










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.16 no.1 Bogotá Jan./Apr. 2011
ANÁLISIS DE ESTs DE YUCA (Manihot esculenta): UNA HERRAMIENTA PARA EL DESCUBRIMIENTO DE GENES
Analysis of Cassava (Manihot esculenta) ESTs: A Tool for the Discovery of Genes
ANDRES ZAPATA1, Ingeniero Mecánico; RAFIK NEME1, Biólogo; CAROLINA SANABRIA1, Biólogo; CAMILO LÓPEZ1, Ph. D. 1 Grupo de Fitopatología Molecular, Departamento de Biología, Facultad de Ciencias. Universidad Nacional de Colombia. celopezc@unal.edu.co
Presentado 17 de febrero de 2010, aceptado 14 de febrero de 2011, correcciones 15 de febrero de 2011.
RESUMEN
La yuca (Manihot esculenta) constituye la base de la alimentación para más de 1.000 millones de personas en el mundo, consolidándose como el cuarto cultivo más importante en el mundo después del arroz, el maíz y el trigo. La yuca es considerada como un cultivo relativamente tolerante a condiciones de estrés abiótico y biótico; sin embargo estas características se encuentran principalmente en variedades no comerciales. Las estrategias de mejoramiento genético convencional o mediadas por transformación genética representan una alternativa para introducir las características deseadas dentro de las variedades comerciales. Un paso fundamental con miras a acelerar los procesos de mejoramiento genético en yuca requiere el descubrimiento de los respectivos genes relacionados con las características buscadas, para lo cual los ESTs (del inglés Expressed Sequence Tags) son una vía rápida para este fin. En este estudio se realizó un análisis de la colección completa de ESTs disponibles en yuca, representada por 80.459 secuencias, los cuales fueron ensamblados en un conjunto de 29.231 genes únicos (unigen), representado por 10.945 contigs y 18.286 singletones. Estos 29.231 genes únicos pueden representar cerca del 80% de los genes del genoma de yuca. Entre el 5 y 10% de los unigenes de yuca no presentaron similitud con las secuencias presentes en las bases de datos de NCBI y pueden constituir genes específicos de yuca. A un grupo de secuencias del set unigen (29%) fue posible asignarles una categoría funcional de acuerdo al vocabulario Gene Ontology. El componente función molecular es el mejor representado con 43% de las secuencias, seguido por el componente proceso biológico (38%) y finalmente el componente celular (19%). Dentro de la colección de ESTs de yuca se identificaron 3.709 microsatélites que podrán ser empleados como marcadores moleculares. Este estudio representa una contribución importante al conocimiento de la estructura genómica funcional de la yuca y se constituye en una herramienta para la identificación de genes asociados a características de interés agrícola para posteriores programas de mejoramiento genético.
Palabras clave: yuca, ESTs, genómica funcional, anotación, mejoramiento genético.
ABSTRACT
Cassava (Manihot esculenta) is the main source of calories for more than 1,000 millions of people around the world and has been consolidated as the fourth most important crop after rice, corn and wheat. Cassava is considered tolerant to abiotic and biotic stress conditions; nevertheless these characteristics are mainly present in non-commercial varieties. Genetic breeding strategies represent an alternative to introduce the desirable characteristics into commercial varieties. A fundamental step for accelerating the genetic breeding process in cassava requires the identification of genes associated to these characteristics. One rapid strategy for the identification of genes is the possibility to have a large collection of ESTs (Expressed Sequence Tag). In this study, a complete analysis of cassava ESTs was done. The cassava ESTs represent 80,459 sequences which were assembled in a set of 29,231 unique genes (unigen), comprising 10,945 contigs and 18,286 singletones. These 29,231 unique genes represent about 80% of the genes of the cassava’s genome. Between 5% and 10% of the unigenes of cassava not show similarity to any sequences present in the NCBI database and could be consider as cassava specific genes. A functional category was assigned to a group of sequences of the unigen set (29%) following the Gene Ontology vocabulary. The molecular function component was the best represented with 43% of the sequences, followed by the biological process component (38%) and finally the cellular component with 19%. In the cassava ESTs collection, 3,709 microsatellites were identified and they could be use as molecular markers. This study represents an important contribution to the knowledge of the functional genomic structure of cassava and constitutes an important tool for the identification of genes associated to agricultural characteristics of interest that could be employed in cassava breeding programs.
Key words: cassava, ESTs, functional genomics, annotation, molecular breeding.
