










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Biológica Colombiana
versão impressa ISSN 0120-548X
Acta biol.Colomb. vol.17 no.3 Bogotá set./dez. 2012
MÉTODOS PROTEÓMICOS APLICADOS AL ESTUDIO DE LA MALARIA: Plasmodium falciparum
Proteomics Methods Applied to Malaria: Plasmodium falciparum
YESID CUESTA ASTROZ1, Biólogo; CESAR SEGURA LATORRE1, Ph. D.
1Grupo Malaria, Universidad de Antioquia, Sede de Investigación Universitaria-SIU, Medellín, Colombia. Correspondencia: Cesar Segura. Grupo Malaria. Sede de Investigación Universitaria, Universidad de Antioquia, carrera 53 # 61-30, laboratorio 610, Medellín, Colombia. Telefax: (574) 219 64 87 cesar.segura@siu.udea.edu.co.
Presentado el 14 de junio de 2012, aceptado el 7 de septiembre de 2012, correcciones 11 de septiembre de 2012.
RESUMEN
La malaria es una de las enfermedades parasitarias que causa mayor impacto en la salud pública en países en desarrollo. La secuenciación del genoma de Plasmodium falciparum y el desarrollo de la proteómica han permitido un gran avance en el conocimiento de la biología del parásito. La proteómica ha permitido caracterizar cualitativa y cuantitativamente la expresión de proteínas del parásito y ha proveído información de la expresión relativa de proteínas bajo condiciones de estrés como presión por antimaláricos. Dada la complejidad de su ciclo de vida, el cual se lleva a cabo en el hospedero vertebrado y el mosquito, se ha caracterizado la expresión de proteínas para cada estadio del parásito con el fin de determinar el proteoma que media diversos procesos metabólicos, fisiológicos y energéticos. Técnicas de electroforesis bidimensional, cromatografía líquida y espectrometría de masas, han sido útiles para evaluar los efectos de antimaláricos sobre la expresión de proteínas del parásito y caracterizar el proteoma de diferentes formas y organelas de P. falciparum. El propósito de esta revisión es presentar el estado del arte de los avances en proteómica aplicada al estudio de la malaria, y plantear las diferentes estrategias experimentales empleadas para el estudio del proteoma del parásito con el fin de describir las ventajas y desventajas de cada una de estas metodologías.
Palabras clave: Antimaláricos, bioinformática, Plasmodium falciparum, proteómica.
ABSTRACT
Malaria is a parasitic disease that has a high impact on public health in developing countries. The sequencing of the Plasmodium falciparum genome and the development of proteomics have enabled a breakthrough in understanding the biology of the parasite. Proteomics have allowed to characterize qualitatively and quantitatively the parasite s expression of proteins and has provided information on protein expression under conditions of estrés induced by antimalarials. Given the complexity of their life cycle, which takes place in the vertebrate host and mosquito vector. It has proven difficult to characterize the protein expression during each stage throughout the infection process in order to determine the proteome that mediates several metabolic, physiological and energetic processes. Two dimensional electrophoresis, liquid chromatography and mass spectrometry have been useful to assess the effects of antimalarials on parasite protein expression and to characterize the proteomic profile of differentt P. falciparum stages and organelles. The purpose of this review is to present state of the art tools and advances in proteomics applied to the study of malaria, and to present different experimental strategies used to study the parasite's proteome in order to show the advantages and disadvantages of each one.
Keywords: Antimalarials, bioinformatics, Plasmodium falciparum, proteomics.
INTRODUCCIÓN
La malaria en humanos es causada por parásitos del género Plasmodium (P. falciparum,P. vivax, P. ovale, P. knowlesi y P. malariae), la cual compromete la salud y el desarrollo socioeconómico de comunidades localizadas en regiones tropicales y subtropicales. Actualmente, en el mundo se presentan más de 255 millones de casos, cerca de 800 mil muertes anuales y 2400 millones de personas en riesgo de contraer la infección, ocasionando pérdidas humanas y económicas (WHO, 2010; Fidock et al., 2004).
Los programas de control de la malaria en Sur América han fallado en parte debido a la resistencia del parásito a los antimaláricos (Vieira et al., 2004). En P. falciparum el mecanismo de resistencia a ciertos medicamentos como por ejemplo sulfadoxina- pirimetamina y 4-aminoquinolinas es conocido, hay evidencia que proteínas transportadoras y proteasas están involucradas (Vieira et al., 2004). Además, que bajo presión con cloroquina y otros antimaláricos P. falciparum expresa muchas más proteínas, sugiriendo que procesos metabólicos pueden afectarse y se abre la posibilidad para la identificación de blancos terapéuticos (Graves et al., 2002; Becker et al., 2004).
