










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Biológica Colombiana
versão impressa ISSN 0120-548X
Acta biol.Colomb. v.15 n.3 Bogotá set./dez. 2010
MECANISMOS DE SALIDA DE PARÁSITOS INTRACELULARES DE SU CÉLULA HOSPEDERA
Exit Mechanisms of Intracellular Parasites from their Host Cells
MARÍA DEL PILAR QUINTANA1,2, M.Sc; SONIA LEÓN2,3, MARÍA ELISA FORERO2, M.Sc; MARCELA CAMACHO2,3, M.D., Ph. D.
1Departamento de Química, Facultad de de Ciencias, Universidad Nacional de Colombia. Carrera 45 # 26-85, Bogotá, Colombia. 2Laboratorio de Biofísica, Centro Internacional de Física, Universidad Nacional de Colombia. Carrera 45 # 26-85, Bogotá, Colombia. 3Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia. Carrera 45 # 26-85, Bogotá, Colombia mmcamachon@bt.unal.edu.co.
Presentado 25 de junio de 2010, aceptado 2 de agosto de 2010, correcciones 10 de octubre de 2010.
RESUMEN
Algunos parásitos intracelulares durante la infección en hospederos vertebrados se localizan al interior de sus células hospederas en un compartimiento intracelular rodeado por membrana denominado vacuola parasitófora. Para el sostenimiento e incremento de las infecciones causadas por estos parásitos es necesario que se dé un evento de liberación/salida de las formas infectivas, para que estas reinicien la infección en nuevas células. Para dicho fenómeno de liberación se han planteado dos mecanismos básicos: 1. la salida se da por eventos de ruptura de la membrana de la vacuola parasitófora (MVP) y de la membrana plasmática de la célula hospedera y/o 2. la salida se da por un proceso de fusión entre la MVP y la membrana de la célula hospedera, de forma que la luz de la VP y el espacio extracelular se hacen continuos, permitiendo la liberación del parásito. En esta revisión se presenta la evidencia que apoya estos modelos en bacterias y protozoarios intracelulares obligatorios, con especial énfasis en la salida de Leishmania.
Palabras clave: parásito intracelular, salida, lisis de membranas, fusión de membranas.
ABSTRACT
Some intracellular parasites inhabit intracellular compartments known as parasitophorous vacuoles. To maintain and amplify infection, infective forms of the parasite must exit from the host cell to infect new cells. During parasite egress, two main mechanisms have been proposed: lysis of the membranes of the parasitophorus vacuole and the plasma membrane of the cell host, or fusion of these two membranes providing continuity between the lumen of the parasitophorous vacuole and the extracellular compartment. In this review we present evidence supporting these models for intracellular parasites of human importance with particular emphasis on Leishmania exit.
Key words: intracellular parasite, exit, membrane lysis, membrane fusion.
INTRODUCCIÓN
Dentro del ciclo de vida de muchos parásitos, se incluyen estadios intracelulares que se albergan en tipos celulares específicos al interior de hospederos vertebrados. Dentro de sus células hospederas, éstos establecen una infección al interior de compartimientos intracelulares rodeados de membrana, denominados: fagosomas, fagolisosomas o vacuolas parasitóforas (VP, denominación que será usada en esta revisión), compartimientos dentro de los cuales los parásitos intracelulares se multiplican para posteriormente abandonar la célula hospedera y reiniciar la infección en nuevas células, lo que finalmente permite el sostenimiento y amplificación de la infección al interior del hospedero vertebrado. Los mecanismos por los cuales bacterias y protozoarios intracelulares son liberados de sus células hospederas han sido poco estudiados, pese a la importancia de dichos mecanismos en la habilidad del parásito para infectar células adicionales, manteniendo la infección dentro del hospedero y generando disponibilidad de parásitos que puedan ser transmitidos a un nuevo hospedero.
En general, se ha asumido que la salida de parásitos de sus células hospederas involucra eventos de lisis de las membranas de la VP (MVP) y la membrana de la célula hospedera. Sin embargo, la evidencia que apoya este mecanismo es fragmentada aceptándose que este sería un proceso organizado con la participación del parásito, de la célula hospedera y de moléculas específicas entre las que se mencionan proteasas y porinas. Otros modelos de salida, recientemente sugeridos, plantean que la salida de parásitos podría ser por fusión de membranas y por tanto más silenciosa (-exocitosis- del contenido de la VP o -extrusión- de la totalidad de la VP), dejando intacta la célula hospedera, dando como resultado parásitos libres en el espacio extracelular (en el caso de la -exocitosis-) o rodeados por membrana (en el caso de la -extrusión-). Estos modelos respecto a la salida de los parásitos de sus células hospederas, plantean que este evento además de ser importante en la continuación de un determinado ciclo de vida, podría tener implicaciones en la forma como el sistema inmune del hospedero reacciona frente a la presencia del parásito y podría representar una estrategia de supervivencia del mismo al interior de su hospedero.
