican que este es madurado en la región C terminal del ORF2 (Hossain et al., 1995). Una región del RNA del RL es poliadenilada (Hossain et al., 1995) y estudios realizados por Devireddy y Jones (1998) demostraron que esta región recibe una maduración alternativa (splicing alternativo), la cual resulta en la presentación de isoformas proteicas con funciones biológicas diversas y especializadas, las cuales podrían ser importantes durante la latencia (Devireddy y Jones, 1998); adicionalmente un anticuerpo dirigido contra la región N terminal del ORF-2 reconoce una proteína codificada por el gen RL (Jiang et al., 1998; Jiang et al., 2004; Fig. 3). Para evaluar la función de estos transcritos alternativos, Devireddy et al. (2003) clonaron un cDNA (a partir del RNA completo) identificado en ganglio trigémino de un bovino a los siete días posinfección y compararon este cDNA con el gen RL. Como resultado de la maduración alternativa, el cDNA contiene un nuevo ORF el cual posee parte del ORF2 fusionado con el ORF1 viral y la sobreexpresión de este nuevo ORF inhibe el promotor de la unidad de transcripción IT (ITut1) del HVB-1, además el producto de este ORF estimula el promotor de RL en células transitoriamente transfectadas, reduciendo así la expresión de genes IT durante el establecimiento y posible mantenimiento de la latencia (Devireddy et al., 2003). Hossain et al. (1995) demostraron que el gen RL codifica una proteína de aproximadamente 41 kDa, la cual en estudios de inmunoprecipitación, se ha visto que se asocia con complejos de ciclinas dependientes de quinasas (cdk2) promoviendo así la supervivencia celular (Jiang et al., 1998). De forma similar, estudios de inmunoprecipitación y estudios in vivo en conejos infectados con el HVB-1, demostraron que los productos proteicos del gen RL interactúan también con la ciclina A, inhibiendo la entrada de la célula infectada a fase "S" aumentando así la supervivencia de las células infectadas (Schang et al., 1996). Ciacci-Zanella et al. (1999) reportaron que los productos del gen RL inhiben o disminuyen considerablemente la muerte celular programada inducida químicamente (tratamiento con Fumonisina B1 o C6-ceramida) en cultivos de células de neuroblastoma (Neuro-2A), células de riñón de mono y fibroblastos humanos transfectados transitoriamente con este gen (Ciacci-Zanella et al., 1999), sugiriendo que el RNA de RL y el producto proteico de este gen actúan en conjunto inhibiendo la expresión de genes de ciclo lítico y disminuyendo la muerte neuronal durante el establecimiento y mantenimiento de la latencia.
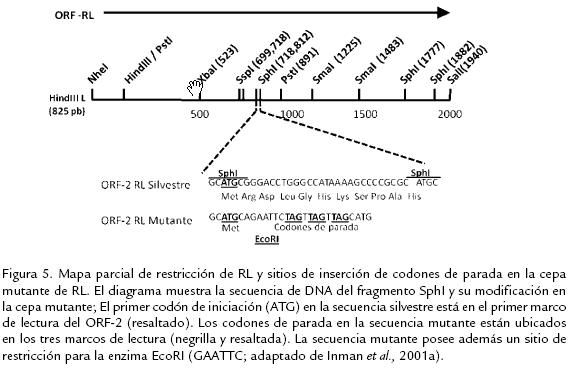
Estudios de cotransfección de bICP0 y ADN genómico de HVB-1 en células bovinas, han mostrado que bICP0 induce la infección lítica del HVB-1 y que la capacidad de bICP0 para activar la expresión génica se correlaciona con la estimulación de dicha fase de la infección (Inman et al., 2001b). Con el objetivo de evaluar el papel del producto del gen RL sobre la infección productiva, Geiser et al. (2002) realizaron estudios, usando plásmidos que contenían el gen completo de RL, bICP0 y ADN genómico de HVB-1 cotransfectando células epidermales bovinas. Los autores encontraron que el gen RL inhibía la capacidad de bICP0 para estimular la infección productiva en una manera dosis dependiente. Interesantemente, la inserción de un codón de parada en la región animo terminal del ORF1 no disminuía la represión sobre bICP0, indicando que no es requerida la expresión de la proteína de RL para inhibir la función de bICP0 (Geiser et al., 2002). Un estudio más reciente de este RNA llevó a la introducción de un codón de parada en el fragmento XbaI-PstI, el cual tampoco tuvo efecto represor sobre bICP0; además se encontró que la expresión del RNA de RL tipo silvestre y mutado (con codones de parada) se correlaciona de manera positiva con la inhibición de la replicación viral en fibroblastos bovinos y en células de Neuro-2A (Geiser y Jones, 2005). Teniendo en cuenta que el gen RL se transcribe en antisentido de bICP0 este resultado permite postular la existencia de un sistema de silenciamiento del RNA, el cual no ha sido completamente dilucidado hasta el momento. Para evaluar el papel de la expresión de las proteínas RL en la infección viral en un modelo in vivo, Inman et al. (2001a), desarrollaron una cepa de HVB-1 la cual contiene tres codones de parada cerca al comienzo del ORF2 del gen RL (Fig. 5). Los terneros infectados con dicha cepa mutante de RL presentaron menor sintomatología clínica y disminución de la excreción de partículas virales infecciosas en secreciones oculares comparados con terneros infectados con la cepa silvestre y con una cepa mutante en la cual se revirtió dicha mutación. Usando hibridación in situ, también se demostró una mayor cantidad de genoma viral en ganglio trigémino y tonsilas en individuos infectados con la cepa silvestre; hallazgo que fue corroborado al demostrar la presencia del RNA de RL usando RT-PCR semicuantitativo (Inman et al., 2001a; Inman et al., 2002; Perez et al., 2005). Contradictoriamente, aunque el mutante RL posee características similares de crecimiento en células MDBK infectadas y en cavidad nasal de los terneros durante la infección