INTRODUCCIÓN
La yuca (Manihot esculenta) constituye la base de la alimentación para cerca de 1.000 millones de personas en el mundo (FAO, 2008). Las raíces de yuca se caracterizan por tener un alto contenido de almidón, el cual es empleado en una amplia gama de procesos agroindustriales. En particular, a partir del almidón se puede obtener bioetanol, uno de los biocarburantes que están cobrando mayor importancia en la última década como una fuente de energía alternativa (MADR, 2006). La yuca tiene una notable tolerancia al estrés abiótico, se puede cultivar en suelos ácidos de baja fertilidad y es muy tolerante a la sequía (Ceballos, 2002). Aunque la yuca es tolerante a la mayoría de enfermedades y plagas, su producción puede verse seriamente comprometida, principal-mente por enfermedades bacterianas como la bacteriosis vascular, ocasionada por Xanthomonas axonopodis pv. manihotis. Esta enfermedad se encuentra distribuida en todas las regiones en donde se cultiva la yuca (Verdier, 2002). Dentro de las enfermedades virales más importantes se encuentra el mosaico africano, sin embargo ésta enfermedad solo se ha reportado en África (Patil y Fauquet, 2009). A pesar de ser un cultivo rústico y relativamente tolerante a diferentes tipos de estrés abiótico y biótico, estas características se encuentran principalmente en variedades no comerciales y no adaptadas a las diferentes regiones agroecológicas en donde la yuca puede ser cultivada (Ceballos, 2002). Las estrategias de mejoramiento genético convencional o mediadas por transformación genética representan una alternativa para introducir las características deseadas dentro de las variedades comerciales. Sin embargo, un paso imprescindible para el desarrollo de este tipo de estrategias requiere, o pueden ser aceleradas, con el descubrimiento de genes particulares asociados a características de interés agronómico. Una vía rápida y económica para el descubrimiento de genes es a través de la obtención de ESTs (del inglés Expressed Sequences Tags; Rudd, 2003). Los ESTs se obtienen a partir de la secuenciación de clones de librerías de ADNc. Este tipo de secuencias representan los transcritos que son producidos por una célula en un momento particular y bajo las circunstancias específicas en las cuales se obtuvo el tejido con el cual se construyeron las librerías de ADNc. Por esta razón para obtener el repertorio completo de genes de un organismo se requiere contar con un conjunto amplio de librerías de ADNc obtenidas bajo diferentes condiciones. Las bases de datos de ESTs son unas de las de mayor crecimiento, particularmente para plantas cuyos genomas son muy complejos y se dificulta la secuenciación genómica completa. Hasta el momento se ha reportado la secuencia del genoma completo para Arabidopsis (AGI, 2000), arroz (Goff et al., 2002) y álamo (Tuskan et al., 2006), aunque existen otros genomas de plantas secuenciados o prontos a ser completamente secuenciados (http://www.ncbi.nlm.nih.gov/genomes/PLANTS/PlantList.html). Los ESTs, además de proveer una vía expedita para el descubrimiento de genes, son también esenciales para la anotación correcta del genoma, y se constituyen en una fuente importante para la identificación de marcadores moleculares. Los ESTs han permitido identificar genes relacionados con el deterioro poscosecha en frutos (Vizoso et al., 2009), rutas de sabor en cacao (Argout et al., 2008), genes de tolerancia a salinidad (Jha, et al., 2009) y en respuesta a diversos tipos de estrés biótico y abiótico (Galbraith y Birnbaum, 2006). La colección de ESTs en yuca ha crecido en los últimos años. A finales del año 2000, el número de ESTs reportados en la base de datos del GenBank no superaba los 900. A mediados del 2005 el número aumentó a alrededor de 20.000 (López et al., 2004) y, recientemente, a partir de una librería de ADNc de longitud completa, se obtuvieron 20.000 nuevos ESTs (Sakurai et al., 2007). En junio de 2009, la colección de ESTs de yuca representaba alrededor de 80.000. Si bien existen análisis para los ESTs previa-mente reportados, no se ha hecho un análisis unificado de la colección total de ESTs disponibles actualmente. En este estudio nosotros buscamos realizar un nuevo análisis del conjunto de ESTs disponible hasta la hora actual de yuca.
MATERIALES Y MÉTODOS
Los ESTs disponibles en la base de datos dbESTs del GenBank para yuca (Manihot esculenta; http://www.ncbi.nlm.nih.gov/dbEST/dbEST_access.html, marzo de 2009) fueron descargados en un archivo multifasta. Para cada EST se extrajo información acerca del tipo de librería de ADNc que se empleó para la generación de los ESTs y el máximo de información reportada para cada una de ellas. El total de ESTs fueron ensamblados en un set de genes únicos (singletones -secuencias únicas- y contigs) usando CAP3 con los parámetros por defecto (Huang y Madan, 1999).
El set unigen fue empleado para realizar un BLASTX (Altschul et al., 1999) contra las secuencias reportadas en la base de datos no redundante del Genebank (nr). Los alineamientos fueron considerados significativos si el valor E fue menor o igual a 1e–5 y se tomó la secuencia del alineamiento con el mayor score como el descriptor que define la característica del EST de yuca.