A pesar del escaso interés que se ha prestado a la salida de bacterias o protozoarios de sus células hospederas (comparado con el interés en otros aspectos de la interacción del parásito con su célula hospedera), existen estudios encaminados a comprender este proceso y dentro de los cuales se encuentran trabajos en la bacteria Chlamydia trachomatis y los protozoarios Toxoplasma gondii, Plasmodium falciparum, Trypanosoma cruzi y Leishmania amazonensis.
SALIDA DE Chlamydia trachomatis DE SU CÉLULA HOSPEDERA
Chlamydia trachomatis es una bacteria que durante parte de su ciclo de vida infecta particularmente células epiteliales de las mucosas, y se localiza al interior de una VP que no se fusiona con lisosomas, pero que permite la adquisición de nutrientes provenientes de la célula hospedera (Beagley y Timms, 2000; Hammerschlag, 2002). Para permitir la salida de C. trachomatis al medio extracelular, se ha asumido un proceso de lisis tanto de la MVP como de la membrana plasmática de la célula epitelial, atribuída a un efecto mecánico dado por el incremento del número de bacterias al interior de la VP. Sin embargo, observaciones ultraestructurales sugieren que la salida resulta de un proceso similar a la exocitosis (Todd y Caldwell, 1985). Recientemente, marcajes con sondas fluorescentes que son excluidas de la VP y estudios de videomicroscopía de fluorescencia en tiempo biológico real, han mostrado que C. trachomatis sale por dos mecanismos mutuamente excluyentes, que se dan con frecuencias equivalentes: 1. lisis de la MVP y de la membrana de la célula hospedera y 2. extrusión: liberación en paquete de la VP. En el primer caso, se da una permeabilización rápida (tomando aproximadamente 15 minutos) y secuencial (evidenciada por la pérdida de la capacidad de excluir o retener el marcador fluorescente), iniciándose con la permeabilización de la MVP, luego de otras membranas de compartimentos intracelulares (como la envoltura nuclear) y finalmente la membrana plasmática de la célula hospedera. En el caso específico de la permeabilización de la MVP, se encuentra involucrada la actividad de proteasas de cisteína, ya que su inhibición utilizando el inhibidor E-64 detiene el proceso, impidiendo que la MVP pierda su capacidad de excluir el marcador fluorescente y que posteriormente se dé la permeabilización de las otras membranas. Adicionalmente, la permeabilización de la membrana plasmática de la célula hospedera parece depender de un incremento de calcio intracelular, proveniente del medio extracelular, ya que la ausencia de calcio extracelular prolonga el proceso de lisis. En el segundo caso en el que se da extrusión de la VP dejando intacta la célula hospedera, se observó que el proceso es lento comparado con el de lisis (tomando aproximadamente 2-3 horas) y puede o no liberar la VP con la totalidad de C. trachomatis que alberga, pudiendo en algunas ocasiones quedar una VP residual dentro de la célula hospedera. Este proceso es dependiente de la polimerización del citoesqueleto de actina, actividad de miosina II y de RhoGTPasas, como lo indica la inhibición con fármacos específicos. Otro aspecto importante del mecanismo de extrusión es que la VP liberada, además de encontrarse rodeada por la MVP, también está rodeada por una porción de citoplasma y de membrana plasmática de la célula hospedera (Hybiske y Stephens, 2007), similar a la secreción de algunas glándulas endocrinas (secreción merocrina).
SALIDA DE Toxoplasma gondii DE SU CÉLULA HOSPEDERA
Toxoplasma gondii es un protozoario perteneciente al Phylum apicomplexa, que durante la parte asexual de su ciclo de vida es capaz de infectar cualquier tipo de célula nucleada de mamíferos y aves (Montoya y Liessenfeld, 2004; Black et al., 2000); células dentro de las cuales se alberga y replica en una VP que no es acidificada ni fusionada con endosomas u otras vesículas citoplasmáticas (Black y Boothroyd, 2000). Luego de que se han dado entre 6-7 ciclos de división por endodiogenia, los parásitos salen e infectan células vecinas. Esta salida fue inicialmente relacionada con la muerte celular de la célula infectada y se asoció con la patología tisular que se presenta en toxoplasmosis (Frenkel, 1973). Estudios posteriores han mostrado que la salida del parásito podría ser dependiente de la temperatura y de un incremento en la concentración de calcio, tanto en el citoplasma de la célula hospedera como en el espacio intravacuolar. Este incremento en la concentración de calcio ha sido inducido por adición a las células infectadas del ionóforo de calcio A23187 (Endo et al., 1982; Pingret et al., 1996; Black et al., 2000), ditioles (Stommel et al., 1997) y microinyección de CaCl2 (Schwab et al., 1994); estos tratamientos inducen la salida de los parásitos, generando inicialmente un incremento en su movilidad y protrusión del conoide (apéndice especializado del citoesqueleto situado en el polo anterior del parásito; Caldas et al., 2007), para posteriormente estimular la secreción del contenido de micronemas (organelos de secreción típicos apicomplexa), liberándose una proteína formadora de poros (TgPLP1) que genera la permeabilización de la MVP y de la membrana de la célula hospedera (Kafsack et al., 2009). La ruptura de la membrana plasmática de la célula hospedera ha sido evidenciada por microscopía de luz, microscopía electrónica de barrido, microscopía electrónica de transmisión y microscopía de fluorescencia, sugiriéndose