










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Biológica Colombiana
versão impressa ISSN 0120-548X
Acta biol.Colomb. v.15 n.2 Bogotá maio/ago. 2010
IDENTIFICACIÓN DE miARNs CONSERVADOS EN YUCA (Manihot esculenta)
Indentification of Conserved miRNAs in Cassava (Manihot esculenta).
ÁLVARO LUIS PÉREZ-QUINTERO1, Pregrado Biología; ANDRÉS ZAPATA1, Ing.; CAMILO LÓPEZ1, Ph. D. 1Universidad Nacional de Colombia, Bogotá, Colombia. alperezq@unal.edu.co
Presentado 25 de noviembre de 2009, aceptado 29 de enero de 2010, correcciones 2 de marzo de 2010.
RESUMEN
Los microARNs (miARNs) son moléculas pequeñas de ARN utilizadas por los eucariotas como un mecanismo de control de la expresión génica. En plantas los miRNAs están implicados en la regulación de distintos aspectos del crecimiento y desarrollo, así como en la tolerancia a estrés biótico y abiótico. Muchos miARNs de plantas se encuentran conservados en todos los grupos de embriófitos, sin embargo aún existen muchas plantas para las que no se conoce el reportorio de miARNs. Asimismo se desconoce el papel que algunos miARNs pueden tener en procesos como defensa contra patógenos. En este trabajo se construyó una librería de ARNs pequeños a partir de muestras de tejidos de Manihot esculenta (yuca) inoculados con la bacteria fitopatógena Xanthomonas axonopodis pv. manihotis (Xam), y se secuenciaron utilizando técnicas de secuenciación de nueva generación (Solexa/Illumina). Se identificaron en la librería 47 familias de miARNs de yuca conservados en otras plantas. Se cuantificó la expresión de estos miARNs, encontrándose similitudes con perfiles de expresión en otras plantas. Se encontró la secuencia de los precursores para algunos miARNs en secuencias de ESTs y GSSs de yuca. Asimismo se predijeron los blancos de estos miARNs en el set de ESTs encontrándose que muchos miARNs están dirigidos contra factores de transcripción, y que existe un gran porcentaje de posibles blancos con función desconocida. Este trabajo es el primer paso hacia entender cómo la vía de miARNs puede estar implicada en la interacción planta-patógeno en el sistema M. esculenta-Xam.
Palabras clave: miARNs, yuca, M. esculenta, Solexa/ Illumina, bacteriosis vascular.
ABSTRACT
microRNAs (miRNAs) are small RNA molecules used by eukaryotes as a control mechanism for gene expression. In plants, miRNAs play a regulatory role in the expression of various genes involved in growth and development, as well in biotic and abiotic stress tolerance. Many plant miRNAs are conserved in all land plants; however the repertoire of miRNAs is still unknown for many plant species. Likewise, the role for some plant miRNAs in pathogen defense is also unknown. In this work we constructed a library of small RNA from tissues of Manihot esculenta (Cassava) infected with the pathogenic bacteria Xanthomonas axonopodis pv. manihotis (Xam). The small RNAs were sequenced using next-generation sequencing (Solexa/Illumina). We identified 47 conserved miRNAs families in Cassava and quantified their expression, finding similarities with expression profiles from other plants. We found sequences for the precursors of some of these families in sets of ESTs and GSSs. We also predicted targets for these miRNAs in a set of ESTs, finding many miRNAs targeting transcription factors, and regions with unknown function. This work constitutes a first step towards understanding the role of the miRNA pathway in plant-pathogen interaction in the M. esculenta-Xam pathosystem.
Key words: microRNAs, cassava, M. esculenta, Solexa/Illumina, cassava bacterial blight.
INTRODUCCIÓN
El silenciamiento génico post-transcripcional (PTGS, del inglés Post Transcriptional Gene Silencing) mediado por ARNs o ARN de interferencia (ARNi) es un mecanismo de defensa utilizado en los eucariotas para proteger sus genomas contra ácidos nucleicos aberrantes o exógenos (Jin, 2008; Voinnet, 2008). El PTGS es un proceso que utiliza ARNs cortos de aproximadamente 20-30 nt (nucleótidos) para reconocer y manipular ácidos nucleicos complementarios (Ding et al., 2004; Obbard et al., 2009). Los microARNs (MIARN) son un tipo de ARN pequeño utilizado en PTGS, inicialmente descubiertos en C. elegans en 1993 (Lee et al., 1993; Wightman et al., 1993), y han llamado la atención en los últimos años por tener un papel crucial en el control de la expresión génica de los eucariotas (Bushati y Cohen, 2007).
Los miARNs son moléculas de aproximadamente 22 nt que se originan a partir de genes nucleares. Un gen nuclear MIARN es primero transcrito por la ARN polimerasa II en un MIARN primario (pri-MIARN) cuyo tamaño puede ser de 100 nt hasta varios kb (kilo-bases). Posteriormente es procesado en un ARN intermediario llamado precursor de MIARN (pre-MIARN) por la enzima Drosha ARNsa II en animales o por Dicer-like 1 (DCL1) en plantas. Los precursores tienen una estructura secundaria característica y estable, con una alta y negativa energía libre de plegamiento, lo cual sirve como criterio para la anotación de miARNs. En plantas los pre-MIARN pueden tener tamaños muy variables, comúnmente entre 70 y 400 nt. Luego, los pre-miARNs son procesados a dúplex MIARN:MIARN* por Dicer en animales y por DCL1 en plantas. Solo la hebra activa del MIARN o MIARN maduro, pero no la hebra pasajera (MIARN*), es incorporada al complejo de silenciamiento inducido por ARN (RISC, del inglés RNA Induced Silencing Complex) el cual contiene la proteína Argonaute 1 (AGO1), este complejo guiará la escisión de ARNs mensajeros complementarios al ARN maduro (blancos o targets) (Tang et al., 2003; Bartel, 2004; Zhang et al., 2006; Meyers et al., 2008; Zhu, 2008).
