










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.16 no.3 Bogotá Sept./Dec. 2011
BUSCANDO AGUJAS EN UN PAJAR: VIAJES DE RNAS PEQUEÑOS IN SILICO E IN VITRO
Finding for a Neadle in a Haystack: Trips of Small RNAs from in silico to in vitro
CLARA ISABEL BERMÚDEZ SANTANA1. 1Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia, Sede Bogotá, Carrera 45 n.º 26-85 - Edificio Uriel Gutiérrez, Bogotá D.C.–Colombia, cibermudezs@unal.edu.co
Presentado 13 de enero de 2011, aceptado 25 de abril de 2011, correcciones 1 de julio de 2011.
RESUMEN
Lo que se conoce tradicionalmente como análisis del transcriptoma comprende la caracterización y cuantificación del conjunto de RNAs transcritos en la célula. En los primeros años, los estudios del transcriptoma se enfocaron principalmente a RNA mensajeros o RNA codificantes. Más recientemente, debido a avances en la tecnología de secuenciammiento de última generación, los estudios del transcriptoma se han extendido a RNAs no codificantes. Estas moléculas presentan gran variedad de funciones en la regulación celular. Una caracterización completa del conjunto de ncRNAs no es alcanzable con las aproximaciones disponibles. Hoy en día la identificación de RNAs no codificantes y de sus RNA pequeños derivados, es un tema de vital importancia en el análisis genético. Uno de los avances recientes en el estudio del transcriptoma por ejemplo, se enfoca en la biología de RNA de interferencia y su aplicación clínica. Durante los últimos años se han desarrollado continuamente la capacidad de cómputo, eficiencia y capacidad de almacenamiento para modelar y procesar grandes volúmenes de información producto de secuenciamiento de última generación. Como consecuencia, esta revisión presenta el análisis del transcriptoma desde una perspectiva histórica unida a la modelación computacional de RNA no codificantes de datos de bibliotecas de RNAs pequeños.
Palabras clave: transcriptoma, RNA pequeños, secuenciamiento de última generación.
ABSTRACT
What is commonly known about transcriptome studies encompass the characterization and quantification of the complete set of RNA transcripts produced by a cell. In its early days these analysis mainly focused on examination of messenger RNAs or coding RNAs. More recently, due to advances in next-generation sequencing technologies, transcriptome analysis has expanded to non-conding RNAs (ncRNAs). These molecules exhibit high variety of functions in cell regulation. Through characterization of complete sets of ncRNAs is not attainable with current approaches. At present de novo identification of ncRNAs and ncRNA-derived small RNAs is a major issue in genetic analysis. One of the most important recent advances of transcriptome analysis focuses, for example, on RNA interference (RNAi) biology and its clinical application. High-performance computing, storage capability and computational modeling have been continuously developed during last years to process and model large amounts of products of next generation sequencing methods. As consequence this review describes transcriptome sequencing analysis from a historical perspective to its link to computational approaches to model ncRNAs from small RNA library data.
Key words: Transcriptome, small RNAs, next-generation sequencing technology.
INTRODUCCIÓN
El transcriptoma se define como el conjunto de todas las moléculas funcionales de RNAs producto de la expresión de genes en una célula o, en una población de células y, con más detalle, como la cuantificación de material transcrito y su relación con la expresión diferencial de genes (Morozova et al., 2009). De esta inmensa población molecular, una primera porción se compone de RNA mensajeros, producto de la transcripción de genes que finalmente son traducidos en proteínas y, una segunda porción, no menos importante para la fisiología celular, se compone de RNAs no codificantes, también producto de la transcripción, con funciones diversas como su papel en la formación de ribonucleoproteínas, adaptación de aminoácidos en la traducción y para el caso particular de los más pequeños (tema principal de esta revisión), la regulación de la expresión génica mediada por RNAs.