El set unigen fue comparado contra el set de ESTs de Arabidopsis thaliana, uva (Vitis vinifera), arroz (Oryza sativa), álamo (Populus trichocarpa), sorgo (Sorghum bicolor) y Brassica napus (descargados de la base de datos de ESTs publicas (http://www.ncbi.nih.gov/dbEST/, diciembre 2009) usando el programa TBLASTX. Los alineamientos similares con un valor E menor o igual a 1e–8 fueron considerados significativos. Un análisis similar se llevo a cabo empleando la base de datos de unigenes de estas especies (http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene).
Para identificar la organización en familias multigénicas y asignar categorías funcionales a los ESTs de acuerdo al vocabulario de Gene Ontology (http://www.geneontology.org/), se utilizó la herramienta UFO (Meinicke, 2009 http://ufo.gobics.de). El set unigen se tradujo a proteínas empleando la herramienta getorf disponible en EMBOSS y se emplearon solamente aquellos marcos de lectura con más de 80 aminoácidos. La secuencia de aminoácidos predicha de estos ESTs (22.281) fue entonces empleada para asignar las categorías funcionales.
Con el objetivo de dar una idea amplia del contenido de los términos de Gene Ontology se identificaron las subcategorías funcionales mediante la construcción de un “GO Slim” empleando la herramienta web CateGOrizer (http://www.animalgenome.org/bioinfo/tools/countgo/) especificando el método de clasificación como Plant_GOslim y método de conteo simple.
El set unigen fue empleado para buscar secuencias que contienen SSR (por sus siglas en ingles: Simple Sequence Repeats), usando el programa Simple Sequence Repeat Identification Tool disponible en http://www.gramene.org/db/markers/ssrtool. El total de genes únicos fueron analizados. Se buscaron secuencias con di, tri y tetra nucleótidos con al menos cinco repeticiones perfectas. Los ESTs conteniendo SSRs fueron agrupados en categorías funcionales empleando el Plant_GOslim como se describió previamente.
RESULTADOS Y DISCUSIÓN
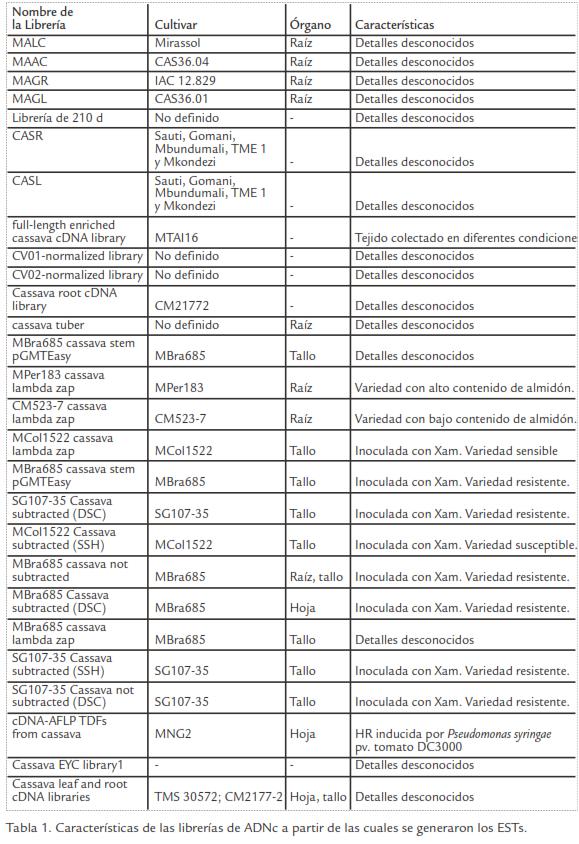
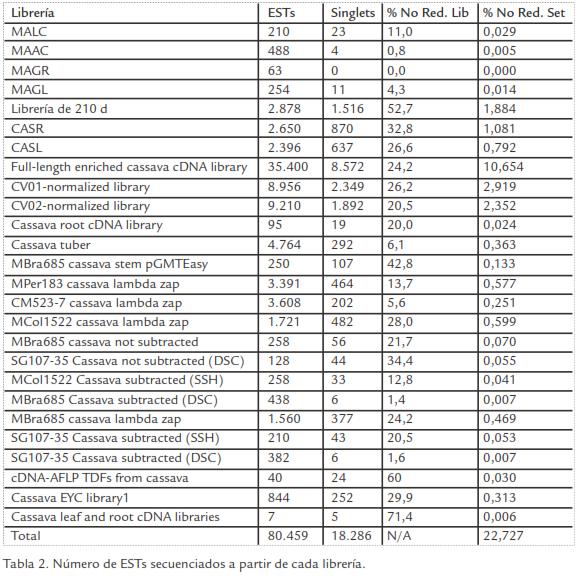
En la base de datos del GenBank existen 80.459 ESTs obtenidos a partir de 26 librerías de ADNc diferentes. Estas librerías provienen de diferentes variedades de yuca, de diferentes tejidos y construidas a partir de plantas sometidas a diferente tipo de estímulo y/o condiciones (Tabla 1). Adicionalmente también se encuentran librerías obtenidas mediante protocolos de sustracción y normalización. La oportunidad de tener ESTs provenientes de diferentes tipos de librerías y construidas por diferentes estrategias incrementa la probabilidad de identificar transcritos expresados a bajos niveles y correspondientes