aguda, los virus mutantes RL no reactivan del estado de latencia luego del tratamiento inicial con dexametasona (100 mg), ni luego de múltiples dosis de dexametasona (25 mg dos y cuatro días después de la primera dosis); dicha reactivación si se evidenció en los terneros infectados, tanto con la cepas silvestre como con la cepa en la cual se revirtió la mutación (Inman et al., 2001a; Inman et al., 2002), sugiriendo de este modo un papel importante del producto del gen RL en el establecimiento y/o mantenimiento del estado de latencia viral. Sin embargo, aunque las cepas con la mutante RL no pueden reactivar del estado de latencia luego del tratamiento con dexametasona en animales infectados, dichas cepas mutantes pueden reactivarse cuando el tejido de la tonsila es sometido a explante. Este tipo de reactivación ex vivo, se caracteriza porque la respuesta inmune del individuo no participa como limitante del crecimiento y dispersión viral. Estos resultados sugieren que el producto del gen RL interfiere con el sistema inmune del hospedero, evitando el reconocimiento de las células infectadas que están entrando en reactivación (Perez et al., 2005). Teniendo en cuenta que los RL favorecen la supervivencia celular por distintas rutas celulares, Lovato et al. (2003) usando la técnica de TUNEL (Terminal dUTP Nick End Labelling) y usando anticuerpos que reconocen el clivaje de la caspasa 3, evaluaron la presencia de apoptosis en neuronas del ganglio trigémino de animales infectados con la cepa mutante de RL y con una cepa silvestre. Luego de 14 días de infección, se encontró que los animales infectados con la cepa mutante presentaron un mayor número de células TUNEL+ y mayor clivaje de caspasa 3 en neuronas trigeminales, comparados con los terneros infectados con la cepa silvestre. Por otro lado, animales infectados con la cepa silvestre tuvieron mayores niveles de DNA viral en ganglio trigémino durante la infección aguda que los infectados con la cepa mutante, confirmando que las propiedades antiapoptóticas del producto de los genes RL inhiben la muerte celular y consecuentemente promueven el establecimiento de la latencia (Lovato et al., 2003). Henderson et al. (2004a) utilizando un sistema de infección in vitro en células Neuro-2A demostraron que el producto del gen RL inhibe el clivaje de las caspasas 3 y 9 durante la infección productiva, sugiriendo que durante ésta fase de la infección se da una regulación negativa de las propiedades antiapoptóticas del producto del gen RL (Henderson et al., 2004a).
Estudios recientes desarrollados por Meyer et al. (2007) usando un sistema doble híbrido bacteriano permitieron identificar que una proteína de fusión RL interactúa con dos proteínas que pueden inducir apoptosis: Bid, la cual une la vía extrínseca con la vía intrínseca de apoptosis y con Cdc42 un miembro de la familia de las Rho-GTPasas, el cual induce apoptosis celular bajo condiciones de estrés, sugiriendo que esta interacción con RL disminuye la muerte celular inducida por vía citotóxica en neuronas infectadas y por el aumento del estrés celular inducido por la infección latente. Adicionalmente, se demostró que la proteína de RL interactúa con C/EBP-α el cual es un potente factor transcripcional encargado de la regulación del ciclo celular y de la diferenciación. Durante la infección productiva, hay una correlación positiva entre la expresión de C/EBP-α y la expresión de genes virales en ganglio trigémino a los cuatro y siete días posinfección, tiempo para el cual la expresión de genes y partículas virales es detectada; además, C/EBP-α aumenta la formación de placas en cultivos celulares, sugiriendo que C/EBP-α aumenta la activación de genes del HVB-1 directa o indirectamente. Por tanto, además de inhibir la apoptosis, la interacción de la proteína de RL con C/EBP-α reprime la infección productiva, promoviendo el establecimiento de latencia (Meyer et al., 2007).
El gen RL del HVB-1 comparte algunas propiedades tanto estructurales como funcionales con los transcritos asociados a latencia (LATs) del HSV-1; entre ellas está que ambos son transcritos en una secuencia antisentido de bICP0 e ICP0, y ambos son abundantemente expresados durante la latencia (Devireddy y Jones, 1998; Hossain et al., 1995; Kutish et al., 1990; Schang y Jones, 1997; Winkler et al., 2000b); además se sabe que al igual que el RL los LATs tienen una función antiapoptótica importante (Branco y Fraser, 2005), y en general los dos transcritos cumplen funciones importantes en el ciclo de latencia y reactivación viral. Sin embargo, a diferencia del RL del HVB-1, hasta la fecha se considera que los LATs de HSV-1 no codifican ninguna proteína.
PAPEL DEL ORF-E EN LA INFECCIÓN LATENTEEl ORF-E es un marco de lectura pequeño que está presente dentro del promotor del RL y codifica para 135 aminoácidos; es un transcrito en dirección antisentido del transcrito RL y se ubica corriente abajo del gen que codifica el activador transcripcional bICP0 pero no lo sobrelapa. El promotor RL contiene múltiples motivos que actúan en cis y tiene un dominio "de unión neuronal" (NBS) que reconoce factores nucleares exclusivamente neuronales (Jones et al., 1990; Bratanich y Jones, 1992; Delhon y Jones, 1997; Fig. 6).