El análisis del transcriptoma, bien sea por la estimación de su tamaño, o por la identificación de los tipos de RNAs, entre otros, es una tarea compleja, aún en desarrollo y compromete múltiples esfuerzos de la investigación experimental (in vitro) y computacional (in silico). La parte experimental comprende aislamiento y secuenciamiento de productos de la transcripción, su tipificación y cuantificación. Desde los trabajos iniciales de Sanger (Sanger y Coulson,1974; Sanger et al.,1977; el único método de secuenciamento usado por muchos años) la biología se ha visto beneficiada por el secuenciamiento de genómas de muchos organismos, y por el conocimiento ganado sobre el análisis del transcriptoma. Sin embargo, la necesidad de disminuir costos e incrementar el rendimiento del secuenciamiento, conllevó al desarrollo de nuevos métodos conocidos hoy como de última generación. Estos han logrado el resecuenciamiento de genomas, lo esperado en la relación costo-eficiencia, una disminución de costos y un incremento en la eficiencia del secuenciamiento. Debido al descubrimiento de nuevos RNAs y de nuevos detalles de análisis de la maquinaria asociada a su biogénesis y función (Tanzer et al., 2010), y hoy por hoy, hemos expandido nuestras fronteras del conocimiento al estudio de los caminos de regulación de la expresión de genes mediada por RNAs pequeños.
DESDE SANGER HASTA LOS MÉTODOS DE SECUENCIAMIENO DE ÚLTIMA GENERACIÓN
En 1977, utilizando el método de Sanger, fue secuenciado en su totalidad el primer genoma, constituido por 5.386 nucleotidos, perteneciente al bacteriófago phiX174 (Sanger et. al., 1977). Este método se conoce como de terminación de cadena o método de secuenciamiento enzimático (Sanger y Coulson,1974; Sanger et al.,1977). Aunque previamente otros métodos de secuenciamiento fueron utilizados (Gilbert y Maxam ,1973), el método de Sanger se convirtió en el predilecto para secuenciamiento de DNA. Modelos utilizados actualmente como Applied Biosystems 3xxx series o GE Healthcare Mega-BACE, utilizan el mismo concepto de secuenciamiento (Kircher y Kelso, 2010) con un ligero incremento del rendimiento por la automatización de algunos pasos, el uso de electroforesis capilar (Swerdlow y Gesteland,1990; George et al.,1997) y el uso de una sola reacción en el proceso (Smith,1986; contrario al método original de Sanger en el cual cuatro reacciones y lecturas separadas se realizaban: una por cada terminador de cadena marcado radioactiva u ópticamente). Sin embargo, el término original -secuenciamiento de alto rendimiento- fue acuñado para el secuenciamiento automatizado del método de Sanger donde la tecnología incluye más capilares, (de 96 y posible hasta 384 capilares) entre otros. Sin embargo, este concepto ha sido completamente reevaluado y quizás en un futuro de nuevo cambiará cuando surjan nuevas tecnologías (Kircher y Kelso, 2010).
Actualmente, el uso métodos de secuenciamiento de últimas generaciones han transformado las ciencias genómicas y muchos campos de la biología (Schuster, 2008). Estas tecnologías, de bajo costo y alto rendimiento se utilizaron por primera vez en 2005 cuando un sistema de secuenciamiento en paralelo, desarollado por -454 Life Sciences-, fue reportado para ensamblar y secuenciar de novo el genoma de Mycoplasma genitallium (Margulies et al., 2005). Simultáneamente, el laboratorio de George Church reportó el protocolo de secuenciamiento -multiplex polony- utilizado por primera vez para resecuenciar el genoma de una cepa de Escherichia coli (Shendure et al.,2005). Otros estudios en biología han utilizado el sistema de secuenciamiento paralelo masivo como la tecnología desarrollada por Solexa (Bentley et al., 2008), hoy conocida como Illumina SBS technology, para identificar modificaciones de histonas en cromatina humana (Barski et al., 2007), o la tecnología -ChIPSeq- (Johnson et al., 2007). Uno de los pasos experimentales de las nuevas tecnologías de secuenciamiento corresponde al reemplazo del clonado in vivo (como es necesario en el secuenciamiento de Sanger) por amplificación in vitro basada en PCR. Las tecnologías desarrolladas (por ejemplo 454, Ilumina y SOLiD), producen una colección clonal de copias que son posteriormente secuenciadas. Un método, que elimine el paso de amplificación y basado en secuenciamiento directo de una única molécula implementado por Helicos y Pacific Biosciences aún en desarrollo es referido como método de tercera generación (Braslavsky, 2003; Morozova, 2009). Este método se proyecta aun más eficiente por la reducción de costos y disminución de pasos en el secuenciamiento de segunda generación.