Los miARNs fueron descritos por primera vez en plantas en Arabidopsis (Llave et al., 2002; Reinhart et al., 2002) y posteriormente en otras especies. A la fecha, en la base de datos miRBase existen 2.222 miARNs agrupados en 519 familias provenientes de 29 especies de plantas disponibles (Griffiths-Jones et al., 2008). Muchos de estos miARNs son altamente conservados en los embriófitos (Axtell y Bowman, 2008).
En plantas debe existir un alto grado de complementariedad entre el MIARN maduro y su blanco para que exista escisión del ARN mensajero. Esta característica ha facilitado la predicción bioinformática de blancos de miARNs en plantas, muchos de los cuales han sido subsecuentemente validados experimentalmente, dándose un crecimiento enorme de las funciones conocidas de los miARNs (Mazière y Enright, 2007; Yang et al., 2007). Entre las diferentes funciones que se han establecido que tienen los miARNs vale la pena destacar aspectos fisiológicos tan importantes como morfogénesis y polaridad de órganos, identidad de órganos florales y tiempo de floración, señalización por hormonas, transición de estados juveniles a adultos, reproducción, y respuesta a distintos tipos de estrés biótico y abiótico (Dugas y Bartel, 2004; Jones-Rhoades et al., 2006; Mallory y Vaucheret, 2006). Recientemente se ha descrito la inducción de algunos miARNs en plantas en respuestas a patógenos, incluyendo bacterias (Navarro et al., 2006; Ruiz-Ferrer y Voinnet, 2009). Así mismo se ha demostrado que las bacterias utilizan efectores como supresores de silenciamiento demostrando que el silenciamiento génico puede ser un mecanismo para regular la expresión de genes durante las interacciones planta-patógeno (Navarro et al., 2008).
La identificación de miARNs suele hacerse mediante métodos clásicos de screening genético o mediante clonación y secuenciación de ARNs pequeños utilizando métodos moleculares tradicionales. Este tipo de estrategias si bien han permitido la identificación de algunos de ellos, no resultan ser las más indicadas porque además de implicar procesos muy largos en ciertos casos llega a ser imposible obtener miARNs, dadas algunas sus propiedades. Otros acercamientos se basaban en la predicción de miARNs conservados por bioinformática pero eventualmente estos debían ser también confirmados por clonación y secuenciación (Berezikov et al., 2006; Xie et al., 2007).
El desarrollo de técnicas de secuenciación de nueva generación, basadas en pirosecuenciación (454 Life Sciences, 2009) o en secuenciación por síntesis o Solexa/Illumina (Illumina, 2009) ha representado una alternativa rápida y económica para la identificación de miARNs en muchas especies (Nobuta et al., 2010). Adicionalmente, estas estrategias permiten indicar los niveles de expresión de miARNs en una muestra, lo cual ha permitido estudiar la manera en que se regulan los miARNs en distintos órganos, en distintos estados de desarrollo o bajo diferentes condiciones de crecimiento en las plantas (Szittya et al., 2008; Zhu et al., 2008; Wei et al., 2009; Zhang et al., 2009). A pesar de estos desarrollos, aún existen muchas especies para las cuales no se conoce el repertorio de miARNs con el que cuentan, y menos el papel que ellos pueden cumplir en la regulación de genes en condiciones particulares.
La yuca (Manihot esculenta) es el cuarto cultivo en importancia a nivel mundial como fuente de calorías para la población humana y está cobrando actualmente una mayor importancia por su potencial utilización en la producción de bioetanol a partir de la fermentación del almidón presente en sus raíces (FAO, 1998). Una de las limitantes más importantes en la producción de este cultivo es la bacteriosis vascular causada por la bacteria gram-negativa Xanthomonas axonopodis pv. manihotis -Xam (Lozano, 1986). En yuca si bien se han reportado tres familias de miARNs predichas por bioinformática, estas aún no se han confirmado experimentalmente (Sunkar y Jagadeswaraan, 2008). Además, la falta, hasta ahora de un genoma completamente secuenciado, dificulta la predicción de muchas familias de miARNs.
Como un paso inicial para entender la regulación de miARNs en respuesta a la infección por esta bacteria, en este trabajo se generó una librería de ARNs pequeños a partir de tejidos de yuca infectada con Xam, los cuales fueron secuenciados con la tecnología de secuenciación Solexa/Illumina.
MATERIALES Y MÉTODOS
MATERIAL VEGETAL Y EXTRACCIÓN DE ARN
Plantas de M. esculenta, variedad MBRA685 fueron mantenidas en invernadero a 26-30 °C, a 80% de humedad. Plantas de cuatro semanas de crecimiento fueron inoculadas mediante perforación en las hojas y punción en tallo con la cepa CIO 151 de Xam a una densidad óptica (O.D.) = 0,2 nm como se describió previamente (Contreras y López, 2007). Muestras de tallo y hojas fueron recolectadas a los tiempos 0 h, 6 h, 24 h, 48 h, post-inoculación, 5, 7 y 15 días post-inoculación.