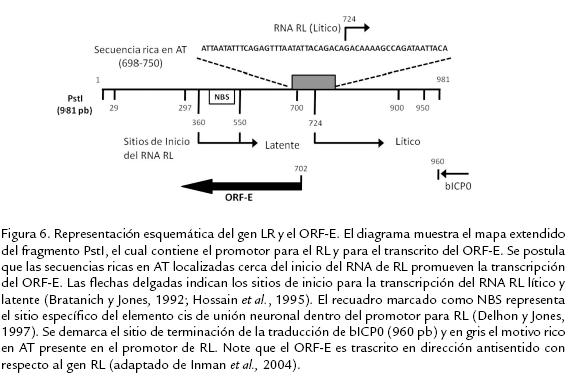
Usando RT-PCR se demostró la presencia de un transcrito poliadenilado codificado por el ORF-E durante la infección latente en ganglio trigémino de animales, en cultivos celulares con infección productiva y en animales con infección aguda. Luego, la secuencia codificante del ORF-E fue clonada en un vector y se expresó como una proteína de fusión unida a la proteína verde fluorescente (GFP). La proteína de fusión ORF-E/GFP se expresó principalmente en el núcleo de células neuronales de ratón o humanas transfectadas; en contraste, esta proteína de fusión se expresa tanto en núcleo como en citoplasma de células no neuronales (Inman et al., 2004). Estos estudios indican que un transcrito del ORF-E es consistentemente expresado durante la latencia; además permiten postular que el producto génico codificado por el ORF-E regula en algún aspecto el ciclo de latencia y reactivación. Recientemente, Perez et al. (2007) desarrollaron un antisuero policlonal dirigido contra la proteína codificada por el ORF-E; dicho antisuero permitió detectar la presencia de la proteína del ORF-E en células Neuro-2A transfectadas y adicionalmente se encontró que la transfección estable, con un plásmido que expresaba el ORF-E en células Neuro-2A, inducía cambios morfológicos principalmente dados por la presencia de proyecciones neuríticas, las cuales no fueron observadas en las mismas células transfectadas con el vector vacío. El antisuero también permitió detectar la presencia de la proteína del ORF-E en el núcleo de neuronas sensoriales de ganglios trigéminos de terneros latentemente infectados; sin embargo, luego de la reactivación con dexametasona no fue posible detectar la presencia de dicha proteína, llevando a los autores a postular que una proteína codificada por el ORF-E tiene el potencial de alterar la fisiología o el metabolismo de células neuronales lo cual cobra gran importancia en el fenómeno de latencia a largo término (Perez et al., 2007). Actualmente los autores se encuentran desarrollando cepas mutante con deleciones del ORF-E y del RL/ORF-E para poder esclarecer el papel de dichos ORFs y sus productos en el ciclo de latencia y reactivación del HVB-1.
LA RESPUESTA INMUNE Y LA LATENCIA DEL HVB-1La presencia de infiltrado linfoide en estrecha asociación con neuronas sensoriales en ganglios trigéminos de animales con infección latente, sugiere la participación del sis-tema inmune del individuo, promoviendo el establecimiento de la latencia probable-mente por inducción de un ambiente no permisivo para la replicación viral. Estudios in vivo demuestran que luego de que se establece la infección latente en terneros, es común encontrar focos de infiltrados linfocíticos en ganglios trigéminos (Inman et al., 2001a; Perez et al., 2005); lo cual ha sido corroborado, usando tinción con hematoxilina-eosina demostrando la presencia de una respuesta inmune celular persistente en ganglios trigéminos de estos individuos (Winkler et al., 2002). Se ha postulado que la persistencia de la inmunidad celular es debida a la expresión de proteínas virales en algunas neuronas o en células satélites del ganglio trigémino, en las cuales debido a la mínima tasa de replicación, es común encontrar la presencia de focos de infiltrados leucocitarios. Esta mínima tasa de transcripción se ha denominado "reactivación molecular espontánea" (Feldman et al., 2002), y resultados de estudios in vivo, permiten postular la existencia de este fenómeno durante la latencia por HVB-1 (Winkler et al., 2002; Jones et al., 2006). Durante la persistencia del sistema inmune en el ganglio trigémino, la expresión de citoquinas puede influir de manera drástica promoviendo tanto la supervivencia como la muerte neuronal, llevando a prevenir la reactivación de la infección en las neuronas infectadas. Algunos estudios de infección por HSV-1 han demostrado que Interferón-α (IFN-α) e IFN-γ juegan un papel importante previniendo la reactivación y controlando la presentación de lesiones herpéticas recurrentes (Mikloska y Cunningham, 2001); estudios realizados por Winkler et al., (2002) reportaron la presencia de IFN-γ e interleuquina 2 en ganglios trigéminos durante la infección latente por HVB-1 en bovinos. Estos resultados indican que la relación virus-huésped puede tener múltiples funciones y factores virales determinantes durante el ciclo de latencia y reactivación viral (Winkler et al., 2002). A diferencia de la regulación negativa del sistema inmune inducida por los LATs de HSV-1, la cual evita la apoptosis de células neuronales infectadas (inducida por las células T CD8+; Branco y Fraser, 2005), hasta la fecha no se ha dilucidado completamente el papel del gen RL y el ORF-E, sus transcritos o sus productos génicos en la regulación de la infiltración leucocitaria y la muerte de células infectadas mediada por el sistema inmune. Recientemente, Perez et al. (2006) usando tinción con hema-toxilina-eosina evaluaron las diferencias en la infiltración de células inflamatorias en ganglios trigéminos de animales infectados con un virus tipo silvestre y un virus con una mutación en el gen RL (codón de parada mencionado anteriormente), encontrando un mayor infiltrado inflamatorio en los ganglios de animales infectados con la cepa silvestre, comparados con los infectados con la cepa RL mutante. Además, usando PCR se encontró una menor cantodad de DNA viral en ganglios de animales infectados con la cepa RL mutante. Estos resultados sugieren que la infiltración de células inflamatorias es el resultado de la replicación y la expresión de genes virales en el ganglio trigémino de animales infectados (Perez et al., 2006). Tomando en cuenta éstos resultados y sabiendo que la infección por la cepa RL mutante induce un aumento de la apoptosis (Lovato et al., 2003), los autores sugieren que el aumento de la muerte celular inducida por el virus mutante lleva a un aumento de la respuesta inflamatoria al término de la infección aguda; consecuentemente, el número de neuronas infectadas se reduce, ocasionando una disminución considerable de la cantidad de DNA viral en ganglios trigéminos y una dramática disminución en la frecuencia de reactivación viral del estado de latencia. Sin embargo, aunque hay una asociación entre la presencia del gen RL y el infiltrado inflamatorio, los estudios actuales no permiten concluir cual es el mecanismo por el cual el gen RL o sus productos regulan la infiltración leucocitaria (Perez et al., 2006).