la posibilidad que después de la salida y dependiendo de la cantidad de parásitos que estén saliendo por una misma zona de la membrana de la célula hospedera, podría darse una recuperación en la continuidad de ésta evitando la liberación de todo el contenido citoplasmático; de hecho, se ha observado que durante la salida del parásito se establece un contacto cercano entre su membrana y la de la célula hospedera (Caldas et al., 2007). Respecto al incremento en la concentración intracelular de calcio (tanto en el citoplasma de la célula hospedera como en el espacio intravacuolar). Hasta el momento no existe ninguna evidencia que demuestre la elevación del calcio libre intracelular durante la salida del parásito en ausencia de algún tipo de inductor ionóforo (Moudy et al., 2001), más aun, las concentraciones de calcio intravacuolar en condiciones fisiológicas hacia las 48 hpi (tiempo cerca del cual se ha sugerido la salida del parásito) son bastante más bajas que aquellas inducidas por el tratamiento con A23187 lo cual podría sugerir que este incremento es un evento rápido y transitorio (Pingret et al., 1996). Estudios paralelos indican que previo a la salida del parásito (sin inducción por A23187), aumenta inespecíficamente la permeabilidad de la membrana de la célula hospedera provocando una disminución de la concentración intracelular de potasio (proceso que puede ser simulado por la adición del ionóforo de potasio nigericina o por la permeabilización de las células con la toxina α de Staphylococcus aureus), la cual a su vez es detectada por el parásito, generándose dentro de éste un incremento de calcio intracelular, mediado por la actividad de la fosfolipasa C (Moudy et al., 2001; Fruth y Arrizabalaga, 2007), esto implica que previo a la permeabilización mediada por la TgPLP1, hay cambios en la membrana de la célula hospedera por un mecanismo desconocido, que son los que finalmente generan la señalización que lleva a la inducción de la salida del parásito.
SALIDA DE Plasmodium falciparum DEL ERITROCITO
En el caso de Plasmodium falciparum los estudios acerca de los mecanismos de salida han sido más extensos, planteándose dos mecanismos básicos: el primero conduciría a una lisis de la membrana de la célula hospedera y la MVP (Dvorak et al., 1975; Salmon et al., 2001; Trager 2002; Wickham et al., 2003; Glushakova et al., 2005; Glushakova et al., 2007; revisado por Rayner , 2006). El segundo mecanismo estaría dado por la fusión entre la membrana de la célula hospedera y la MVP (Clavijo et al., 1998; Winograd et al., 1999; Cortés y Caldas 2005; Cortés et al., 2007). Para el mecanismo de ruptura, la discusión se ha centrado sobre el orden secuencial de la ruptura de las membranas, si es simultáneo (Glushakova et al., 2005), o si se da primero la ruptura de la membrana de la célula hospedera (Salmon et al., 2001; Soni et al., 2005) o de la MVP (Wickham et al., 2003). En estos estudios se ha demostrado por videomicroscopía usando marcadores fluorescentes, que la salida del parásito implica una ruptura de las membranas que lo contienen con una rápida descarga radial de los merozoitos, dejando fragmentos vesiculares de membrana en el sitio de liberación del parásito (Glushakova et al., 2005). En el proceso de lisis se han involucrado proteasas (Salmon et al., 2001; Wickham et al., 2003). Se sugiere además desagregación del citoesqueleto previo a la salida dePlasmodium (Pei et al. 2007).
Para el segundo mecanismo, que involucra fusión de membranas, existe evidencia por videomicroscopía de contraste de fases en la que se muestra que durante la salida del parásito del eritrocito, no se dan cambios significativos en el contenido citoplasmático y en la integridad de éste (medido por densidad óptica), descartando ruptura de membranas para permitir la salida del parásito (Winograd et al., 1999); además se han detectado estructuras de membrana que aparecen periódicamente durante la infección en los momentos en que se da la salida del parásito (48 hpi), y que podrían formar una especie de ducto entre la MVP y la ME, promoviendo el contacto entre las dos, permitiendo su fusión y haciendo que el compartimiento que contiene al parásito al interior del eritrocito se haga continuo con el espacio extracelular, favoreciendo la salida del parásito (Clavijo et al., 1998).
SALIDA AL CITOPLASMA DE LA CÉLULA HOSPEDERA DE Trypanosoma cruzi
Para el caso de Trypanosoma cruzi, protozoario causante del mal de Chagas en humanos, se ha demostrado que rápidamente después de la invasión o infección en monocitos humanos, el parásito (en su estadio amastigote o formas más tempranas como tripomastigotes y formas intermedias) sale de la VP y se hace citoplasmático (Ley et al., 1988; Ley et al., 1990). Se sugiere que la salida del parásito de la VP al citoplasma para multiplicarse allí como amastigote, se encuentra mediada por la proteína Tc-TOX, una porina activa a pH ácido, secretada por el parásito y que al insertarse en la MVP generaría su lisis (Ley et al., 1990). Esta proteína es reconocida por anticuerpos anti-C9 (noveno componente del complemento, C9) y antiperforina (Andrews y Withlow, 1989; Andrews et al., 1990). Respecto al mecanismo que el parásito usa para abandonar el citoplasma de la célula hospedera, una vez se ha multiplicado, no ha sido estudiado; pero en general, como en otros parásitos se cree que genera la ruptura de la célula para lograr su salida al espacio extracelular.