Se realizaron extracciones de ARN para cada tejido en cada tiempo utilizando LiCl y fenol ácido:cloroformo. Brevemente, el tejido se maceró y se agregó buffer de extracción (acetato de sodio 100mM, EDTA 1mM, SDS 4%) precalentado a 65 °C. Posteriormente se realizó una extracción con fenol ácido:cloroformo:isoamil (25:24:1) (pH fenol = 4,5± 0,2) y un lavado con cloroformo:isoamil (1:1). El sobrenadante se precipitó con LiCl (3M concentración final) y luego con etanol al 70%. El pellet resultante se resuspendió en agua libre de ARNsas. La calidad del ARN fue evaluada por electroforesis en gel de agarosa, y se cuantificó en un espectrofotómetro (BioRad). Las diferentes extracciones de ARN (de los diferentes tejidos y de los diferentes tiempos post-inoculación) se juntaron en un pool de ARN a una concentración de 150µg/ml.
Al ARN obtenido se le ligaron los adaptadores para la posterior amplificación (5’-ATCTCGTATGCCGTCTTCTGCTTG-3’) y se sintetizó el ADNc empleando la transcriptasa reversa (Invitrogen). A partir de este ADNc se realizó una amplificación por PCR utilizando primers correspondientes al adaptador. El producto de PCR se separó en un gel denaturante de poliacrilamida, y se extrajo la fracción correspondiente a los ARN pequeños (10-50 nt). Estos fueron secuenciados directamente mediante tecnología Solexa/ Illumina (Illumina, 2009) en el BC Cancer Agency’s Michael Smith Genome Sciences Centre http://www.bcgsc.ca.
ANÁLISIS DE DATOS
A partir del archivo de texto conteniendo todas las lecturas de la secuenciación, se eliminaron las secuencias de baja calidad. Las secuencias de baja calidad fueron consideradas aquellas que en dos o más bases de la primeras 25 no tuvieran un nivel de pureza mayor a 0,6 (determinado a partir de las secuencias generadas). En las secuencias que cumplieron con el criterio de calidad se identificó el adaptador utilizando la herramientawordfinder del paquete EMBOSS (Rice etal., 2000), se utilizaron distintas combinaciones de parámetros para obtener las distintas variaciones posibles del adaptador, se usaron dos combinaciones de gap opening (GO) y gap extention (GE) (GO 0,0 + GE 10, GO 500 + GE 10), el parámetro Minimun match score se fijó en 70, el parámetro Alignment width se fijó en 6. La eliminación del adaptador se realizó empleando un programa en C++. Posteriormente se eliminaron secuencias menores a 15 nucleótidos. Las secuencias idénticas fueron colapsadas (se creó un set de secuencias no redundantes) utilizando MySQL y contabilizadas usando un programa en C++.
Para buscar miARNs conservados se realizó un BLASTN (Altschul et al., 1990) empleando los ARN pequeños obtenidos de nuestra librería de secuencias únicas contra todos los miARNs de plantas previamente identificados y con evidencia experimental disponibles en miRBase (Griffiths-Jones et al., 2008). Los criterios para considerar similitud con otro MIARN fueron i) tener menos de dos mismatches en la longitud del MIARN maduro, ii) no presentar gaps y iii) tener un valor E máximo de 0,001 (Meyers et al., 2008; Zhang et al., 2009). Los miARNs conservados candidatos fueron agrupados utilizando cd-hit considerando miembros de una misma familia aquellos que presentaron una similitud mayor al 90% para evitar redundancias (Li y Godzik, 2006).
Para buscar los precursores de los miARNs en el genoma de yuca se realizó un BLASTN (Altschul et al., 1990) de los miARNs homólogos candidatos contra un Set Unigen (Zapata, Neme, Sanabria y López, resultados sin publicar) que contiene 29.231 genes únicos obtenidos a partir de la colección de 80459 ESTs de yuca disponibles en el genbank NCBI y contra secuencias GSS (Genome Survey Sequences) de M. esculenta extraídas del NCBI (Benson et al., 2005). Los criterios para considerar los alineamientos del BLASTN significativos fueron poseer mínimo 20 nt que contuvieran el MIARN maduro, 100% de identidad, no gaps, un valor E máximo de 0,001. Se extrajeron regiones desde 100 nt curso arriba hasta 100 nt curso abajo de la porción alineada de los ESTs o los GSSs empleando un programa en C++. Se analizó la estructura secundaria de ARN de esta región y se calculó la energía de plegamiento (MFE, Minimun folding energy) utilizando Mfold, RNA 3.0, con los parámetros predeterminados (Zuker, 2003). Se calculó la significancia de la energía de plegamiento (p-value), para esto se generaron 1.000 secuencias aleatorias con la misma composición de bases y longitud de cada precursor con el programa uShuffle (Jiang et al., 2008), el p-value se calculó como la proporción de estructuras con MFE igual o menor al precursor original (Freyhult et al., 2005). Se consideraron como precursores válidos aquellos que tuvieran menos de seis mismatches o desapareamientos entre la región del MIARN maduro y la hebra complementaria, pocos abultamientos asimétricos en la estructura (máximo tres) y de existir que fuesen de tamaño corto (menos de tres bases) (Ambros et al., 2003; Meyers et al., 2008) Debían además tener una MFE baja (<-30 kcal/mol) y un p-value menor a 0,05.