ESTABLECIMIENTO DE LATENCIA POSTVACUNACIÓNEn la profilaxis y el control de la infección por el HVB-1, el uso de vacunas de virus vivo atenuado se ha constituido en uno de los principales factores de riesgo para el establecimiento de portadores en estado latente. En un estudio realizado por Castrucci et al. (2002) se evaluó el potencial de cinco vacunas comerciales de virus vivo para establecer infección latente en individuos posvacunación y reactivar luego del tratamiento con dexametasona (0,1 mg/kg, vía intravenosa durante cinco días). En este estudio fue posible aislar el virus vacunal en tres de las cinco vacunas estudiadas, preparadas a partir de virus vivo modificado; sin embargo, ninguno de los animales presentó signos clínicos de la infección luego de la reactivación experimental, o luego de un nuevo reto experimental con una cepa de campo. Además, usando análisis de polimorfismos de restricción, los autores encontraron que los virus reexcretados diferían ligeramente de la vacunal parental posiblemente debido a un fenómeno de recombinación endógena del virus (Castrucci et al., 2002). El uso de vacunas vivas marcadoras (con deleción en glicoproteínas, por ejemplo la gE) se ha extendido en el ámbito internacional, principalmente en países en vía de erradicación de la enfermedad. Sin embargo, múltiples estudios han mostrado que dichas cepas vacunales pueden generar latencia en individuos vacunados (Kaashoek et al., 1998; Mars et al., 2001; Castrucci et al., 2002), generarando portadores virales latentes seronegativos (Lemaire et al., 2001) y luego de procesos de recombinación intraespecífica con cepas silvestres del virus se pueden generar cepas que in vivo, poseen una virulencia mayor que sus cepas parentales vacunales, demostrando que estos fenómenos de recombinación pueden restaurar la virulencia de las cepas vacunales gE negativas (Muylkens et al., 2006). De igual forma, vacunas atenuadas termosensibles pueden generar infección latente (Hage et al., 1998). Estudios realizados por Lemaire et al. (2000a) demuestran que es posible generar infección latente luego de la administración de una cepa vacunal en individuos inmunizados pasivamente o con anticuerpos maternales. Además, estos autores reportaron que estos individuos pueden reactivar el virus y ser fuente de infección para individuos seronegativos (Lemaire et al., 2000b). Teniendo en cuenta que toda vacuna de virus vivo es susceptible de inducir estados de latencia, países con programas de erradicación han introducido el uso de vacunas recombinantes basadas en subunidades virales, principalmente usando la gD viral, la cual es altamente inmunogénica y siendo solo proteína no es factible el desarrollo de infección y latencia (van Drunen Littel-van den Hurk, 2006).
CONCLUSIONESEl fenómeno de latencia de la infección por el HVB-1 es el punto más crítico en los planes de prevención, control y erradicación de la infección, ya que la biología propia del virus le permite evadir la respuesta inmune del individuo, permanecer durante toda la vida del animal y reactivar en algunas situaciones para alcanzar nuevos hospederos. Este fenómeno de latencia se desarrolla principalmente en el ganglio trigémino de los individuos luego de la primoinfección. En estos ganglios infectados hay una expresión abundante de los genes RL y ORF-E, los cuales se postulan como los encargados de regular el complejo ciclo de latencia y reactivación de la infección. Basados en el conocimiento sobre los genes implicados en este proceso, Jones et al., (2006) plantearon el siguiente modelo de cómo estos genes regulan el ciclo de reactivación y latencia en el hospedero (Fig. 7).