Finalmente, la tecnología de secuenciamiento de última generación ha sido utilizada para descifrar la complejidad de la expresión celular. En combinación con métodos para cuantificar transcritos y sus isoformas, la aproximación RNA-seq (Wang et al., 2009) ha permitido el mapeo y cuantificación de transcriptomas de mamíferos (Mortazavi et al., 2008), caracterización de transcriptomas de células madre, de líneas celulares de cáncer de seno, de células HeLa (Marioni et al., 2008; Cloonan et al., 2008; Zhao et al., 2009) y de levadura entre otros. Adicionalmente, esta nueva herramienta ha permitido el surgimiento de una de las principales áreas de la genética moderna: el estudio de promotores alternativos (Strausberg y Levy, 2007).
ANÁLISIS DEL TRANSCRIPTOMA: UNA BREVE PERSPECTIVA HISTÓRICA
Los primeros estudios del transcriptoma se basaron en aislamiento de RNA total de diferentes tejidos y el uso de la técnica de -Northern blot- para la detección de transcritos (Alwine, 1977). Las limitaciones dadas por el requerimiento de volúmenes grandes de RNA para el análisis, fueron posteriormente superadas por el uso de PCR cuantitativa de transcripción reversa o (RT-qPCR; Becker-Andre y Hahlbrock, 1989). Sin embargo, aún después de su amplio uso, esta no permite analizar un transcriptoma a gran escala (Morozova, 2009). Posteriormente, la era de los chips de microarreglos de DNAs (microarrays; Schena, 1995), ha logrado analizar la expresión diferencial de genes en líneas celulares y estudiar patrones del transcriptoma a gran escala. Aunque es una técnica robusta, la medición de la expresión es indirecta y no corresponde a una medida explícita de expresión génica. Algunas alternativas complementarias a los chips de DNAs son los métodos basados en secuenciamiento que permiten directamente la detección de transcritos y más recientemente, la estimación de la abundancia de la expresión (Morozova, 2009). Estos métodos varían desde la construcción y secuenciamiento de librerías de DNA complementario o cDNA, el uso de secuenciamiento de EST (Expressed Sequence Tags por su sigla en inglés) hasta los métodos SAGE (Serial Analysis of Gene Expression), ofreciendo ventajas sobre las técnicas de chips de DNA por que su uso facilita el secuenciamiento tipo Sanger para estudiar expresión génica (Velculescu et al.,1995). Finalmente, como se mencionó en la sección anterior, los últimos estudios de transcriptoma a gran escala actualmente se basan en la tecnología de secuenciamiento de última generación como técnica de secuenciamiento directo del transcriptoma.
SOBRE LAS DESVENTAJAS DEL SECUENCIAMIENO Y LOS GRANDES BENEFICIADOS: lOS RNAS PEQUEÑOS
Los métodos de secuenciamiento de última generación producen lecturas, secuencias producto de la secuenciación conocidas como reads, que varían desde 20 hasta 500 nucleótidos. Sin embargo, el volumen de lecturas de tamaño pequeño producto del secuenciamiento puede superar el orden de 106 por ronda de secuenciación (Kircher y Kelso, 2010) y este manejo de datos fue considerado una desventaja y fuente de crítica en los primeros años de su utilización; primero, por la dificultad de almacenamiento, y segundo por la gran demanda de recursos que dificultó el trabajo computacional para realizar el ensamble, por ejemplo, de un genoma en corto tiempo. Como resultado también se incrementaron los requerimientos de nuevos algoritmos para el análisis.