Para determinar los posibles blancos de los miARNs conservados se empleó el set unigen de ESTs y los GSSs de M. esculenta utilizando una versión modificada del software miRANDA (Enright et al., 2003), para adecuarse mejor a la complementariedad MIARN: blanco requerida en plantas. Los criterios para considerar un blanco candidato fueron: cuatro o menos mismatches (pares de bases no complementarias) en total, solo uno o menos mismatches entre las posiciones 1 a 12, no más de dos mismatches consecutivos entre las posiciones 13 a 21, ningún mismatch en las posiciones 10 y 11, el par MIARN: blanco debía tener baja energía libre de unión (mínimo 23 kcal/mol) (Schwab et al., 2005; Zhang et al., 2006b). Se escogieron los mejores blancos para cada secuencia utilizando el puntaje del programa miRANDA (mínimo 150).
Las secuencias de blancos candidatos fueron anotadas realizando búsquedas con BLASTX (Altschul et al., 1990) contra la base de datos Uniref (Suzek et al., 2007), para lo cual se emplearon los parámetros por defecto y con un valor E máximo de 1x10-5. El programa UFO se empleo para la anotación automática en categorías funcionales (Meinicke, 2009).
RESULTADOS Y DISCUSIÓN
Plantas de yuca de la variedad resistente MBRA685 fueron inoculadas con la cepa de Xam CIO151 y se colectó el tejido (hojas y tallo) a 0, 6, 24, y 48 hpi y 5, 7 y 15 dpi. A partir de este tejido se realizó la extracción de ARN para posteriormente purificar y construir una librería de miARNs de yuca. Esta librería de pequeños ARNs consistió de 15’968.516 lecturas obtenidas por secuenciación mediante la tecnología Solexa/ Illumina (Illumina, 2009). Después de aplicar un filtro de calidad bajo un umbral de pureza de 0,6 en las primeras 25 bases se obtuvieron 12’748.722 secuencias. Al eliminar el adaptador y filtrar las secuencias menores a 15 nt se obtuvieron 11’871.330 secuencias, que correspondían a 1’644.261 secuencias únicas. Este fue el set de secuencias con que se trabajó.
A partir de las secuencias únicas se realizó una búsqueda de similitud con los miARNs conservados en otras plantas y reportados en la base de datos miRBase, resultando en 3.643 miARNs candidatos, esto es el número de secuencias únicas que contienen un posible MIARN maduro. Estas fueron agrupadas por similitud de 90% en la porción correspondiente al MIARN maduro. En total se obtuvieron 118 representantes correspondientes a 47 familias de miARNs de las 519 identificadas en el grupo de las plantas verdes. Esto ubica a M. esculenta como la sexta planta con más familias de miARNs identificadas (Fig. 1). Las plantas con las que M. esculenta comparte más familias de miARNs son en su orden Arabidopsis thaliana (29 familias), Populus tricocarpa (24 familias), Vitis vinifera (23 familias) y Oryza sativa (22 familias). Además se identificaron familias con un amplio rango filogenético no reportado previamente. Por ejemplo, las familias 894, 896 y 1.030 están conservadas desde el linaje entre M. esculenta y Physcomitrella patens y la familia 1.310 se encuentra en M. esculenta y Pinus taeda, una gimnosperma. Algunas de estas familias han sido predichas por bioinformática en otras especies (Zhang et al., 2010).
El número de lecturas de una secuencia en una muestra de miARNs secuenciada por Solexa/Illumina puede utilizarse como una medida del nivel de expresión de la secuencia en cuestión (Fahlgreen et al., 2007; Zhang et al., 2009). Según este criterio, las familias de miARNs de yuca más expresadas en nuestras condiciones fueron en su orden, las familias 166/165 (las familias con dos nombres corresponden a familias que por razones históricas fueron designadas diferente en algunas especies y luego fueron reagrupadas (Meyers et al., 2008)), 159/319, 167 y 396 todas con más de 10.000 lecturas en la secuenciación (Fig. 2). Estas familias se encuentran también entre las más conservadas y han sido identificadas por lo menos en 15 especies de plantas (Griffiths-Jones et al., 2008). Sin embargo otras familias altamente expresadas parecen estar distribuidas en un rango filogenético más estrecho, como la familia 2.911 identificada hasta ahora únicamente en Populus euphratica (Li et al., 2009), el mismo orden taxonómico en el que se encuentra la yuca (Malpighiales).
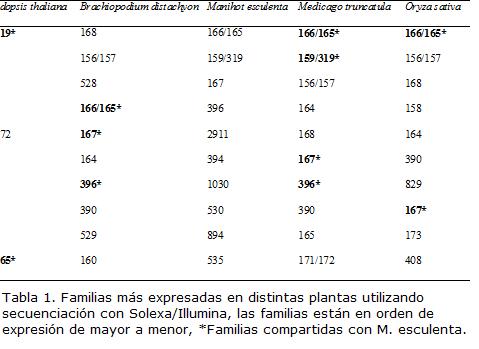
Se compararon estos resultados con otros perfiles de expresión obtenidos por secuenciación con Solexa/Illumina en diferentes plantas. Para la comparación se emplearon las diez familias más expresadas en cada planta (Fahlgreen etal., 2007; Backman etal., 2008; Sunkar et al., 2008; Szittya et al., 2008; Zhang et al., 2009). Ciertas familias se encuentran entre las más expresadas, por ejemplo, las familias 166/165 y 167 están entre las más expresadas en todas las plantas analizadas (Tabla 1). Sin embargo los perfiles son diferentes para cada especie y dependen altamente de las condiciones de crecimiento en las cuales se encontraban las plantas en el momento de la extracción del ARN. Entre las plantas analizadas el perfil de M. esculenta es más similar al de M. truncatula, tanto en número de familias compartidas entre las más expresadas (cuatro familias, igual que en el caso de A. thaliana) como en el orden de las familias de la más expresada a la menos expresada (Tabla 1), ambas hacen parte del grupo de Eurósidas I, fábidas.