a genes diferentes. Como se observa en la Tabla 2, algunas de las librerías poseen un bajo número de ESTs y pueden corresponder a secuencias de genes expresadas bajo condiciones muy particulares. La librería con mayor número de ESTs corresponde a aquella que presenta clones de ADN de longitud completa (Tabla 2), lo cual constituye una herramienta valiosa para la anotación del genoma de yuca. El número total de ESTs obtenidos a partir de la totalidad de librerías de ADNc es de 80.459. A pesar de los grandes esfuerzos llevados a cabo recientemente para ampliar el número de secuencias de ESTs de yuca este número sigue siendo relativamente bajo si se considera el número de ESTs presentes en otras especies vegetales. Las plantas con mayor número de ESTs son maíz (2’018.798), Arabidopsis (1’527.298), soya (1’422.604) y arroz (1’249.110). Sin embargo para plantas modelos como el álamo (Populus trichocarpa) cuyo genoma ha sido completamente secuenciado, solo se han reportado 89.943 ESTs en el GenBank. El número de ESTs en yuca es similar al reportado para otras especies de plantas de interés agrícola como fríjol (83.847), papaya (77.393) o pimentón (118.054).
El total de ESTs de yuca fue ensamblado en un conjunto de 29.231 secuencias únicas, representada por 10.945 contigs (secuencias sobrelapantes o similares) y 18.286 singletones. Del total de 80.459 ESTs, 62.173 hicieron parte de los contigs (77,3%). El tamaño promedio de los contigs fue de 795 pb y el de los singletones fue de 504 pb.
Los genomas de plantas completamente secuenciados han permitido identificar que el genoma de Arabidopsis contiene 25.498 genes (AGI, 2000), el de arroz entre 46.000 a 56.000 genes (Goff et al., 2002) y Populus trichocarpa 45.000 genes (Tuskan et al., 2006). La reciente liberación del genoma de yuca estima el número de genes a 35.000 (http://www.phytozome.net/cassava.php). De esta manera, el conjunto de 29.231 secuencias únicas, puede representar cerca del 83,5% de los genes totales de yuca.
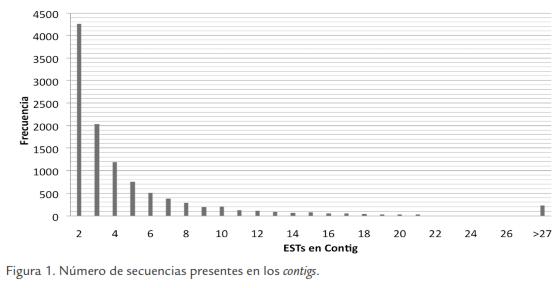
El número de secuencias en cada contig se muestra en la Figura 1, en donde se puede observar que la mayoría de los contigs contiene pocas secuencias (dos o tres). El 75% contiene entre dos y cinco secuencias por contig. El número de ESTs en los contigs varió de dos a 626. Los contigs con mayor número de lecturas fueron el 9.119 con 626 ESTs, el contig 685 con 314 secuencias, el 10.861 con 217 secuencias. Estos contigs presentan similitud con una proteína predicha de Populus trichocarpa, una proteína tipo auxinreprimida ARP1 de Manihot esculenta, y con la ribuolosa 1,5 bifosfato carboxilasa de Manihot esculenta, respectivamente.