SALIDA Leishmania amazonensis DEL MACRÓFAGO
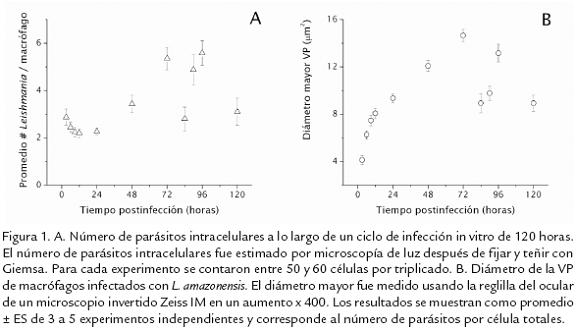
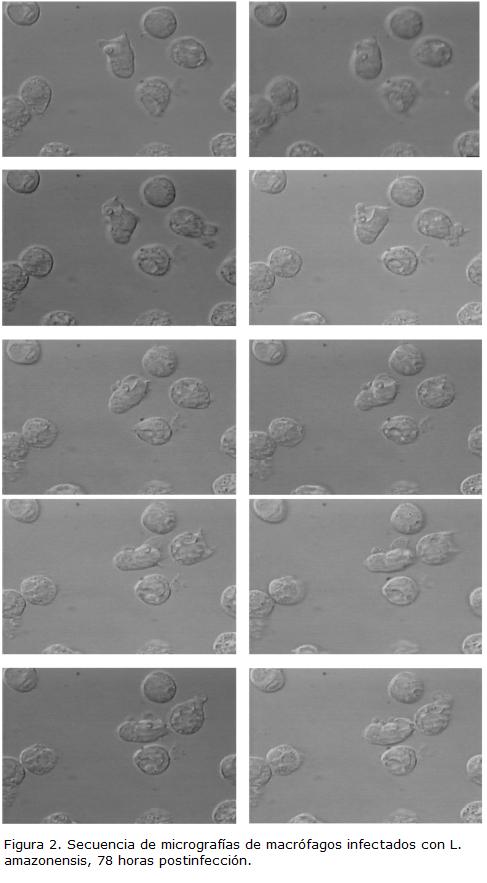
Leishmania es un protozoario obligado de macrófagos y células dendríticas, confinado en una VP con pH ácido que se fusiona con endosomas y lisosomas. En el caso deLeishmania se ha aceptado que la liberación de amastigotes ocurre como consecuencia de la ruptura mecánica del macrófago, generada por el incremento en el número de parásitos intracelulares (Handman y Bullen, 2002), afirmación basada en observaciones de macrófagos dañados con amastigotes libres, durante los estudios iniciales del crecimiento de los parásitos en cultivo (Hawking, 1948; Handman y Spira, 1977). En el caso específico deL. amazonensis se ha estudiado una proteína llamada leishporina (nombre dado por su posible capacidad de formar poros) con actividad óptima a pH ácido (5,0 -5,5) y a 37º C, que es expresada tanto por la forma promastigote como la amastigote. Esta proteína además muestra actividad lítica sobre eritrocitos y macrófagos, produciendo un incremento en la permeabilidad de la membrana y un incremento en la liberación de proteínas citosólicas (Noronha et al., 1994; Noronha et al., 1996; Noronha et al., 2000; Almeida-Campos y Horta, 2000). A pesar de que esta proteína presenta actividad óptima a pH ácido se ha observado que la actividad lítica se conserva hasta valores de pH de 7,5, dando lugar a la sugerencia que esta proteína se secretaría dentro de la VP para luego insertarse en la MVP provocando su lisis (Noronha et al., 1996) y tendría también actividad en el citoplasma del macrófago para insertarse en su membrana (MPM) generando también su lisis, permitiéndose así la salida de los amastigotes (Horta, 1997; Almeida-Campos et al., 2002). Sin embargo, los resultados muestran que a pH 4,5 la actividad de la leishporina se reduce drásticamente, siendo la más baja en el rango de pHs estudiado (Noronha et al., 1996), lo que pone en duda su acción a nivel de la VP ya que ésta presenta pHs entre 4,5-5,2 en infecciones establecidas (revisado por Russell, 1995). A pesar de estos resultados, es posible que el papel de esta leishporina no sea durante el proceso de salida de los amastigotes del macrófago, sino que funcione en otros procesos como la adquisición de moléculas provenientes del citoplasma de la célula hospedera como ha sido sugerido para Plasmodium (Desai et al., 1993; Desai y Rosenberg, 1997; Desai et al., 2000) y Toxoplasma (Schwab et al., 1994). Leishporina al igual que TcTOX, es reconocida por anticuerpos contra C9 y perforina, lo que apoya la idea de su papel como proteína formadora de poros, que junto a una proteasa de serina podría romper una proteína precursora o un péptido inhibidor unido a la proteína, permitiendo así su activación (Almeida-Campos y Horta, 2000). También se ha demostrado que la actividad lítica de esta proteína depende de la presencia de un alto contenido de fosfolípidos en la membrana blanco, mientras que la presencia de otros posibles receptores como proteínas, carbohidratos y colesterol no parecen tener un papel en la acción de esta porina (Castro-Gomes et al., 2009). De estos estudios es importante destacar que no existe ninguna evidencia que demuestre que el parásito en efecto secrete e inserte esta proteína durante el proceso de infección y salida del macrófago y esta leishporina sería especie específica en vista que no se ha encontrado en todas las especies de Leishmania (Almeida-Campos y Horta, 2000). A pesar de que se asume que el amastigote se libera después de lisis de las membranas que lo rodean, recientemente se ha propuesto que su salida es consecuencia de la fusión entre la MVP y la MPM. Observaciones por microscopía de luz de macrófagos infectados han mostrado que no se da una ruptura de su membrana después de la acumulación en su periferia de amastigotes liberados, sugiriéndose la posibilidad de que el parásito reclute la maquinaria exocítica del macrófago como estrategia