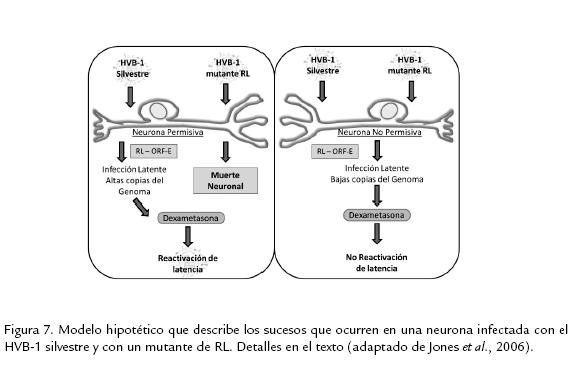
Cuando un virus mutante de RL infecta una neurona que tiene todos los factores celulares necesarios para apoyar la amplificación del genoma y la expresión de genes virales (neurona permisiva), frecuentemente, esta neurona muere por apoptosis inducida por el daño viral y/o por citotoxicidad mediada por linfocitos. Dicha neurona permisiva, tiene mayor probabilidad de sobrevivir si la infección ocurre por una cepa silvestre del HVB-1 debido a la acción reguladora de las proteínas y quizás de los RNAs expresados por el gen RL y posiblemente el ORF-E. Luego de la interacción con esta neurona permisiva se establece una latencia en la cual es posible encontrar alto número de copias de genoma viral en ellas, y las cuales pueden reactivar de su estado de latencia luego del tratamiento con dexametasona. Cuando un HVB-1 o una cepa mutante de RL infectan una neurona "no permisiva", no se genera una amplificación genómica eficiente, ni expresión génica; consecutivamente, no se da el daño neuronal y se establece el estado de latencia, aun en ausencia de expresión de las proteínas de RL. Contrario a lo que ocurre en la neurona permisiva, luego del tratamiento con dexametasona, no se da la reactivación de la infección latente en la neurona no permisiva, debido a los bajos niveles de DNA presentes y a que el ambiente neuronal no soporta la expresión de genes necesarios para que se de la reactivación. Aunque los productos proteicos del gen RL regulan el ciclo de replicación y latencia, los estudios mencionados en esta revisión sugieren que los RNAs codificados por el gen RL inhiben la expresión de bICP0 y previene la muerte celular, dicha inhibición promueve el establecimiento y mantenimiento de la latencia; además, este gen y sus productos interactúan con ciclinas en neuronas de ganglios trigéminos infectados prolongando la supervivencia de la neurona infectada.
BIBLIOGRAFÍABABIUK LA, VAN DRUNEN LITTEL-VAN DEN HURK S, TIKOO SK. Inmunology of Bovine herpesvirus 1 infection. Vet Microbiol. 1996;53:31-42. [ Links ]
BIELANSKI A, LOEWEN KG, HARE WCD. Inactivation of Bovine Herpesvirus1 (BHV-1) From in vitro Infected Bovine Semen. Theriogenology 1988;30:649-656. [ Links ]
BIELANSKI A, DUBUC C. In-Vitro Fertilization and Culture of Ova From Heifers Infected with Bovine Herpesvirus-1 (BHV-1). Theriogenology 1994;41:1211-1217. [ Links ]
BRANCO FJ, FRASER NW. Herpes Simplex Virus Type 1 Latency-Associated Transcript Expression Protects Trigeminal Ganglion Neurons From Apoptosis. J Virol. 2005;79:9019-9025. [ Links ]
BRATANICH AC, JONES C. Localization of Cis-Acting Sequences in the Latency-Related Promoter of Bovine Herpesvirus 1 Which are Regulated by Neuronal Cell Type Factors and Immediate-Early Genes. J Virol. 1992;68:6099-6106. [ Links ]
BÜCHEN-OSMOND C. 1995. Alphabetical List of ICTV Approved Virus Families and Genera. Última actualización: 8 de abril de 1998. Revisado febrero de 2007. [ Links ]
CASTRUCCI G, FRIGERI F, SALVATORI D, FERRARI M, LO DICO M, ROTOLA, et al. A Study on Latency in Calves by Five Vaccines Against Bovine Herpesvirus-1 Infection. Comp Immun Microbiol Infect Dis. 2002;25:205-215. [ Links ]
CIACCI-ZANELLA J, STONE M, HENDERSON G, JONES C. The Latency-Related Gene of Bovine Herpesvirus 1 Inhibits Programmed Cell Death. J Virol. 1999;73:9734-9740. [ Links ]
CIAT -Centro Internacional de Agricultura Tropical. Informe anual de salud animal; 1972. [ Links ]
CIAT -Centro Internacional de Agricultura Tropical. Informe anual de salud animal; 1975. [ Links ]
CONNOLLY SA, WHITBECK JJ, RUX AH, KRUMMENACHER C, VAN DRUNEN LITTEL-VAN DEN HURK S, COHEN GH, et al. Glycoprotein D Homologuesin Herpes Simplex Virus Type 1, Pseudorabies Virus and Bovine Herpesvirus-1 Bind Directly to Human HveC (nectin-1) whit Different Affinities. Virology. 2001;280:7-18. [ Links ]
DASIKA DK, LETCHWORTH III GJ. Cellular Expression of Bovine Herpesvirus1 gD Inhibits Cell-To-Cell Spread of Two Related Viruses Without Blocking Their Primary Infection. Virology. 1999;254:24-36. [ Links ]
DASIKA DK, LETCHWORTH III GJ. Homologous and Heterologous Interference Requires Bovine Herpesvirus-1 gD on the Cell Surface During Virus Entry. J Gen Virol. 2000;81:1041-1049. [ Links ]
DELHON G, JONES C. Identification of DNA Sequences in the Latency Related Promoter of Bovine Herpes Virus Type 1 Which are Bound by Neuronal Specific Factors. Virus Res. 1997;51:93-103. [ Links ]
DEVIREDDY LR, JONES C. Alternative Splicing of the Latency Related Transcript of Bovine Herpesvirus 1 Yields RNAs Containing Unique Open Reading Frames. J Virol. 1998;72:7294-7301. [ Links ]
DEVIREDDY LR, JONES CJ. Activation of Caspases and p53 by Bovine Herpesvirus 1 Infection Results in Programmed Cell Death and Efficient Virus Release. J Virol. 1999;73:3778-378. [ Links ]
DEVIREDDY LR, ZANG Y, JONES C. Cloning and Initial Characterization of An Alternatively Spliced Transcript Encoded by the Bovine Herpes Virus 1 Latency-Related Gene. J Neurovirol. 2003;9:612-622. [ Links ]
FRAEFEL C, ZENG J, CHOFFAT Y, ENGELS M, SCHWYZER M, ACKERMANN M. Identification and Zinc Dependence of the Bovine Herpesvirus 1 Transactivator Protein BICP0. J Virol. 1994;68:3154-3162. [ Links ]
FUCHS M, HUBERT P, DETTERER J, RZIHA HJ. Detection of bovine herpesvirus type 1 in blood from naturally infected cattle by using a sensitive PCR that discriminates between wild-type virus and virus lacking glycoprotein E. J Clin Microbiol. 1999;37:2498-2507. [ Links ]
GEISER V, INMAN M, ZHANG Y, JONES C. The latency-related gene of bovine herpesvirus-1 can inhibit the ability of bICP0 to activate productive infection. J Gen Virol. 2002;83:2965-2971. [ Links ]
GEISER V, JONES C. Localization of sequences within the latency-related gene of bovine herpesvirus 1 that inhibit mammalian cell growth. J Neurovirol. 2005;11:563-570.