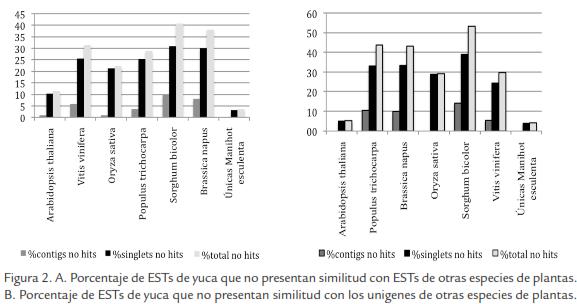
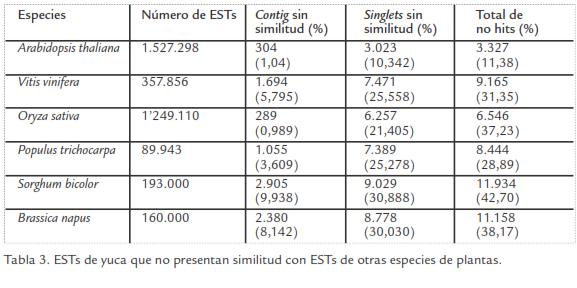
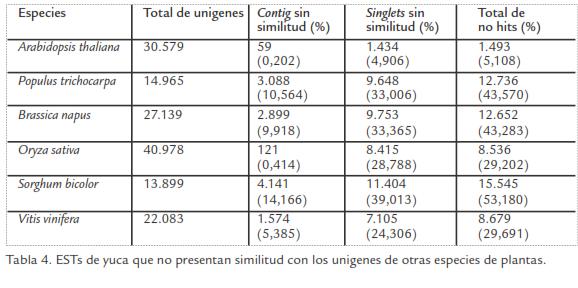
Con el fin de realizar una anotación funcional de genes únicos, se realizó en primer lugar un BLASTX contra la base de datos no redundante (nr) del GenBank. De 29.231 secuencias analizadas, 26.888 (92%) mostraron similitud significativa con alguna de las proteínas presentes en la base de datos. Las secuencias restantes (2.343) no mostraron similitud significativa con ninguna de las proteínas presentes en la base de datos y pueden representar secuencias específicas de yuca. Para determinar la presencia de secuencias específicas de yuca se realizó un TBLASTX de los ESTs de yuca contra las bases de datos de las especies con mayor número de ESTs reportados y para algunas especies cercanas evolutivamente con la yuca (Tabla 3; Figura 2A). El porcentaje de secuencias diferenciales entre yuca y las especies comparadas oscila entre 11% y 40% (Tabla 3; Figura 2A). La especie con la cual la yuca comparte mayor número de secuencias es Arabidopsis, en donde casi el 90% de los ESTs de yuca tienen una similitud significativa con esta planta (Tabla 3; Figura 2A). En otros estudios se han reportado similitudes importantes entre estas dos especies (López et al., 2004; Sakurai et al., 2007), lo cual no sólo debe considerarse a la luz de la cercanía evolutiva entre estas especies sino también al alto número de EST reportados en Arabidopsis, lo que incrementa la probabilidad de encontrar secuencias similares. Entre las especies con menor número de ESTs compartidos con yuca se encuentran soya (40%) y arroz (22%), lo cual es entendible dada la ancestral separación entre monocotiledóneas y dicotiledóneas. Dentro de las especies estudiadas se encuentra Populus trichocarpa, la cual es la especie más cercana evolutivamente con la yuca, perteneciendo al mismo orden taxonómico, Malpighiales. Con esta especie se presentaron relativamente pocos ESTs comunes (29%), aunque es necesario resaltar que en esta especie también el número de ESTs reportados es bajo (89.943) a pesar de que su genoma ha sido completamente secuenciado. Una situación similar se presentó cuando se consideraron las secuencias de las base de datos de unigenes (Tabla 4; Figura 2B). En total, los ESTs de yuca que no presentaron similitud con ESTs de las especies seleccionadas fue de 11% o de 5% cuando se consideran los ESTs o los unigenes, respectivamente. Estas 670 secuencias pueden representar genes únicos de yuca. Sin embargo algunas de ellas pueden ser demasiado cortas como para no presentar similitud significativa o correspondan a secuencias que contienen UTRs (del ingles untranslated regions) demasiado largos, lo que dificulta la identificación de similitudes con otras secuencias presentes en la base de datos.