Para algunas de las familias de miARNs altamente expresados se ha reportado expresión diferencial en respuesta a distintos tipos de estrés en otras plantas. Por ejemplo, la expresión de la familia 171 disminuye en respuesta a estrés biótico (Naqvi et al., 2008); mientras que los miARNs de las familias 159/319, 169 aumentan (Bazzini et al., 2007). Los miARNs de las familias 156/157, 160 y 164 pueden ser inducidos o reprimidos por patógenos específicos (Bazzini et al., 2007; Lu et al., 2007; Tagami et al., 2007). Además, la familia 156/157 es inducida en condiciones de hipoxia (Moldovan et al., 2009). Las familias 395 y 399 son inducidas en respuesta a deficiencias de nutrientes y la familia 169 es reprimida en respuesta a deficiencias de nitrógeno e inducida en condiciones de estrés por salinidad e hipoxia (Moldovan et al., 2009; Pant et al., 2009; Zhao et al., 2009). En M. esculenta nosotros detectamos la expresión, aunque a bajos niveles, de la familia 393, la cual juega un papel importante en defensa contra bacterias en Arabidopsis (Navarro et al., 2006). La familia 168, que en M. esculenta presentó una muy baja expresión comparada con otras plantas, se ha reportado como inducida en respuesta a estrés por frío y por patógenos (Lu et al., 2008; Ruiz-Ferrer y Voinnet, 2009).
Cada familia de miARNs se encuentra representada por diferentes secuencias, que corresponden probablemente a diferentes loci de genes de miARNs en el genoma de yuca. Las familias con mayor diversidad de secuencias fueron, en su orden, las familias 166/165 (15 secuencias), 159/319 (13 secuencias), 156/157 (11 secuencias), 170/171 (9 secuencias), 169 (8 secuencias) (Fig. 3). Estas son también las familias representadas por más loci en otras plantas, por ejemplo, en A. thaliana (A) y P. tricocarpa (P) el número de loci para estas familias son respectivamente 166/165 = 11A y 17P, 159/319 = 6A y 15P, 170/171 = 4A y 14P, y 169 = 15A y 32P (Griffiths-Jones et al., 2008). Estos datos pueden estar subestimados, pues pueden existir varios loci en yuca conteniendo miARNs con la misma secuencia. Para confirmarlos in silico se deben identificar todos los genes de miARNs en el genoma, lo cual es imposible si no se cuenta con la secuencia completa del genoma; la confirmación in vivo supone otras dificultades dadas por las posibles duplicaciones de genes MIARN. A pesar de esto, la coherencia con los datos de otras especies sugiere que nuestra aproximación es correcta. Como es de esperarse, las familias más diversas suelen también tener un alto nivel de expresión, sugiriendo que la alta expresión de ciertas familias está dada por múltiples genes expresados simultáneamente.
La hebra complementaria o pasajera corresponde a aquella que hace parte del dúplex de 22 pares de bases de ARN procesado a partir del precursor de MIARN pero que no es incorporada al complejo RISC, es decir, carece de actividad de silenciamiento, y por tanto permanece poco tiempo en la célula (Rajagopalan et al., 2006). Es usual encontrar ambas hebras en muestras de ARNs pequeños, y es una evidencia adicional de la expresión de las distintas familias de miARNs. Usualmente se determina que el MIARN maduro es aquél con mayor expresión (Rajagopalan et al., 2006; Meyers et al., 2008). Nuestros datos muestran que en yuca, para la mayoría de familias se detectó la hebra complementaria o pasajera (MIARN*), la cual normalmente se encontraba expresada en una cantidad mucho menor que el MIARN maduro como es de esperarse (Fig. 4). Sin embargo, una excepción son las familias 476 y 414, para las cuales solo se identificó la hebra pasajera y no el MIARN maduro, sugiriendo que en M. esculenta la hebra complementaria para estas familias puede ser aquella que realice la actividad catalítica, experimentos (detectando la unión de alguna de las hebras al complejo RISC) permitirán corroborar esta hipótesis.
Los posibles precursores de los miARNs encontrados se buscaron en las secuencias de ESTs y GSSs reportadas para M. esculenta. Se identificaron 12 regiones que forman una estructura secundaria característica de un pre-MIARN. Los precursores corresponden a miARNs de seis familias. Se encontraron cuatro precursores de la familia 166, la cual mostró también la mayor expresión y mayor diversidad de secuencias. Curiosamente se encontraron tres precursores de la familia 399, una de las menos expresadas (Tabla 2). La figura 5 muestra el plegamiento característico de algunos de los precursores de miARNs encontrados. Se encontraron precursores de la misma familia ubicados en una misma región uno junto al otro, esto sucedió para la familia 166 con dos precursores en el EST DV457519 y para la familia 169 con dos precursores en el GSS BH793692. Este tipo de organización de genes de miARNs, puede ser resultado de los procesos de duplicación en tándem, una forma usual de evolución de los miARNs (Shabalina y Koonin, 2008).