No obstante, la disponibilidad de lecturas de tamaño pequeño desarrolló una nueva área de investigación para la identificación de RNAs pequeños. El temor de manejar, depurar y anotar (asignar un locus correspondiente) a millones de productos, fue rápidamente reemplazado por el uso eficiente de recursos computacionales. Cientos de laboratorios de biología computacional y bioinformática se han consolidado a nivel mundial con el fin de desarrollar un software comercial o de uso libre con fines académicos. Los laboratorios altamente tecnificados poseen repositorios para bases de datos de productos de secuenciamiento, secuencias de genomas, códigos de programación en desarrollo, interfaces de internet para análisis de -ómicas-, y una comunidad científica que trabaja interdisciplinariamente para estructurar redes de intercambio de información, elaborar y depurar algoritmos para interpretar cuál es el destino final de los productos de secuenciamiento, ensamblar genomas en unos casos y, en otros, como en el análisis de transcriptoma para establecer valores de referencia de expresión y hacer la respectiva anotación genómica. Para los RNAs pequeños queda un mundo por descubrir, un reto continuo para los investigadores en ciencias de la computación: desarrollo de herramientas computacionales para la identificación de genes blanco, de su regulación, identificación de las regiones genómicas que los codifican, predicción automatizada de novo, normalización de datos de su expresión, investigación de su potencial en nuevas terapias y diagnóstico de enfermedades. En palabras de Schuster, 2008, -biólogos y científicos de la computación continúan enfrentándose a nuevos retos para transformar la biología de hoy. Este objetivo será alcanzado a través del desarrollo de modelos computacionales estructuralmente enlazados con la bioquímica, la biofísica y el manejo de los datos experimentales. Una vez desarrollados estos modelos, por medio de ingeniería moderna y computacional se podrán simular, analizar y comprender los datos para de nuevo avanzar en el diseño de nuevos experimentos-.
EL TRANSCRIPTOMA Y LAS CIENCIAS DE LA COMPUTACIÓN: ANOTACIÓN E IDENTIFICACIÓN DE RNAS PEQUEÑOS
Aunque fue superado el problema del manejo de volumen de productos del secuenciamiento, una nueva dificultad surgió debido a errores incorporados en las lecturas. Dependiendo de la técnica usada para secuenciar, errores son incorporados en las lecturas -inserciones, deleciones o sustituciones. Por ejemplo en Solexa, el tipo más común de error es sustitucióm, y en la tecnología 454 inserción o deleción (Hoffmann et al., 2009). Entonces, la demanda para el desarrollo de nuevos métodos computacionales de mapeo (es decir el asignamiento de las regiones genómicas de las cuales son derivadas las lecturas; ver Fig. 1 como ejemplo), se ha incrementado en los últimos años y debe responder de forma robusta al manejo de errores incorporados en las lecturas. Es decir, las nuevas herramientas computaciones dirigidas a resolver este problema han sido desarrolladas para permitir apareamientos no correspondientes entre la secuencia de la lectura y la secuencia de la región genómica blanco. La mayor parte de las herramientas actuales son modificaciones o adaptaciones de técnicas de alineamiento, que deben permitir de forma óptima la incorporación de pequeños huecos y errores mínimos de no apareamiento entre secuencias, para lograr finalmente un mapeo óptimo de lecturas y secuencia genómica de referencia (en otras palabras, cuando una lectura es mapeada al genóma lo esperado es encontrar correspondencias únicas entre nucleótidos, A con A, G con G, etc). Algunos de los programas actuales como SOAP (Li et al., 2008) y Maq (Li et al., 2008) mapean resultados de SOLEXA o SOLiD. SOAP realiza alineamientos con o sin huecos, pero no permite la manipulación de inserciones, deleciones y sustituciones. Actualmente, el programa segemehl (Hoffmann et al., 2009) permite mapeo evaluando estadísticamente todas las fuentes de error posibles del producto del secuenciamiento.