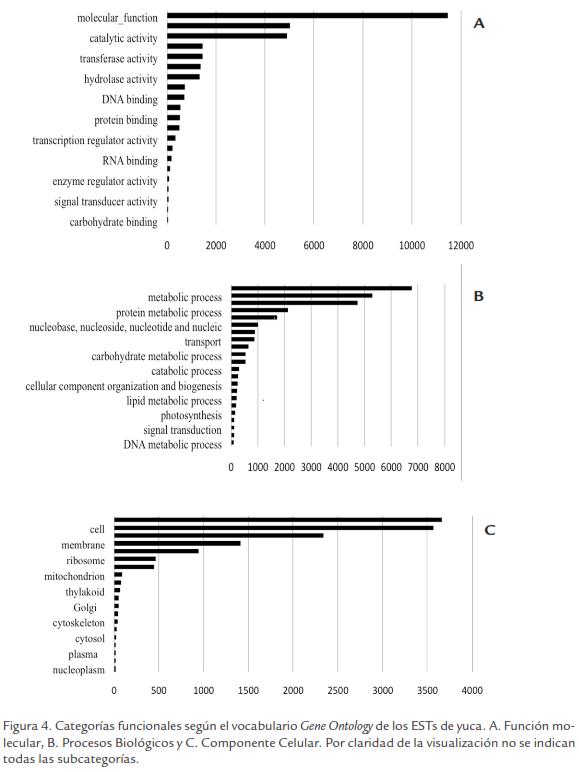
Los ESTs del set unigen fueron agrupados en categorías funcionales previamente definidas empleando el vocabulario de Gene Ontology. Para ello, primero se identificaron aquellas secuencias que presentaron marcos abiertos de lectura mayores a 80 aminoácidos, obteniéndose en total 22.281, de las cuales solo a 8.494 (29%) fue posible asignarles una categoría funcional. La distribución dentro de las tres grandes categorías funcionales se muestra en la (Figura 3), en donde se observa que el componente función molecular es el mejor representado con 43% de las secuencias, seguido por el componente proceso biológico (38%) y finalmente el componente celular (19%). Los números de los ESTs en cada categoría funcional no son aditivos puesto que un EST puede ser ubicado en dos o más categorías funcionales, ya que es posible encontrar múltiples relaciones dentro de las categorías jerárquicas de los términos de Gene Ontology. Dentro de la categoría función molecular, la mayoría de secuencias codifican para proteínas implicadas en unión a diferentes sustratos (5.027 secuencias) y en actividades enzi-máticas (4.904; Figura 4A). Dentro de los procesos biológicos la mayor parte de los ESTs codifican proteínas involucradas en procesos metabólicos (5.293) y celulares (4.736), reflejando la importancia que tienen estos procesos en el funcionamiento celular básico (Figura 4B). La categoría de respuestas a estrés solo esta representado por 176 secuencias. Las subcategorías mejor representadas dentro del componente celular se encuentran el celular (3.567 secuencias) y el intracelular (2.340; Figura 4C).

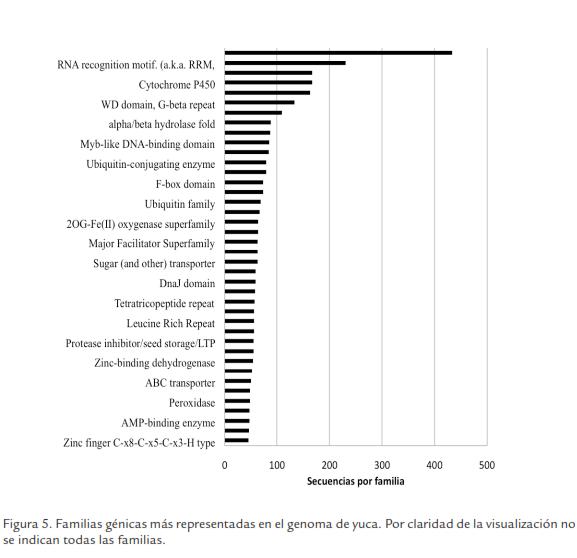
Los genomas de células eucariota superiores están formados en algunos casos por un conjunto amplio de genes organizados en familias multigénicas (AGI, 2000; Goff et al., 2002). Con el fin de identificar la presencia de familias multigénicas en yuca, se identificaron aquellas secuencias que presentarán dominios conservados presentes en la base de datos Pfam. Aquellas secuencias que compartiesen dominios conservados fueron consideradas miembros de la misma familia génica. Las familias multigénicas mejor representadas fueron las de los genes que codifican para proteínas que presentan un dominio kinasa, las que presentan dominios de unión al ARN, citocromo P450 y factores de transcripción de la familia dedos de zinc (Figura 5). Este tipo de proteínas han sido previamente descritas como ampliamente distribuidas en el genoma de varias especies de plantas (AGI, 2000; van der Hoeven et al., 2002; Flinn et al., 2005; Goff et al., 2002). Estas proteínas están implicadas en una amplia gama de funciones que van desde actividades de metabolismo funcional como aspectos de respuesta a estímulos específicos.