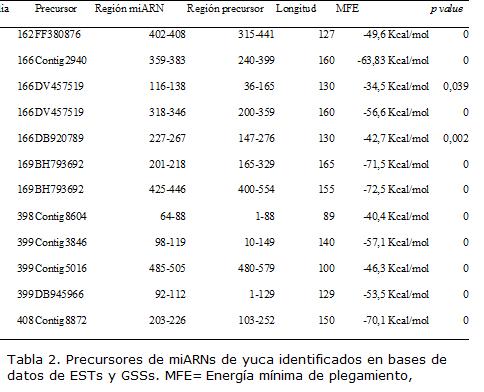
Se espera una baja representación de precursores de miARNs en las bases de datos de ESTs, debido a que muchos de estos precursores no son secuenciados en las librerías de cDNAs ya que la mayoría de las veces se encuentran en intrones o por ser muy cortos. Los valores aquí encontrados son coherentes con lo reportado en otros trabajos (Xie et al., 2007 Zhang et al., 2006c), en donde aproximadamente de 10.000 ESTs debe encontrarse un precursor de MIARN’. En consecuencia, la razón encontrada de 10 precursores /80.000 ESTs es adecuada. Se espera que en las secuencias GSSs se encuentren precursores en mayor cantidad, sin embargo para yuca solo existen 1821 secuencias en el NCBI genbank (Benson et al., 2005).
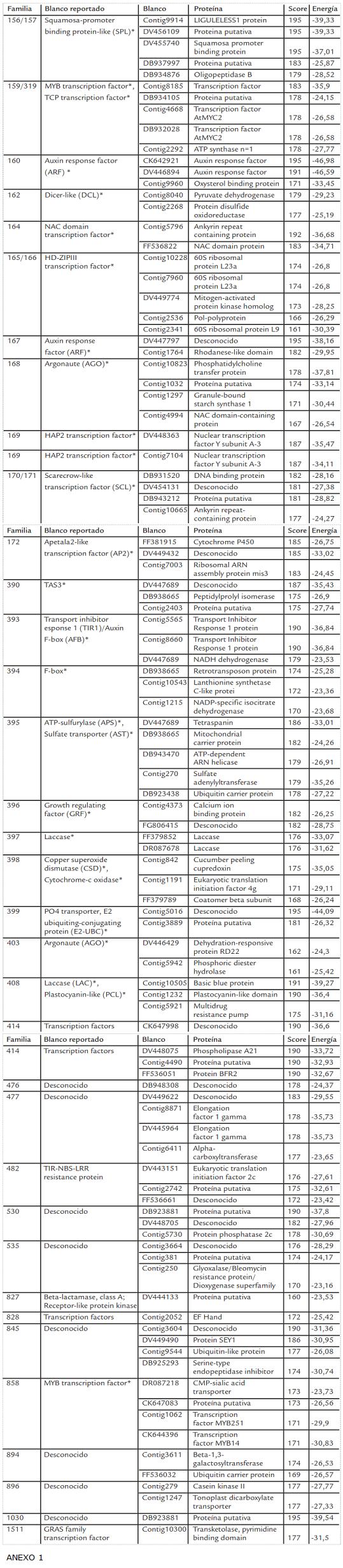
Para identificar los posibles blancos de los miARNs de las 47 familias se realizó una búsqueda de complementariedad contra las secuencias de ESTs de Manihot esculenta utilizando el software de predicción de blancos miRANDA (Anexo 1). Para muchas familias no se logró identificar el blanco o este corresponde a una proteína de función desconocida (30%) (Fig. 6). Muchos de ellos corresponden a familias también sin blancos validados experimentalmente en otras plantas (Fahlgreen et al., 2007; Garcia, 2008; Zhang et al., 2010), mostrando que existen muchos procesos desconocidos regulados por miARNs en plantas. Para varias de las familias conservadas se logró identificar en yuca los blancos previamente identificados y verificados experimentalmente en otras plantas (Anexo 1). Como se ha reportado anteriormente, una gran parte de los miARNs están dirigidos contra factores de transcripción corroborando su importancia en la actividad reguladora de la expresión génica (Mallory y Vaucheret, 2006).
Muchos de los miARNs descubiertos en este trabajo, además de ser importantes en el desarrollo fisiológico de la planta, pueden estar cumpliendo un rol determinante en la interacción M.esculenta-Xam. Para dilucidar estos roles es importante continuar identificando el repertorio de miARNs de la yuca y realizar comparaciones entre los perfiles de expresión de plantas inoculadas con la bacteria y plantas sin inocular, asimismo entre los perfiles de variedades resistentes y variedades susceptibles a la bacteriosis vascular. La inminente publicación del genoma secuenciado de yuca permitirá la identificación de los precursores de los miARNs maduros acá encontrados, y posiblemente el descubrimiento de unos nuevos exclusivos de esta especie. Además se necesita analizar la expresión de estos miARNs en plantas de yuca no inoculadas con la bacteria y en variedades no resistentes inoculadas. Las diferencias entre estos perfiles podrán dar indicios acerca de los procesos mediados por miARNs que ocurren durante la interacción en reacciones de resistencia y/o suceptibilidad.
Nota: durante la preparación del manuscrito fue liberado el primer borrador de la secuencia del genoma de yuca.
AGRADECIMIENTOS
Este proyecto fue financiado parcialmente por apoyo de la DIB, ficha quipu N.º 201010011438.
BILIOGRAFÍA
454 LIFE SCIENCES ROCHE, 2009.http://www.454.com/index.asp.
[ Links ]ALTSCHUL SF, GISH W, MILLER W, MYERS EW, LIPMAN DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-410.