Se han diseñado estrategias para disminuir la alta mortalidad y morbilidad ocasionada por la malaria, las cuales incluyen la búsqueda de nuevos antimaláricos, la identificación de blancos y el establecimiento de esquemas terapéuticos combinados, los cuales han sido implementados con un éxito relativo; al igual que se ha insistido en la mejora del acceso a los servicios de salud en las poblaciones vulnerables de las zonas maláricas. Sin embargo debido al complejo ciclo de vida del parásito, involucrando su vector y las condiciones de poblaciones humanas en regiones endémicas para malaria, hace que esta enfermedad mantenga altos índices de prevalencia (Gardner et al., 2002).
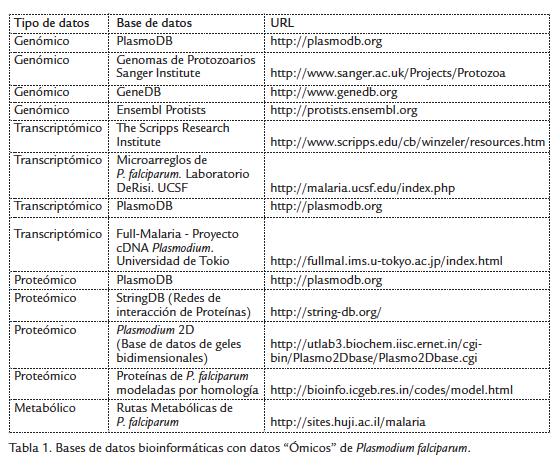
La secuenciación del genoma de P. falciparum (Gardner et al., 2002) junto con la descripción del transcriptoma (Bozdech et al., 2003) y el proteoma (Florens et al., 2002; Lasonder et al., 2002) posibilitaron una comprensión más sistémica de la enfermedad (Gardner et al., 2002), porque permiten interpretar la información relacionada con los distintos niveles de expresión de miles de genes y proteínas del parásito (Bozdech et al., 2003), bajo diferentes condiciones, como las diversas etapas del ciclo de vida o durante el tratamiento con un determinado fármaco o potencial antimalárico (Gardner et al., 2002). De esta manera con el desarrollo de las "ómicas" se ha contribuido al entendimiento de la biología del parásito, generando una gran cantidad de bases de datos (Tabla 1), y apuntando hacia la integración del conocimiento de la biología de P. falciparum a nivel del genoma, transcriptoma, proteoma, metaboloma (Yeh et al., 2004) e interactoma (Date et al., 2006).
El término proteómica es derivado de la palabra proteoma acuñada por Wilkins (1995), y hace referencia al conjunto de proteínas expresadas por un genoma o por un tipo de célula-tejido. Aunque la genómica es un aspecto de suma importancia en la comprensión biológica de los organismos, las proteínas son en última instancia, las responsables del control de muchos aspectos de la función celular. Es así como la proteómica se ha convertido en una de las la principales herramientas para el estudio de la patogénesis de diversas enfermedades (List et al., 2008), y en la comprensión de los eventos moleculares que ocurren en el parásito ante el tratamiento con antimaláricos o durante las distintas etapas del ciclo de vida (Nirmalan et al., 2004).
El análisis proteómico usando electroforesis bidimensional, es actualmente el enfoque más utilizado para observar la expresión de proteínas del parásito (Nirmalan et al., 2004; Makanga et al., 2005; Gelhaus et al., 2005; Aly et al., 2007; Radfar et al., 2008). Igualmente, existen diversas metodologías para estudiar el proteoma del parásito, las cuales tienen distintos fundamentos metodológicos en cuanto a la cantidad y preparación de la muestra, métodos de identificación y cuantificación de la expresión de proteínas. El propósito de esta revisión es ofrecer una visión del estado del arte de la proteómica aplicada al estudio de la malaria, específicamente al estudio de diferentes antimaláricos y al estudio de la biología del parásito P. falciparum.
IMPORTANCIA DEL ESTUDIO DEL PROTEOMA DE Plasmodium falciparum
Dada la dinámica del ciclo de vida del parásito, se podría plantear que existen diversos proteomas dependiendo del estadio en el cual se encuentre el parásito, por tanto el proteoma de P. falciparum es polifacético y específico de estadio, con un alto grado de especialización con el fin de apoyar los cambios biológicos y metabólicos asociados con cada estadio (Sims et al., 2006).