[ Links ]GEISER V, ZHANG Y, JONES C. Characterization of a BHV-1 strain that does not express the major regulatory protein, bICP0. J Gen Virol. 2005;86:1987-1996. [ Links ]
GERAGHTY RJ, KRUMMENACHER C, COHEN GH, EISENBERG RJ, SPEAR, P.G. Entry of alfaherpesvirus mediated by poliovirus receptor-related protein-1 and poliovirus receptor. Science. 1998;280:1618-1620. [ Links ]
HAGE JJ, GLAS RD, WESTRA HH, MARIS-VELDHUIS MA, VAN OIRSCHOT JT, RIJSEWIJK FA. Reactivation of latent bovine herpesvirus 1 in cattle seronegative to glycoproteins gB and gE. Vet Microbiol. 1998;60:87-98. [ Links ]
HANON E, KEIL G, VAN DRUNEN LITTEL-VAN DEN HURK S, GABRIEL P, VANDERPALSSCHEN A, RIJSEWIJK FA, et al. Bovine herpesvirus-1-induced apoptotic cell death: role of glycoprotein D. Virology. 1999;257:191-197. [ Links ]
HANON E, MEYER G, VANDERPALSSCHEN A, DESSY-DOIZE C, THIRTY E, PASTORET PP. Attachment but not penetration of bovine herpesvirus-1 is necessary to induce apoptosis in target cells. J Virol. 1998;72:7638-41. [ Links ]
HENDERSON G, PERNG GC, NESBURN AB, WECHSLER SL, CLINTON C. The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol. 2004a;10:64-70. [ Links ]
HENDERSON G, ZHANG Y, INMAN M, JONES D, JONES C. Infected cell protein 0 encoded by bovine herpesvirus 1 can activate caspase 3 when overexpressed in transfected cells. J Gen Virol. 2004b;85:3511-3516. [ Links ]
HIGAKI S, GEBHARDT BM, LUKIW WJ, THOMPSON HW, HILL JM. Effect of Immunosuppression on Gene Expression in the HSV-1 Latently Infected Mouse Trigeminal Ganglion. Invest Ophthalmol Vis Sci. 2002;43:1862-1869. [ Links ]
HOSSAIN A, SCHANG LM, JONES C. Identification of gene products encoded by the latency-related gene of bovine herpesvirus 1. J Virol. 1995;69:5345-5352. [ Links ]
HUNTER E. Virus Assembly. Chapter 8. In: Knipe, DM., Howley, PM., Fields' Virology edition 4th, Vol. 1. Lippincott Williams & Wilkins, Philadelphia . 2001. p. 171-197. [ Links ]
INMAN M, LOVATO L, DOSTER A, JONES C. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J Virol. 2001a;75:8507-8515. [ Links ]
INMAN M, ZHANG Y, GEISER V, JONES C. The zinc ring finger in the bICP0 protein encoded by bovine herpes virus-1 mediates toxicity and activates productive infection. J Gen Virol. 2001b;82:483-492. [ Links ]
INMAN M, LOVATO L, DOSTER A, JONES C. A mutation in the latency related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. J Virol. 2002;76:6771-6779. [ Links ]
INMAN M, ZHOU J, WEBB H, JONES C. Identification of a Novel Transcript Containing a Small Open Reading Frame That Is Expressed During Latency, And Is Antisense To The Latency Related Gene Of Bovine Herpes Virus 1 (Bhv-1). J Virol. 2004;78:5438-5447. [ Links ]
JIANG Y, HOSSAIN A, WINKLER MT, HOLT T, DOSTER A, JONES C. A protein encoded by the latency-related gene of bovine herpesvirus 1 is expressed in trigeminal ganglionic neurons of latently infected cattle and interacts with cyclin-dependent kinase 2 during productive infection. J Virol. 1998;72:8133-8142. [ Links ]
JIANG Y, INMAN M, ZHANG Y, POSADAS NA, JONES C. A mutation in the latency related gene of bovine herpesvirus 1 (BHV-1) inhibits protein expression of a protein from open reading frame 2 (ORF-2) and an adjacent reading frame during productive infection. J Virol. 2004;78:3184-3189. [ Links ]
JONES C, DELHON G, BRATANICH A, KUTISH G, ROCK D. Analysis of the transcriptional promoter which regulates the latency-related transcript of bovine herpesvirus 1. J Virol. 1990;64:1164-1170. [ Links ]
JONES C. Herpes Simplex Virus Type 1 and Bovine Herpesvirus 1 Latency. Clin Microbiol Rev. 2003;16:79-95. [ Links ]
JONES C, GEISER V, HENDERSON G, JIANG Y, MEYER F, PEREZ S, et al. Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Vet. Microbiol. 2006;113:199-210. [ Links ]
JUBB, K. Pathology of domestic animals. 4th edition. Academic Press Inc. San Diego, Calfornia, 1993. p. 177-179. [ Links ]
KAASHOEK MJ, RIJSEWIJK FA, RUULS RC, KEIL GM, THIRTY E, PASTORET PP, et al. Virulence, immunogenicity and reactivation of bovine herpesvirus 1 mutants with a deletion in the gC, gG, gI, gE, or in both the gI and gE gene. Vaccine. 1998;16:802-809. [ Links ]