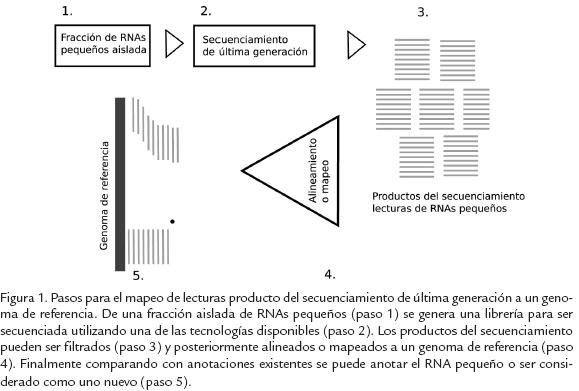
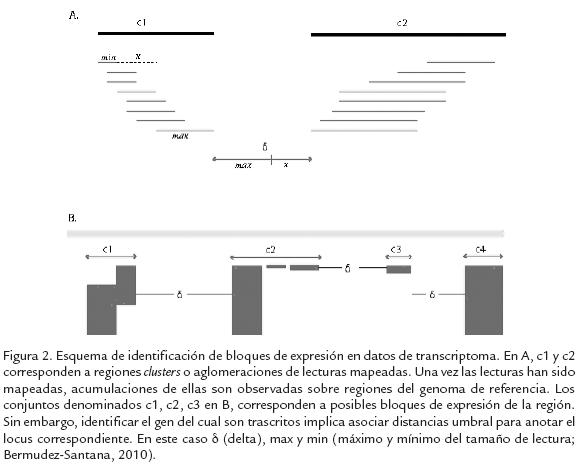
Un resultado a primera vista del mapeo es la observación de lecturas acumuladas en ciertas regiones del genoma. Estos bloques de expresión (ver como ejemplo Fig. 2) son anotados computacionalmente y a su vez son candidatos potenciales para identificar la expresión de RNA pequeños aún desconocidos. Por otro lado, el procesamiento de RNAs y el estudio de producción de nuevas fuentes de RNAs pequeños, es posible gracias al trabajo en conjunto de experimentalistas y científicos de las ciencias de la computación (ver como ejemplo Fig. 3).
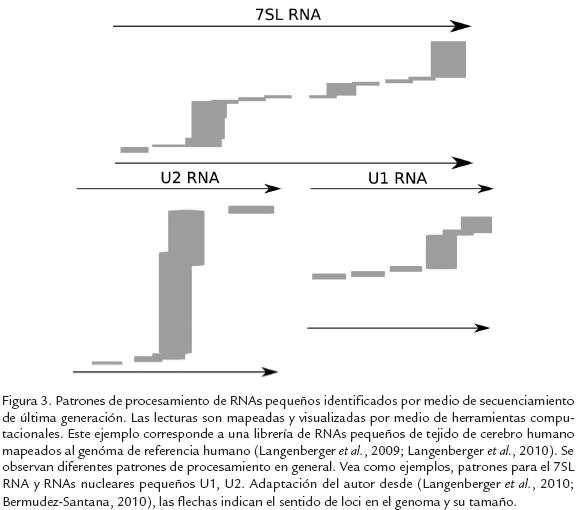
Entonces, el uso de herramientas computacionales permiten hoy en día analizar de forma eficiente el transcriptoma y anotar de manera automática nuevas regiones del genoma que producen RNAs pequeños, así como la visualización de patrones de procesamiento de los RNAs. Aun está abierta la pregunta sobre conocer si estos patrones de expresión pueden corresponder a productos de caminos de control de calidad de los RNAs o a procesamiento de RNAs que posteriormente pueden producir RNAs pequeños funcionales (Langenberger et al., 2010). En la Fig. 3 se observan algunos ejemplos de patrones de expresión asociados a RNAs.