para salir de este y reiniciar la infección (Rittig et al., 1998; Rittig y Bogdan, 2000). Nuestro grupo ha hecho estudios durante el ciclo infectivo de L. amazonensis, evaluando el porcentaje de infección (Hoyos et al., 2009), el número de parásitos por célula (Fig. 1A) y el diámetro de la VP (Fig. 1B), en donde se observa un incremento tanto en el número de parásitos por célula como en el diámetro de la VP, alcanzándose un máximo a las 72 hpi, y luego observándose un descenso en los dos parámetros hasta las 76-78 hpi, para luego registrarse un nuevo incremento a las 96 hpi, alcanzándose valores similares a los observados a las 72 hpi. Estos datos nos estarían indicando que en el periodo de tiempo analizado se están observando dos ciclos de infección, uno que se inicia desde las 0 hpi hasta las 72 hpi, tiempo en el cual el parásito en forma promastigote entra al macrófago, se diferencia a la forma amastigote y se replica, luego, entre las 72 hpi y las 78 hpi se daría la salida de los amastigotes, que a su vez reinfectarían nuevos macrófagos generando un número de parásitos por célula y un volumen de la VP similares a los de 72 hpi en un menor tiempo, debido probablemente a que ya no es necesaria la transformación de promastigote a amastigote (Díaz, 2002; León, 2005; León et al., 2005). Esta interpretación de dos ciclos es apoyada por el hecho de que la viabilidad de parásitos y macrófagos, medida con diacetato de fluoresceína y yoduro de propidio, no se altera en el periodo en el cual se observa el descenso en el promedio del número de parásitos por macrófago y el promedio del diámetro de la VP (León, 2005; Hoyos et al., 2009), descartando que esta disminución se de por muerte del parásito. Así mismo, para los tiempos en que se sugiere la salida del parásito en este doble ciclo infectivo hemos observado alteraciones en la forma de los macrófagos, el movimiento de amastigotes y la salida de parásitos (Fig. 2). Más aún, mediciones de capacitancia (Cm; medida indirecta del área de la membrana) de la MPM a lo largo del ciclo infectivo con L. amazonensis indican incrementos de Cm que alcanzan un máximo a las 72 horas post infección (hpi) y presentan una caída a las 96 hpi. Estos cambios son significativos y podrían estar relacionados con un proceso de incorporación de nueva membrana a la MPM a las 72 hpi (Corrales et al., 2000), indicando un evento de fusión entre la MVP y la MPM.
Díaz, 2002, realizó estudios del comportamiento del ciclo infectivo de L. amazonensis en presencia de leupeptina (implicada por Flaherty y Swann, 1993 en inhibición de fusión de membranas en la reacción acrosomal) y forbol miristato acetato (PMA; reportado por Kielian y Cohn, 1981 como promotor de fusión entre fagosomas y lisosomas en macrófagos), evidenciando que se presentan cambios en el ciclo infectivo que sugieren que los amastigotes salen del macrófago por un mecanismo que involucra fusión de membranas. La generación de células gigantes (macrófagos fusionados) en enfermedades crónicas que generan granulomas, implica fusión por parte de macrófagos, siendo una respuesta normal de estas células (Vignery, 2000; McNally y Anderson 2005; Vignery, 2005; Cui et al., 2006); este proceso de fusión de macrófagos podría ser usado por Leishmania en su salida. En otros tipos celulares como miocitos, que se fusionan para generar fibras musculares, se ha visto que éstos se hiperpolarizan debido a la expresión de canales de potasio Kir2.1 cuya inhibición a nivel de expresión (Fischer-Lougheed et al., 2001) y a nivel funcional (Liu et al., 1998) previene la fusión celular. La hiperpolarización del potencial de reposo facilitaría la entrada de calcio necesario para el proceso de fusión. En el caso de la infección por Leishmania se observa hiperpolarización del macrófago asociada con un aumento de corrientes de entrada de potasio sensibles a Ba2+ (Forero et al., 1999) en el momento previo a la salida de Leishmania (Forero et al., 1999; Corrales et al., 2000).