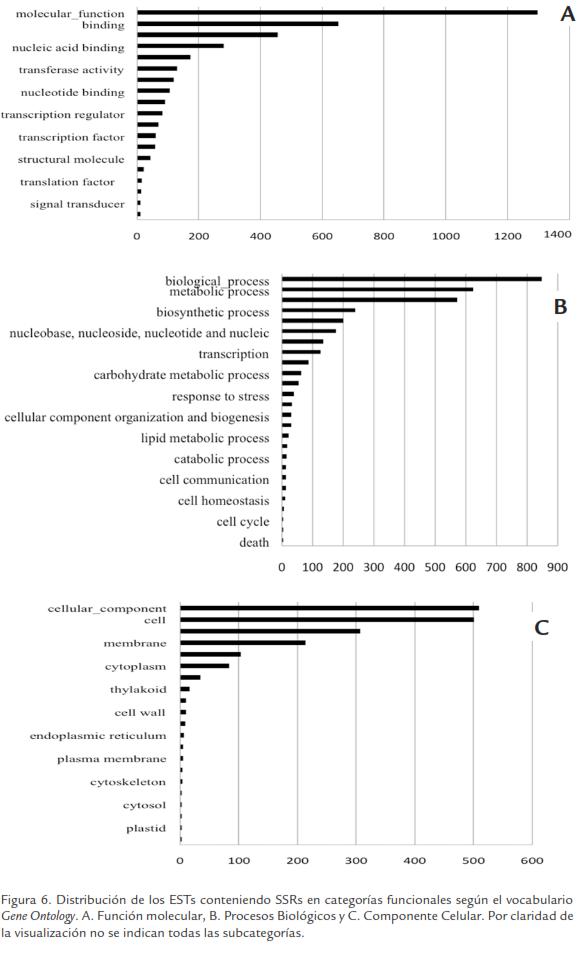
Uno de los marcadores moleculares más ampliamente empleados en estudios de diversidad y de mapeo genético son los microsatélites, los cuales están formados por secuencias repetitivas de un corto motivo (de dos a cinco nucleótidos) (Agarwal et al., 2008). Por mucho tiempo se consideró que los SSRs están asociados principalmente con ADN no codificante, pero recientemente se ha establecido que este tipo de secuencias están incluso mejor representadas en regiones transcritas (Varshney et al., 2005). En consecuencia, las bases de datos de ESTs se constituyen en una fuente ideal para identificar este tipo de secuencias repetitivas. La utilización de secuencias codificantes como marcadores tiene la gran ventaja que en estudios de mapeo genético permiten establecer asociaciones mas directas entre fenotipos y marcadores moleculares. Del conjunto de 29.231 secuencias de ESTs se encontró que en 3.709 de ellas se presentan microsatélites. Los microsatélites más frecuentes fueron los dinucleótidos (2.600, 70%), seguido por los trinucleótidos (1.037, 28%) y los tetranucleótidos (72,2%; Tabla 3; Figura 5). Un estudio previo en yuca, analizando 8.577 ESTs reportó la presencia de un total de 846 microsatélites, de los cuales 68,7% representan dinucleótidos, 30,4% de trinucleótidos y solo se identificaron 0,6% y 0,3% de tetra y pentanucleótidos respectivamente (Raji et al., 2009). Estos números son muy similares a nuestro estudio que incluye aproximadamente 10 veces más ESTs. Los microsatélites derivados de estos ESTs pueden ser empleados para mapeo, estudios de diversidad o pueden ser transferidos a otras especies relacionadas. Los microsatélites dentro de secuencias codificantes pueden tener un efecto fundamental en la actividad génica, ya que la expansión o contracción en el número de unidades repetitivas puede alterar el producto génico, produciendo proteínas disfuncionales o con nuevas funciones, lo que en últimas puede cambiar significativamente el fenotipo. Dentro de los microsatélites encontrados, la mayoría de ellos se encuentran en los ESTs asignados en la categoría del Pfam PF00076 motivo de reconocimiento de ARN (RNA recognition motif: RRM, RBD, or RNP domain), seguido por PF00097 dedos de zinc (Zinc finger, C3HC4 type (RING finger). La identificación de SSRs en secuencias que codifican proteínas específicas permitirá desarrollar estrategias dirigidas hacia el mapeo de genes implicados en una función particular. De esta forma los ESTs conteniendo SSRs se agruparon en categorías funcionales empleando la información disponible según el Gene Ontology (Fig. 6A; Fig. 6B; Fig. 6C). En este estudio hemos unificado la información generada sobre ESTs en yuca logrando realizar un análisis comprensivo de 29.231 genes expresados. Este tipo de información será valiosa para la identificación dirigida de grupos de genes de interés agronómico con miras a estudiar en mayor profundidad su función y ser incorporados dentro de los programas de mejoramiento genético de yuca. Al mismo tiempo la anotación de este grupo de genes facilitará la anotación de la secuencia completa del genoma de yuca liberada recientemente.
AGRADECIMIENTOS
Los autores agradecen el apoyo económico de la Dirección de Investigaciones de la Universidad Nacional, sede Bogota (DIB) y de Colciencias.
BIBLIOGRAFÍA
ALTSCHUL S, MADDEN T, SCHÄFFER A, ZHANG J, ZHANG Z, MILLER W y LIPMAN D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 1999;25:3389-3402.
[ Links ]AGARWAL M, SHRIVASTAVA N, PADH H. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 2008;27:617-631.