RNAS PEQUEÑOS, CAMINOS DE REGULACIÓN Y SECUENCIAMIENTO DE ÚLTIMA GENERACIÓN
Uno de los principales impactos del uso de nuevas tecnologías de secuenciamiento responde a la caracterización de la regulación de la expresión génica mediada por RNAs pequeños proceso conocido como RNA de interferencia (RNAi). Este proceso biológico es principalmente basado en la función de tres clases de RNA pequeños, microRNAs (miRNAs), RNA pequeños de interferencia (siRNA) y piwiRNAs (piRNAs). El camino fue descubierto entre 1986 y 1990 a través de observaciones de inhibición transcripcional por RNAs antisentido expresados en plantas transgénicas (Ecker y Davis, 1986) y resultados experimentales en inicios de 1990 (Napoli et al., 1990). Aunque el descubrimiento del camino RNAi precede las nuevas tecnologías de secuenciamiento, hoy en día los estudios más detallados de caminos de silenciamiento de genes mediados por el RNAi, son exclusivamente posibles por la existencia de esta tecnología (Haussecker et al., 2010). El RNAi describe una variedad de procesos de silenciamiento de genes mediados por RNAs pequeños que guían complejos macromoleculares para completar su función. Estos caminos de silenciamiento asociados a diferentes proteínas se clasifican en tres mecanismos principales para la regulación de genes: el primero es la inhibición traduccional, donde miRNAs se enlazan a RNA mensajeros e inhiben la traducción, el segundo comprende RNAs pequeños que marcan RNAs mensajeros blanco y causan degradación por medio de la activación del sistema de silenciamiento inducido por RNA. Algunos RNAs pequeños que hacen parte de este camino son los miRNAs, siRNA, piRNAs entre otros. El tercer camino corresponde al silenciamiento de genes por medio de metilación guiada por RNAs pequeños. Una revisión más detallada de los caminos puede consultarse en Mette et al., 2002, Bhattacharyya et al., 2008 y Tanzer et al., 2010.
DATOS SOBRE RNAS PARA CONSULTAR
Finalmente, existen muchos motivos para continuar profundizando sobre la biología de RNAs pequeños y su unión con el mundo RNA. Familias de RNAs están disponibles en http://rfam.sanger.ac.uk/ (Gardner, 2009). Utilizando herramientas computacionales y secuencias de RNAs validadas experimentalmente, actualmente son reportadas 1446 familias de RNAs. El consorcio internacional de RNA RNA Ontology Consortium (ROC) http://roc.bgsu.edu/, se encuentra disponible para definir un marco conceptual sobre la investigación de RNA, programas de cómputo disponibles y de uso en diferentes plataformas como Linux, Windows están disponibles bajo licencia o de uso libre por laboratorios computacionales.
CONCLUSIONES
Los estudios en genómica y transcriptómica se han desarrollado principalmente por nuevos métodos de secuenciamiento y desarrollo computacional. Desde los trabajos pioneros de aplicación de técnicas de secuenciamiento y otros métodos, nuestro conocimiento sobre la expresión diferencial de genes y la organización genómica de diversos organismos se ha incrementado. Históricamente, el incremento en la eficiencia del secuenciamiento requerido por los investigadores conllevó paralelamente al encuentro entre disciplinas de las ciencias de la computación y la biología debido a la necesidad de procesar, almacenar de forma eficiente, sistematizada y computacional millones de productos de secuenciamiento y a las necesidades de construir bases de datos, desarrollar e implementar nuevos algoritmos para alinear los productos de secuenciamiento a los genomas de referencia. Nuevas preguntas de investigación relacionadas con el análisis del transcriptoma han surgido y paralelamente han nutrido el desarrollo de diferentes métodos computacionales. Muchos de los algoritmos disponibles y próximos a implementar, han incrementado nuestro conocimiento de los componentes del transcriptoma y a su vez han ampliado nuestro conocimiento sobre los componentes de menor tamaño o RNAs pequeños. Estos pequeños RNAs nos han sorprendido y nos seguirán sorprendiendo una vez su importancia en la regulación de la expresión genética mediada por RNAs sea conocida en más detalle y nuevas funciones sean descubiertas.
AGRADECIMIENTOS
La autora agradece a Peter F. Satdler, David Langenberger y Steve Hoffmann de la Universidad de Leipzig y a Martin Kircher de MPI-EVA de Leipzig por sus discusiones y bibliografía que aportaron al marco conceptual de esta revisión. A los organizadores de la Cátedra José Celestino Mutis -Todo lo que Usted quizo saber de genética y nunca se atrevió a preguntar.-
BIBLIOGRAFÍA
ALWINE JC, KEMP DJ, STARK GR. Method for Detection of Specific RNAs in Agarose Gels by Transfer to Diazobenzyloxymethyl-paper and Hybridization with DNA Probes. Proc Natl Acad Sci U S A. 1997;74:5350-5354.