[ Links ]AMBROS V, BARTEL B, BARTEL DP, BURGE CB, CARRINGTON JC, CHEN X. et al. A uniform system for microRNA annotation. RNA. 2003;9:277-279.
[ Links ]AXTELL MJ, BOWMAN JL. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008;13:343-349
[ Links ]BACKMAN TW, SULLIVAN CM, CUMBIE JS, MILLER ZA, CHAPMAN EJ, FAHLGREN N. et al. Update of ASRP: the Arabidopsis Small RNA Project database. Nucleic Acids Res. 2008;36,D:982-985.
[ Links ]BARTEL DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297.
[ Links ]BAZZINI AA, HOPP HE, BEACHY RN, ASURMENDI S. Infection and coaccumulation of tobacco mosaic virus proteins alter microRNA levels, correlating with symptom and plant development. Proc Natl Acad Sci USA, 2007;104:12157-12162.
[ Links ]BENSON D, KARSCH-MIZRACHI I, LIPMAN D, OSTELL J, WHEELE D. GenBank. Nucleic Acids Res. 2005;33:D34-D38.
[ Links ]BEREZIKOV E, CUPPEN E, PLASTERK RH. Approaches to microRNA discovery. Nat Genet. 2006;38:S2-S7.
[ Links ]BUSHATI N, COHEN S. microRNA functions. Annu Rev Cell Dev. 2007;23:175-205
[ Links ]CONTRERAS NE, LÓPEZ CCE. Expresión de dos genes candidatos a resistencia contra la bacteriosis vascular en yuca. Acta biol Colomb. 2008;13(2):175-188
[ Links ]DING SW, LI H, LU R, LI F, LI WX. RNA silencing: a conserved antiviral immunity of plants and animals. Virus Res. 2004;102:109-115.
[ Links ]DUGAS D, BARTEL B. MicroRNA regulation of gene expression in plants. Curr Opin Plant Biol. 2004;7:512-520
[ Links ]ENRIGHT AJ, JOHN B, GAUL U, TUSCHL T, SANDER C, MARKS DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
[ Links ]FAHLGREN N, HOWELL MD, KASSCHAU KD, CHAPMAN EJ, SULLIVAN CM, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE. 2007;2:e219.
[ Links ]FAO Food and agriculture Organization of the United Nations (FAO). Agricultural commodity Projections. Rome, Italy: FAO; 1998.
[ Links ]FREYHULT E, GARDNER PP, MOULTON V. Comparison of RNA folding measures. BMC Bioinformatics. 2005;6:241.
[ Links ]GARCIA D. A miRacle in plant development: role of microRNAs in cell differentiation and patterning. Semin Cell Dev Biol. 2008;19(6):586-95.
[ Links ]GRIFFITHS-JONES S, SAINI HK, VAN DONGEN S, ENRIGHT AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36 D154-D158 (http://www.mirbase.org/).
[ Links ]ILLUMINA Inc. 2009. http://www.illumina.com/ .
[ Links ]JIANG M, ANDERSON J, GILLESPIE J, MAYNE M. uShuffle: A useful tool for shuffling biological sequences while preserving the k-let counts. BMC Bioinformatics. 2008;9:192.
[ Links ]JIN H. Endogenous small RNAs and antibacterial immunity in plants. FEBS Lett. 2008;582(18):2679-84.
[ Links ]JONES-RHOADES M, BARTEL D, BARTEL B. MicroRNAs and Their Regulatory Roles in Plants. Annu Rev Plant Biol. 2006;57:19-53.
[ Links ]LEE RC, FEINBAUM RL, AMBROS V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-54.
[ Links ]LI W, GODZIK A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658-9.
[ Links ]LI B, YIN W, XIA X. Identification of microRNAs and their targets from Populus euphratica. Biochem Biophys Res Commun. 2009;388:272-277.
[ Links ]LLAVE C, KASSCHAU K, RECTOR M, CARRINGTON J. Endogenous and Silencing-Associated Small RNAs in Plants. The Plant Cell. 2002;14:1605-1619.
[ Links ]LOZANO JC. Cassava bacterial blight: a manageable disease. Plant Dis. 1986;70:1089-1093.
[ Links ]LU S, SUN Y H, AMERSON H, CHIANG V L. MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007;51:1077-1098.
[ Links ]LU S, SUN Y, CHIANG V. Stress-responsive microRNAs in Populus. Plant J. 2008;55,131-151.
[ Links ]MALLORY A, VAUCHERET H. Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38(7):850.
[ Links ]MAZIERE P, ENRIGHT A. Prediction of microRNA targets. Drug Discov Today. 2007;12(11/12).
[ Links ]MEINICKE P. UFO: a web server for ultra-fast functional profiling of whole genome protein sequences. BMC Genomics. 2009;10:409.
[ Links ]MEYERS B, AXTELL M, BARTEL B, BARTEL D, BAULCOMBE D, BOWMAN J. et al. Criteria for Annotation of Plant MicroRNAs. Plant Cell. 2008;20:3186-3190.
[ Links ]MOLDOVAN D, SPRIGGS A, YANG J, POGSON B, DENNIS E S, WILSON W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J Exp Bot. (EN PRENSA), 2009. doi:10.1093/jxb/erp296.
[ Links ]NAQVI A, CHOUDHURY N, MOHD Q, HAQ R, MUKHERJEE S. MicroRNAs as biomarkers in Tomato Leaf Curl Virus (ToLCV) disease. Nucleic Acids Symp Ser. 2008;52:507-508.