[ Links ]AGI. Arabidopsis Genome Initiative. Analyses of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:769-815.
[ Links ]ARGOUT X, FOUET O, WINCKER P, GRAMACHO K, LEGAVRE T, SABAU X, et al. Towards the understanding of the cocoa transcriptome: Production and analysis of an exhaustive dataset of ESTs of Theobroma cacao L. generated from various tissues and under various conditions. BMC Genomics. 2008;9:512.
[ Links ]CEBALLOS H. La yuca en Colombia y el mundo: nuevas perspectivas para un cultivo milenario. En: CIAT (eds.). La yuca en el Tercer Milenio: Sistemas modernos de producción, procesamiento, utilización y comercialización; 2002.
[ Links ]FAO. Crop prospects and food situation, FAO corporate document repository. Oct 2008. Disponible en URL: http://www.fao.org/docrep/011/ai473e/ai473e03.htm
[ Links ]FLINN B, ROTHWELL C, GRIFFITHS R, LÄGUE M, DEKOEYER D, SARDANA R, AUDY P, GOYER C, LI XQ, WANG-PRUSKI G, REGAN S. Potato expressed sequence tag generation and analysis using standard and unique cDNA libraries. Plant Mol Biol. 2005;59:407-433.
[ Links ]GALBRAITH DW, BIRNBAUM K. Global studies of cell type-specific gene expression in plants. Annu Rev Plant Biol. 2006;57:451-475.
[ Links ]GOFF SA, RICKE D, LAN TH, PRESTING G, WANG R, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002;296:92-100.
[ Links ]HUANG X, MADAN A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868-877.
[ Links ]JHA B, AGARWAL PK, REDDY PS, LAL S, SOPORY SK, REDDY MK. Identification of salt-induced genes from Salicornia brachiata, an extreme halophyte through expressed sequence tags analysis. Genes Genet Syst. 2009;84:111-120.
[ Links ]LÓPEZ C, JORGE V, PIÉGU B, MBA C, CORTES D, RESTREPO S, et al. A unigene catalogue of 5700 expressed genes in cassava (Manihot esculenta). Plant Mol Biol. 2004;54:541-554.
[ Links ]MADR. Ministerio de Agricultura y Desarrollo Rural. Apuesta Exportadora Agropecuaria. Biocombustibles. http://www.minagricultura.gov.co/inicio/default.aspx; 2006.
[ Links ]MEINICKE P. UFO: a web server for ultra-fast functional profiling of whole genome protein sequences. BMC Genomics. 2009;10:409.
[ Links ]PATIL BL Y FAUQUET CM. Cassava mosaic Geminiviruses: actual knowledge and perspectives. Mol. Plant Pathol. 2009;10:685-701.
[ Links ]RAJI AA, ANDERSON JV, KOLADE OA, UGWU CD, DIXON AG, INGELBRECHT IL. Gene-based microsatellites for cassava (Manihot esculenta Crantz): prevalence, polymorphisms, and cross-taxa utility. BMC Plant Biol. 2009;9:118.
[ Links ]RUDD S. Expressed sequence tags: alternative or complement to whole genome sequences? Trends in Plant Science. 2003;8:321-329.
[ Links ]SAKURAI T, PLATA G, RODRÍGUEZ-ZAPATA F, SEKI M, SALCEDO, A TOYODA, A, ISHIWATA, A, et al. Sequencing analysis of 20,000 full-length cDNA clones from cassava reveals lineage specific expansions in gene families related to stress response. BMC Plant Biol. 2007;7:66.
[ Links ]TUSKAN GA, DIFAZIO S, JANSSON S, BOHLMANN J, GRIGORIEV I, HELLSTEN U, PUTNAM, N, RALPH S, et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596-1604.
[ Links ]VAN DER HOEVEN R, RONNING C, GIOVANNONI J, MARTIN G, TANKSLEY S. Deductions about the number, organization, and evolution of genes in the tomato genome based on analysis of a large expressed sequence tag collection and selective genomic sequencing. Plant Cell. 2002;14:1441-1456.
[ Links ]VARSHNEY RK, GRANER A, SORRELLS ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48-55.
[ Links ]VERDIER V. Bacteriosis vascular (o añublo bacteriano) de la yuca causada por Xanthomonas axonopodis pv. manihotis. En: CIAT (eds.). La yuca en el Tercer Milenio: Sistemas modernos de producción, procesamiento, utilización y comercialización; 2002. p. 148-159.
[ Links ]VIZOSO P, MEISEL L, TITTARELLI A, LATORRE M, SABA J, CAROCA R, MALDONADO J, et al. Comparative EST transcript profiling of peach fruits under different post-harvest conditions reveals candidate genes associated with peach fruit quality. BMC Genomics. 2009;10:423.
[ Links ]