[ Links ]BARSKI A, CUDDAPAH S, CUI K, ROH TY, SCHONES DE, WANG Z, et al. High-resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129(4):823-837.
[ Links ]BERMUDEZ-SANTANA C. tRNomics: Genomic Organization and Processing Patterns of tRNAs. Tesis de Doctorado. Leipzig (Alemania). Facultad de Matemáticas e Informática, Universidad de Leipzig; 2010
[ Links ]FILIPOWICZ W, BHATTACHARYYA SN, SONENBERG, N. Mechanisms of Posttranscriptional Regulation by MicroRNAs: Are the Answers in Sight? Nat Rev Genet. 2008;9:102-114.
[ Links ]BRASLAVSKY I, HEBERT B, KARTALOV E, QUAKE SR. Sequence Information can be Obtained from Single DNA Molecules. Proc Natl Acad Sci U S A. 2003;100:3960-3964.
[ Links ]BECKER-ANDRE M, HAHLBROCK K. Absolute mRNA Quantification Using the Polymerase Chain Reaction (PCR). A Novel Approach by a PCR Aided Transcript Titration Assay (PATTY). Nucleic Acids Res. 1989;17:9437-9446.
[ Links ]BENTLEY DR, BALASUBRAMANIAN S, SWERDLOW HP, SMITH GP, MILTON J, BROWN CG, et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature. 2008;456:53-59.
[ Links ]CLOONAN N, FORREST A, KOLLE G, GARDINER B, FAULKNER G, BROWN M, et al. Stem Cell Transcriptome Profiling Via Massive-scale mRNA Sequencing. Nat Methods. 2008;5(7):613-619.
[ Links ]ECKER JR, DAVIS RW. Inhibition of Gene Expression in Plant Cells by Expression of Antisense RNA-. Proc Natl Acad Sci U S A. 1986:83(15):5372-5376. doi:10.1073/pnas.83.15.5372.
[ Links ]GARDNER PP, DAUB J, TATE JG, NAWROCKI EP, KOLBE DL, LINDGREEN S, et al. Rfam: Updates to the RNA Families Database. Nucleic. Acids Res. 2009: 37:D136-D140.
[ Links ]GEORGE KS, ZHAO X, GALLAHAN D, et al. Capillary Electrophoresis Methodology for Identification of Cancer Related Gene Expression Patterns of Fluorescent Differential Display Polymerase Chain Reaction. J Chromatogr B Biomed Sci Appl. 1997;695:93-102.
[ Links ]GILBERT W, MAXAM A. The Nucleotid Sequence of the Lac Operator. Proc Natl Acad Sci U S A. 1973;70:3581-3584.
[ Links ]HAUSSECKER D, HUANG Y, LAU A, PARAMESWARAN P, FIRE A, KAY M. Human tRNA-derived Small RNAs in the Global Regulation of RNA Silencing. RNA. 2010(1):1-5.
[ Links ]HOFFMANN S, OTTO C, KURTZ S, SHARMA C, KHAITOVICH P, STADLER PF et al. Fast Mapping of Short Sequences with Mismatches, Insertions and Deletions Using Index Structures. PLoS Comp Biol. 2009;5(9):1-10.
[ Links ]JOHNSON DS, MORTAZAVI A, MYERS R, WOLD.B Genome-wide Mapping of in vivo Protein-DNA Interactions. Science. 2007;316(5830):1497-1502.
[ Links ]KIRCHER, M, KELSO, J. High-throughput DNA Sequencing -Concepts and Limitations. Bioessays. 2010;32:524-536.
[ Links ]LANGENBERGER D, BERMUDEZ-SANTANA C, HERTEL J, HOFFMANN S, KHAITOVICH P, STADLER PF. Evidence for Human MicroRNA-offset RNAs in Small RNA Sequencing Data. Bioinformatics. 2009;25:2298-2301.
[ Links ]LANGENBERGER D, BERMUDEZ-SANTANA C, STADLER PF, HOFFMANN S. Identification and Classification of Small RNAs in Transcriptome Sequence Data. Pac Symp Biocomput. 2010;15:80-87.