[ Links ]NAVARRO L, DUNOYER P, JAY F, ARNOLD B, DHARMASIRI N, ESTELLE M. et al. A Plant miRNA Contributes to Antibacterial Resistance by Repressing Auxin Signaling. Science. 2006;312:436-439.
[ Links ]NAVARRO L, JAY F, NOMURA K, HE SY, VOINNET O. Suppression of the microRNA pathway by bacterial effector proteins. Science. 2008;321(5891):964-967.
[ Links ]NOBUTA K, MCCORMICK K, NAKANO M, MEYERS BC. Bioinformatics Analysis of Small RNAs in Plants Using Next Generation Sequencing Technologies. Methods Mol Biol. 2010;592:89-106. [ Links ]
OBBARD DJ, GORDO KHJ, BUCK AH, JIGGINS FM. The evolution of RNAi as a defense against viruses and transposable elements. Phil Trans R Soc B. 2009;364:99-115.
[ Links ]PANT BD, MUSIALAK-LANGE M, NUC P, MAY P, BUHTZ A, KEHR J. et al. Identification of Nutrient-Responsive Arabidopsis and Rapeseed MicroRNAs by Comprehensive Real-Time Polymerase Chain Reaction Profiling and Small RNA Sequencing. Plant Physiol. 2009;150:1541-1555.
[ Links ]RAJAGOPALAN R, VAUCHERET H, TREJO J, BARTEL DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20:3407-3425.
[ Links ]REINHART B, WEINSTEIN E, RHOADES M, BARTEL B, BARTEL D. Micrornas in plants. Genes Dev. 2002;16:1616-1626.
[ Links ]RICE P, LONGDEN I, BLEASBY A. EMBOSS: the European Molecular Biology Open software Suite. Trends Genet. 2000;16(6):276-7.
[ Links ]RUIZ-FERRER V, VOINNET O. Roles of Plant Small RNAs in Biotic Stress Responses. Annu Rev Plant Biol. 2009;60:485-510.
[ Links ]SCHWAB R, PALATNIK J, RIESTER M, SCHIMMER C, SCHMID M, WEIGELL D. Specific Effects of MicroRNA on the Plant Transcriptom. Dev Cell. 2005;8:517-527.
[ Links ]SHABALINA A, KOONIN E. Origins and evolution of eukaryotic RNA interference. Trends Ecol and Evol. 2008;23(10):578-587.
[ Links ]SUNKAR R, JAGADEESWARAN G. In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol. 2008;8:37.
[ Links ]SUNKAR R, ZHOU X, ZHENG Y, ZHANG W, ZHU J. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8:25.
[ Links ]SUZEK BE, HUANG H, MCGARVEY P, MAZUMDER R, WU CH. UniRef: comprehensive and non redundant UniProt reference clusters. Bioinformatics. 2007;23(10):1282-8.
[ Links ]SZITTYA G, MOXON S, SANTOS DM, JING R, FEREVEIRO MPS, MOULTON V, DALMAY T. High-throughput sequencing of Medicago truncatula. BMC Genomics. 2008;9:593.
[ Links ]TAGAMI Y, INABA N, KUTSUNA N, KURIHARA Y, WATANABE Y. Specific enrichment of miRNAs in Arabidopsis thaliana infected with tobacco mosaic virus. DNA Res. 2007;14:227-233.
[ Links ]TANG GL, REINHART BJ, BARTEL DP, ZAMORE PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17:49-63.
[ Links ]VOINNET O. Post-transcriptional RNA silencing in plant-microbe interactions: a touch of robustness and versatility. Curr Opin Plant Biol. 2008;11(4):464-70.
[ Links ]WEI B, CAI T, ZHANG R, LI A, HUO N, LI S, et al. Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat (Triticum aestivum) and Brachypodium distachyon. Funct Integr Genomics. 2009;9(4):499-511.
[ Links ]WIGHTMAN B, HA I, RUVKUN G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855-62.
[ Links ]XIE F, HUANG S, GUO K, XIANG A, ZHU Y, NIE L, YANG Z. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007;581:1464-1474.
[ Links ]YANG T, XUE L, AN L. Functional diversity of miRNA in plants. Plant Sci. 2007;172:423-432.
[ Links ]ZHANG B, PAN X, COBB GP, ANDERSON T. Plant microRNA: A small regulatory molecule with big impact. Dev Biol. 2006;289:3-16.
[ Links ]ZHANG B, PAN X, WANG Q, COBBA G, ANDERSON T. Computational identification of microRNAs and their targets. Comput Biol Chem. 2006b;30:395-407.
[ Links ]ZHANG B, PAN X, CANNON CH, COBB GP, ANDERSON TA. Conservation and divergence of plant microRNA genes. Plant J. 2006c;46:243-259.
[ Links ]ZHANG J, XU Y, HUAN Q, CHONG K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10:449.
[ Links ]ZHANG Z, YU J, LI D, ZHANG Z, LIO F, ZHOU X, WANG T, LING Y, SU Z. PMRD: plant microRNA database. Nucleic Acids Res. 2010;38:D806-D813.
[ Links ]ZHAO B, GE L, LIANG R, LI W, LIN KH, JIN Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Molecular Biology. 2009;10:29.
[ Links ]ZHU J. Reconstituting plant miRNA biogenesis. Proc Natl Acad Sci U S A. 2008;105(29):9851-9852.
[ Links ]ZHU Q, SPRIGGS A, MATHEW L, FAN L, KENNEDY G, GUBLER F, HELLIWELL C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008;18:1456-1465.
[ Links ]ZUKER M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406-15.
[ Links ]