KUTISH G, MAINPRIZE T, ROCK D. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J Virol. 1990;64:5730-5737. [ Links ]
LEMAIRE M, MEYER G, BRANOWSKY E, SCHYNTS F, WELLEMANS G, KERKHOFS P, et al. Production of Bovine Herpesvirus Type 1-Seronegative Latent Carriers by Administration of a Live-Attenuated Vaccine in Passively Immunized Calves. J Clin Microbiol. 2000a;38:4233-4238. [ Links ]
LEMAIRE M, WEYNANTS V, GODFROID J, SCHYNTS F, MEYER G, LETESSON JJ, et al. Effects of Bovine Herpesvirus Type 1 Infection in Calves with Maternal Antibodies on Immune Response and Virus Latency. J Clin Microbiol. 2000b;38:1885-1894. [ Links ]
LEMAIRE M, SCHYNTS F, MEYER G, GEORGIN GP, BRANOWSKY E, GABRIEL A, et al. Latency and reactivation of a glycoprotein E negative bovine herpesvirus type 1 vaccine: influence of virus load and effect of specific maternal antibodies. Vaccine. 2001;19:4795-4804. [ Links ]
LI Y, VAN DRUNEN LITTEL-VAN DEN HURK S, BABIUK LA, LIANG X. Characterization of Cell Binding Properties of Bovine Herpesvirus-1 Glycoprotein B, C and D: Identification of a Dual Cell Binding Cell Function of gB. J Virol. 1995;69:4758-4768. [ Links ]
LI Y, LIANG X, VAN DRUNEN LITTEL-VAN DEN HURK S, ATTAH-POKU S, BABIUK LA. Glycoprotein Bb, N-terminal subunit of bovine herpesvirus-1 glycoprotein B, can bind to heparan sulfate on the surfaces of Madin-Darby bovine kidney cells. J Virol. 1996;70:2032-2037. [ Links ]
LOVATO L, INMAN M, HENDERSON G, DOSTER A, JONES C. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J Virol. 2003;77:4848-4857. [ Links ]
MARS MH, DE JONG MCM, FRANKEN P, VAN OIRSCHOT JT. Efficacy of a live glycoprotein E-negative bovine herpesvirus 1 vaccine in cattle in the field. Vaccine. 2001;19:1924-1930. [ Links ]
MEURENS F, SCHYNTS F, KEIL GM, MUYLKENS B, VANDERPLASSCHEN, A, GALLEGO P, et al. Superinfection prevents recombination of the alphaherpesvirus bovine herpesvirus-1. J Virol. 2004;78:3872-3879. [ Links ]
MEYER F, PEREZ S, GEISER V, SINTEK M, INMAN M, JONES C. A protein encoded by the bovine herpesvirus 1 latency-related gene interacts with specific cellular regulatory proteins, including CCAAT enhancer binding protein alpha. J Virol. 2007;81:59-67. [ Links ]
MIKLOSKA Z, CUNNINGHAM AL. Alpha and Gamma Interferons Inhibit Herpes Simplex Virus Type 1 Infection and Spread in Epidermal Cells after Axonal Transmission. J Virol. 2001;75:11821-11826. [ Links ]
MISRA V, WALKER S, HAYES S, O'HARE P. The bovine herpesvirus alpha gene transinducing factor activates transcription by mechanisms different from those of its herpes simplex virus type 1 counterpart VP16. J Virol. 1995;69:5209-5216. [ Links ]
MOLANO D, RODRÍGUEZ JL, RAMÍREZ G, VILLAMIL LC. Caracterización electroforética e inmunológica de una cepa de campo del virus de la rinotraqueitis infecciosa bovina y su comparación con cepas de referencia. Rev Med Vet Zootec. 1996;44:35-8. [ Links ]
MUYLKENS B, MEURENS F, SCHYNTS F, FARNIR F, POURCHET A, BARDIAU M, et al. Intraspecific bovine herpesvirus 1 recombinants carrying glycoprotein E deletion as a vaccine marker are virulent in cattle. J Gen Virol. 2006; 87:2149-2154. [ Links ]
MWEENE AS, OKAZAKI K, KIDA H. Detection of viral genome in non-neural tissues of cattle experimentally infected with bovine herpesvirus 1. Jpn J Vet Res. 1996;44:165-174. [ Links ]
PEREZ S, INMAN M, DOSTER A, JONES C. Latency-realated gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. J Clin Microbiol. 2005;43:393-401. [ Links ]
PEREZ S, LOVATO L, ZHOU J, DOSTER A, JONES C. Comparison of inflammatory infiltrates in trigeminal ganglia of cattle infected with wild-type Bovine herpesvirus 1 versus a virus strain containing a mutation in the LR (latency-related) gene. J NeuroVirol. 2006;12:392-397. [ Links ]
PEREZ S, MEYER F, HENDERSON G, JIANG Y, SHERMAN S, DOSTER A, et al. A protein encoded by the bovine herpesvirus 1 open reading frame E gene induces neurite-like morphological changes in mouse neuroblastoma cells and is expressed in trigeminal ganglionic neurons. J NeuroVirol. 2007;13:139-149. [ Links ]