[ Links ]LI R, LI Y, KRISTIANSEN K, WANG J. SOAP: Short Oligonucleotide Alignment Program. Bioinformatics. 2008;24:713-714.
[ Links ]LI H, RUAN J, DURBIN R. Mapping Short DNA Sequencing Reads and Calling Variants Using Mapping Quality Scores. Genome Res. 2008;18:1851-1858.
[ Links ]MARGULIES M, EGHOLM M, ALTMAN W, ATTIYA S, BADER J, BEMBEN L, et al. Genome Sequencing in Microfabricated High-density Picolitre Reactors. Nature. 2005;437(7057):376-380.
[ Links ]MARIONI J, MASON C, MANE S, STEPHENS M, GILAD Y. RNA-seq: An Assessment of Technical Reproducibility and Comparison with Gene Expression Arrays. Genome Res. 2008;18(9):1509-1517.
[ Links ]METTE MF, VAN DER WINDEN J, MATZKE AJ, AUFSATZ W, MATZKE M. RNA-directed DNA Methylation in Arabidopsis. Proc Natl Acad Sci U S A. 2002;99(NIL):16499-16506.
[ Links ]MOROZOVA O, HIRST M, MARRA M. Applications of New Sequencing Technologies for Transcriptome Analysis. Annu Rev Genomics Hum Genet. 2009;10:135-151.
[ Links ]MORTAZAVI A, WILLIAMS B, MCCUE K, SCHAEFFER L, WOLD B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621-628.
[ Links ]NAPOLI C, LEMIEUX C, JORGENSEN R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell. 1990;2(4):279-289.
[ Links ]SANGER F, COULSON AR. A Rapid Method for Determining Sequences in DNA by Primed Synthesis with DNA Polymerase. J Mol Biol. 1974;94:441-448.
[ Links ]SANGER F, AIR GM, BARRELL BG, et al. Nucleotide Sequence of Bacteriophage phiX174 DNA. Nature. 1977;265:687-695.
[ Links ]SCHENA M, SHALON D, DAVIS RW, BROWN PO. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science, 1995; 270:467-470.
[ Links ]SCHUSTER SC. Next-generation sequencing Transforms Today s Biology. Nat methods. 2008;5(1):16-18.
[ Links ]SHENDURE J, PORRECA G, REPPAS N, LIN X, MCCUTCHEON JP, ROSENBAUM A, et al. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science. 2005;309(5741):1728-1732.
[ Links ]SMITH LM, SANDERS JZ, KAISER RJ, et al. Fluorescence Detection in Automated DNA Sequence Analysis. Nature. 1986;321:674-679.
[ Links ]STRAUSBERG R, LEVY S. Promoting transcriptome diversity. Genome Res. 2007;17(7):965-968.
[ Links ]SWERDLOW H, GESTELAND R. Capillary Gel Electrophoresis for Rapid, High Resolution DNA Sequencing. Nucleic Acids Res. 1990;18:1415-1419.
[ Links ]TANZER A, RIESTER M, HERTEL J, BERMUDEZ-SANTANA C, GORODKIN J, HOFACKER I, STADLER PF. Evolutionary Genomics of MicroRNAs and Their Relatives: En: Evolutionary Genomics and Systems Biology, G. Caetano Anolles, eds. Wiley-Blackwell, Hoboken NJ; 2010. p. 295-327
[ Links ]VELCULESCU VE, ZHANG L, VOGELSTEIN B, KINZLER KW. Serial Analysis of Gene Expression. Science.1995;270:484-487.
[ Links ]WANG, Z, GERSTEIN M, SNYDER M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nature Reviews Genetics. 2009 ;10, 57-63.
[ Links ]ZHAO Q, CABALLERO O, LEVY S, STEVENSON B, CSELI C, DE SOUZA S et al. Transcriptome Guided Characterization of Genomic Rearrangements in a Breast Cancer Cell Line. Proc Natl Acad Sci U S A. 2009;106(6):1886-1891.
[ Links ]