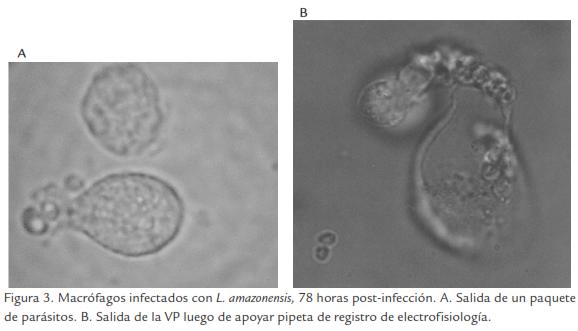
Nuestra evidencia apoya el modelo de salida por fusión de membranas. Sin embargo, hemos observado salida de paquetes de parásitos que podría parecerse a la extrusión descrita para C. tracomatis (Fig. 3A), y salida de la VP coteniendo parásitos asociados con cambios en Cm, por lo tanto no es posible considerar la fusión de membranas como único mecanismo de salida del parásito (Fig. 3B).
AGRADECIMIENTOS
A Colciencias, Programa nacional de ciencia y tecnología de la salud, proyecto código: 110110140820398, a la Universidad Nacional de Colombia, División de investigación sede Bogotá proyecto código: 8003189. A los Departamentos de Química y Biología, Facultad de Ciencias, Universidad Nacional de Colombia, al Centro Internacional de Física en especial a los miembros del Laboratorio de Biofísica, Grupo de Biofísica y Biología de Membranas.
BIBLIOGRAFÍA
ALMEIDA-CAMPOS FR, HORTA MF. Proteolytic activation of leishporin: evidence that Leishmania amazonensis and Leishmania guyanensis have distinct inactive forms. Mol Biochem Parasitol. 2000;111:363-375.
[ Links ]ALMEIDA-CAMPOS FR, NORONHA FS, HORTA MF. The multitalented pore-forming proteins of intracellular pathogens. Microbes Infect. 2002;4(7):741-750.
[ Links ]ANDREWS NW, ABRAMS CK, SLATIN SL, GRIFFITHS G. A. Trypanosoma cruzisecreted protein immunologically related to the complement component C9: Evidence for membrane pore-forming activity at low pH. Cell. 1990;61(7):1277-1287.
[ Links ]ANDREWS NW, WHITLOW MB. Secretion by Trypanosoma cruzi of a hemolysin active at low pH. Mol Biochem Parasitol. 1989;33:249-256.
[ Links ]BEAGLEY KW, TIMMS P. Chlamydia trachomatis infection: Incidence, health costs and prospects for vaccine development. J Reprod Immunol. 2000;48:47-68.
[ Links ]BLACK MW, ARRIZABALAGA G, BOOTHROYD JC. Ionophore-resistant mutant of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol Cell Biol. 2000;20(24):9399-9408.
[ Links ]BLACK MW, BOOTHROYD JC. Lytic cycle of Toxoplasma gondii. Mol Cell Biol Rev. 2000;64(3):607-623.
[ Links ]CALDAS LA, DE SOUZA W, ATTIAS M. Calcium ionophore-induced egress of Toxoplasma gondii shortly after host cell invasion. Vet Parasitol. 2007;147:210-220.
[ Links ]CASTRO-GOMES T, ALMEIDA-CAMPOS FR, CALZAVARA-SILVA CE, DA SILVA RA, FREZARD F, HORTA MF. Membrane binding requirements of the cytolytic activity of Leishmania amazonensis leishporin. FEBS Letter. 2009;583:3209-3214.
[ Links ]CLAVIJO CA, MORA CA, WINOGRAD E. Identification of novel membrane structures in Plasmodium falciparum infected erythrocytes. Mem Inst Oswaldo Cruz 1998;93(1):115-120.
[ Links ]CORRALES A, CAMACHO M, FORERO ME, LLANO I, AGNEW A. Leishmania amazonensis entry and exit from macrophages. Biophys J. 2000;78(1)A1050:179A.
[ Links ]CORTÉS GT, CALDAS ML. Estudio del cuerpo residual de Plasmodium falciparum y su posible relación en la salida de merozoítos de su célula huésped. Biomedica. 2005;S1:138-139.
[ Links ]CORTÉS GT, CALDAS ML, CAMACHO M, WISER MF. The Release of the Malaria Parasite and the transfer of the DIIC(16) (3) from infected erythrocytes to intracytoplasmatic membranes. In: Technological advances in understanding host responses to parasites and parasite manipulation of host responses. Proceedings from the 29th June meeting in Hertfordshire, Reino Unido. 2007:7-8.
[ Links ]CUI W, KE JZ, ZHANG Q, KE HZ, CHALOUNI C, VIGNERY A. The intracellular domain of CD44 promotes the fusion of macrophages. Blood. 2006;107(2):796-805.