PIDONE CL, GALOSI CM, ETCHEVERRIGARAY EM. Herpesvirus Bovinos 1 y 5. Analecta Veterinaria; 1999;19:40-50 [ Links ]
PIEDRAHITA D, RAMÍREZ G, VERA V. Detección y caracterización por métodos moleculares de aislamientos colombianos de Herpesvirus bovino tipo 1. Rev Fac Med Vet Zoo. 2005;52:122-127. [ Links ]
RIJSEWIJK FA, KAASHOEK MJ, LANDEVELD JP, MELOEN R, JUDEK J, BIENKOWSKA-SZEWCZYK K, et al. Epitopes of glycoprotein C of bovine herpesvirus1 that allow differentiation between BHV-1.1 and BHV-1.2 strains. J Gen Virol. 1999;80:1477-83. [ Links ]
ROCK D, LOKENSGARD J, LEWIS T, KUTISH G. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J Virol. 1992;66:2484-2490. [ Links ]
ROIZMAN B, PELLETT PE. The family herpesviridae: A brief introduction. In Fields Virology, Fourth edition. Knipe DM and Howley PM, Editors. Lippincott Williams & Wilkins. USA. 2001. p. 2381-2397. [ Links ]
SCHANG LM, HOSSAIN A, JONES C. The latency-related gene of bovine herpesvirus 1 encodes a product which inhibits cell cycle progression. J Virol. 1996;70:3807-3814. [ Links ]
SCHANG LM, JONES C. Analysis of bovine herpesvirus 1 transcripts during a primary infection of trigeminal ganglia of cattle. J Virol. 1997;71:6786-6795. [ Links ]
SCHRODER C, KEIL GM. Bovine herpesvirus-1 requieres glycoprotein H for infectivity and direct spreading, and glycoproteins gH(W450) and gB for gDindependient cell-to-cell spread. J Gen Virol. 1999;80:57-61. [ Links ]
SCHWYZER M, ACKERMAN M. Molecular virology of ruminant herpesviruses. Vet. Microbiol. 1996;53:17-29. [ Links ]
SHEFFY BE, DAVIES DH. Reactivation of a bovine herpesvirus after corticosteroid treatment. Proc Soc Exp Biol Med. 1972;140:974-976. [ Links ]
TYLER KL, NATHANSON N. Pathogenesis of Viral infections. Chapter 7. In: Knipe, DM., Howley, PM., Fields` Virology edition 4th, Vol. 1. Lippincott Williams & Wilkins, Philadelphia. 2001. p 199-243. [ Links ]
VAN DRUNEN LITTEL-VAN DEN HURK S. Rationale and perspectives on the success of vaccination against bovine herpesvirus-1. Vet Microbiol. 2006;113:275-82. [ Links ]
VOGUEL FSF, FLOREZ EF, WEIBLEN R, WINKELMANN R, MORAES MP, BRAGANÇA JFM. Intrapreputial infection of young bulls with bovine herpesvirus type 1.2 (BHV-1.2): acute balanoposthitis, latent infection and detection of viral DNA in regional neural and non-neural tissues 50 days after experimental reactivation. Vet Microbiol. 2004;85:185-196. [ Links ]
WINKLER MT, DOSTER A, JONES C. Bovine herpesvirus 1 can infect CD4(+) T lymphocytes and induce programmed cell death during acute infection of cattle. J Virol. 1999;73:8657-8668. [ Links ]
WINKLER MT, SCHANG LS, DOSTER A, HOLT T, JONES C. Analysis of cyclins in trigeminal ganglia of calves infected with bovine herpesvirus-1. J Gen Virol. 2000a;81:2993-2998. [ Links ]
WINKLER MTC, DOSTER A, JONES C. Persistence and Reactivation of Bovine Herpesvirus-1 (BHV-1) in Tonsils of Latently Infected Cattle. J Virol. 2000b;74:5337-5346. [ Links ]
WINKLER MT, DOSTER A, SUR JH, JONES C. Analysis of Bovine Trigeminal Ganglia Following Infection with Bovine Herpesvirus 1. Vet Microbiol. 2002;86:139-155. [ Links ]
WIRTH UV, GUNKEL K, ENGELS M, SCHWYZER M. Spatial and temporal distribution of bovine herpesvirus 1 transcripts. J Virol. 1989;63:4882-4889. [ Links ]
WIRTH UV, VOGT B, SCHWYZER M. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J Virol. 1991;65:195-205. [ Links ]
WIRTH UV, FRAEFEL C, VOGT B, VLCEK C, PACES V, SCHWYZER M. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3' coterminal and encode a putative zinc finger transactivator protein. J Virol. 1992;66:2763-2772. [ Links ]
WITTELS M, SPEAR G. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 1990;18:271-290. [ Links ]
WYLER R, ENGELS M, SCHWYZER M. Infectious Bovine Rhinotracheitis/ Vulvovaginitis (BHV1). In: Herpesvirus Diseases of Cattle, Horses and Pigs, Wittmann G., ed. Kluwer Academic Publishers, Boston, USA, 1989. p. 1-72. [ Links ]
WYLER R, ENGELS M, SCHWYZER M. Infectious Bovine Rhinotracheitis/ Vulvovaginitis (BHV1) Institute of Virology. University of Zurich 1995. p. 1-55. [ Links ]