[ Links ]DESAI SA, BEZRUKOV SM, ZIMMERBERG J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature. 2000;406(6799):1001-1005.
[ Links ]DESAI SA, KROGSTAD DJ, McCLESKEY EW. A nutrient-permeable channel on the intraerythrocytic malaria parasite. Nature. 1993;362(6421):643-646.
[ Links ]DESAI SA, ROSENBERG RL. Pore size of the malaria parasite s nutrient channel. Proc Natl Acad Sci USA. 1997;94(5):2045-2049.
[ Links ]DÍAZ JC. Liberación de amastigotes de Leishmania amazonensis de macrófagos murinos (trabajo de grado). Bogotá: Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia; 2002.
[ Links ]DVORAK JA, MILLER LH, WHITEHOUSE WC, SHIROISHI T. Invasion of erythrocytes by malaria merozoites. Science. 1975;187(4178):748-750.
[ Links ]ENDO T, SETHI K, PIERANSKI G. Toxoplasma gondii: Calcium ionophore A23187mediated exit of trophozoites from infected murine macrophages. Exp Parasitol. 1982;53:179-188.
[ Links ]FISCHER-LOUGHEED J, LIU JH, ESPINOS E, MORDASINI D, BADER CR, et al. Human myoblast fusion requires expression of functional inward rectifier Kir2.1 channels. J Cell Biol. 2001;153(4):677-686.
[ Links ]FLAHERTY S, SWANN NJ. Proteases are not involved in the membrane fusion events of the lysolecithin-mediated guinea pig sperm acrosome reaction. J Cell Sci. 1993;104:163-172.
[ Links ]FORERO ME, MARÍN M, LLANO I, MORENO H, CAMACHO M. Leishmania amazonensis infection induces changes in the electrophysiological properties of macrophage-like cells. J Membr Biol. 1999;170:173-180.
[ Links ]FRENKEL JK. Toxoplasmosis: Parasite life cycle, pathology and immunology. En: Hammond DM, Long PL, editors. The Coccidia. Baltimore, MD: Univ. Park Press; 1973.
[ Links ]FRUTH IA, ARRIZABALAGA G. Toxoplasma gondii: Induction of egress by the potassium ionophore nigericin. Int J Parasitol. 2007;37:1559-1567.
[ Links ]GLUSHAKOVA S, YIN D, GARTNER N, ZIMMERBERG J. Quantification of malaria parasite release from infected erythrocytes: inhibition by protein-free media. Malar J. 2007;21(6):61.
[ Links ]GLUSHAKOVA S, YIN D, LI T, ZIMMERBERG J. Membrane transformation during malaria parasite release from human red blood cells. Curr Biol. 2005;15(18):1645-1650.
[ Links ]HAMMERSCHLAG MR. The intracellular life of Chlamydiae. Semin Pediatr Infect Dis. 2002;13(4):239-248.
[ Links ]HANDMAN E, BULLEN D. Interaction of Leishmania with the host macrophage. Trends Parasitol. 2002;5(9):332-334.
[ Links ]HANDMAN E, SPIRA DT. Growth of Leishmania amastigotes in macrophages from normal and immune mice. Z Parasitenkd. 1977;53:75-81.
[ Links ]HAWKING F. Growth of protozoa in tissue culture. V. Leishmania donovani. Trans R Soc Trop Med Hyg. 1948;41:545-554.
[ Links ]HOYOS M, NIÑO A, CAMARGO M, DÍA JC, LEÓN S, CAMACHO M. Separation of Leishmania infected macrophages by step-SPLITT fractionation. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(29):3712-3718.
[ Links ]HORTA MF. Pore-forming proteins in pathogenic protozoan parasites. Trends Microbiol. 1997;5(9):363-366.
[ Links ]HYBISKE K, STEPHENS RS. Mechanisms of host cell exit by the intracelular bacterium Chlamydia. Proc Natl Acad Sci U S A. 2007;104(27):11430-11435.
[ Links ]KAFSACK B, PENA JD, COPPENS I, SANDEEP R, BOOTHTOYD J, CARRUTHERS VB. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science. 2009;323:530-533.
[ Links ]KIELIAN M, COHN Z. Phorbol myristate acetate stimulates phagosome-lysosome fusion in mouse macrophages. J Exp Med. 1981;171:101-111.
[ Links ]LEÓN S. Acercamiento al estudio de la interacción y salida de Leishmania amazonensis en un modelo in vitro con macrófagos murinos de la línea celular J774A.1 (trabajo de grado). Bogotá: Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia; 2005.
[ Links ]LEÓN S, DÍAZ JC, CORRALES A, CAMACHO M. La salida de Leishmania amazonensis de su célula hospedera está asociada con procesos de fusión de membranas. Biomedica. 2005;25(S1):173.
[ Links ]LEY V, ANDREWS NW, ROBBINS ES, NUSSENZWEIG V. Amastigotes of Trypanosoma cruzi sustain an infection cycle in mammalian cells . J Exp Med. 1988;168:649.
[ Links ]LEY V, ROBBINS ES, NUSSENZWEIG V, ANDREWS NW. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J Exp Med. 1990;171(2):401-413.
[ Links ]LIU XK, KATCHMAN A, EBERT SN, WOOSLEY RL. The antiestrogen tamoxifen blocks the delayed rectifier potassium current, IKr, in rabbit ventricular myocytes. J Pharmacol Exp Ther. 1998;287(3):877-883.
[ Links ]MCNALLY AK, ANDERSON JM. Multinucleated giant cell formation exhibits features of phagocytosis with participation of the endoplasmic reticulum. Exp Mol Pathol. 2005;79(2):126-135.
[ Links ]MONTOYA JG, LIESSENFELD O. Toxoplasmosis. Lancet. 2004;363:1965-76.
[ Links ]MOUDY R, MANING TJ, BECKER CJ. The Loss of Cytoplasmic Potassium upon Host Cell Breakdown Triggers Egress of Toxoplasma gondii. J Biol Chem. 2001;276(44):41492-41501.
[ Links ]NORONHA FS, CRUZ J, BEIRAO PS, HORTA MF. Macrophage damage by Leishmania amazonensis cytolysin: evidence of pore formation on cell membrane. Infect Immun. 2000;68(8):4578-4584.
[ Links ]NORONHA FS, RAMALHO-PINTO FJ, HORTA MF. Identification of a putative pore-forming hemolysin active at acid pH in Leishmania amazonensis. Braz J Med Biol Res. 1994;27(2):477-482.
[ Links ]NORONHA FS, RAMALHO-PINTO FJ, HORTA MF. Cytolytic activity in the genus Leishmania: Involvement of a putative pore-forming protein. Infect Immun. 1996;64(10):3975-3982.
[ Links ]PEI X, GUO X, COPPEL R, MOHANDAS N, AN X. Plasmodium falciparum erythrocyte protein 3 (PfEMP3) destabilizes erythrocyte membrane skeleton. J Biol Chem. 2007;282(37):26754-26758.
[ Links ]PINGRET L, MILLOT J-M, SHARONOV S, BONHOMME MM, PINON J-M. Relationship Between Intracellular Free Calcium Concentrations and the Intracellular Development of Toxoplasma gondii. J Histochem Cytochem. 1996;44(10):1123-1129.
[ Links ]RAYNER JC. Erythrocyte exit: Out, damned merozoite! Out I say! Trends Parasitol. 2006;22(5):189-192.
[ Links ]RITTIG MG, BOGDAN C. Leishmania-host-cell interaction: complexities and alternative views. Parasitol Today. 2000;16(7):292-297.
[ Links ]RITTIG MG, SCHROPPEL K, SEACK KH, SANDER U, N DIAYE EN, et al. Coiling phagocytosis of trypanosomatids and fungal cells. Infect Immun. 1998;66(9):4331-4339.
[ Links ]RUSSELL DG. Mycobaterium and Leishmania: stowaways in the endosomal network. Trends Cell Biol. 1995;5:125-128.
[ Links ]SALMON BL, OKSMAN A, GOLDBERG DE. Malaria parasite exit from the host erythrocyte: a two-step process requiring extraerythrocytic proteolysis. Proc Natl Acad Sci USA. 2001;98(1):271-276.
[ Links ]SCHWAB JC, BECKERS CJ, JOINER KA. The parasitophorous vacuole membrane surrounding intracellular Toxoplasma gondii functions as a molecular sieve. Proc Natl Acad Sci USA. 1994;18;91(2):509-513.
[ Links ]SONI S, DHAWAN S, ROSEN KM, CHAFEL M, CHISHTI AH, HANSPAL M. Characterization of events preceding the release of malaria parasite from the host red blood cell. Blood Cells Mol Dis. 2005;35:201-211.
[ Links ]STOMMEL EW, SCHWARTZMAN JP, KASPER LH. Toxoplasma gondii: Dithiolinduced Ca+2 flux causes egress of parasites from the parasitophorous vacuole. Exp Parasitol. 1997;87:88-97.
[ Links ]TODD W, CALDWELL HD. The interaction of Chlamydia trachomatis with host cells: ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J Infect Dis. 1985;151(6):1037-1044.
[ Links ]TRAGER W. On the release of malaria merozoites. Trends Parasitol. 2002;18:60-61.
[ Links ]VIGNERY A. Macrophage fusion: the making of osteoclasts and giant cells. J Exp Med. 2005;202:337-340.
[ Links ]VIGNERY A. Osteoclasts and giant cells: macrophage-macrophage fusion mechanism. Int J Exp Pathol. 2000;81:291-304.
[ Links ]WICKHAM ME, CULVENOR JG, COWMAN AF. Selective inhibition of a two-step egress of malaria parasites from the host erythrocyte. J Biol Chem. 2003;278(39):37658-37663.
[ Links ]WINOGRAD E, CLAVIJO CA, BUSTAMANTE LY, JARAMILLO M. Release of merozoites from Plasmodium falciparum infected erythrocytes could be mediated by a non explosive event. Parasitol Res. 1999;85:621-624.
[ Links ]