Bozdech et al., 2003, describieron métodos para anotación funcional del genoma de P. falciparum utilizando agrupamiento de acuerdo a la expresión génica, seguido por patrones de correlación temporal de expresión relacionado a procesos celulares. Mientras que el estudio de Le Roch et al., 2003, mostró que el 75 % de los genes son activados solamente una vez durante el ciclo intraeritrocítico y que están relacionados con procesos biológicos específicos. De acuerdo a estos análisis también se encontró que la expresión de genes caracterizada por medio de microarreglos muestra una baja correspondencia entre transcripción y traducción en Plasmodium (Bozdech et al., 2003; Le Roch et al., 2003), por lo que los datos de transcripción no soportan la existencia de patrones particulares de expresión bajo presión con drogas en Plasmodium, por tanto, la caracterización del proteoma es la estrategia para establecer las diferencias fenotípicas en P. falciparum (Wu et al., 2006). De aquí que el estudio del proteoma de P. falciparum se plantee como prioritario en el estudio de la malaria, avances tecnológicos en proteómica han posibilitado el análisis global de proteínas a partir de extractos celulares del parásito. El objetivo principal de estos análisis es la obtención del perfil de expresión proteíca con el que se pretende maximizar la identificación y cuantificación de proteínas que contribuyen a la patogénesis de la enfermedad, incluyendo mecanismos regulatorios y la habilidad de responder a perturbaciones externas (Smit et al., 2010).
El estudio del proteoma ahora supera el objetivo de la identificación de proteínas y pretende determinar patrones proteícos o fenotipos que caracterizan la respuesta del parásito al medio ambiente. Antimaláricos como quinina y mefloquina comparten con la cloroquina su estructura química, no obstante se espera que tengan diferentes mecanismos de acción y por lo tanto una particular respuesta del parásito a nivel de su proteoma (Ciach et al., 2003). Este tipo de inquietudes son las que se espera resuelva el estudio del proteoma del parásito.
METODOLOGÍAS UTILIZADAS EN EL ESTUDIO DE PROTEOMAS
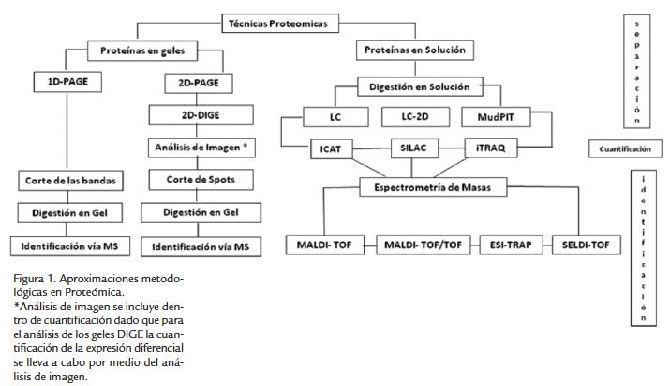
La proteómica en general aplica separación de péptidos o de proteínas por medio de electroforesis o cromatografía líquida (LC) y su posterior identificación y cuantificación por espectrometría de masas (Fig.1). Estos métodos parten de fundamentos diferentes y la implementación de cada una de estas tecnologías dependerá en gran parte de la pregunta a resolver y de la resolución que se pretende alcanzar.
ELECTROFORESIS UNI Y BIDIMENSIONAL
La electroforesis es una técnica analítica que permite la separación de proteínas según sus propiedades fisicoquímicas tales como tamaño y carga. En el sistema electroforético denominado SDS-PAGE (Sodium Dodecyl Sulfate- Polyacrylamide Gel Electrophoresis) las proteínas se separan según su tamaño, porque el SDS desnaturaliza y carga negativamente todas las proteínas. La electroforesis SDS-PAGE refleja la calidad de una mezcla de proteínas por lo que su uso en proteómica ha sido aplicado con éxito (Lasonder et al., 2002).
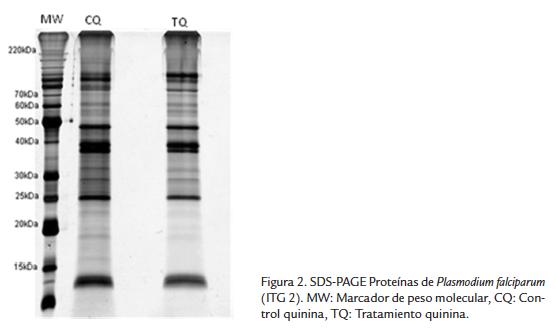
La figura 2 muestra el perfil electroforético SDS-PAGE de extractos proteícos de trofozoítos de P. falciparum cepa ITG2 bajo tratamiento con quinina. Nótese la aparente integridad de las proteínas dada la distribución de bandas desde bajos a altos pesos moleculares, la calidad de la muestra proteica utilizada en esta electroforesis se evidenció en la figura 3 donde se muestra su correspondiente electroforesis bidimensional, allí las proteínas se presentan a lo largo y ancho del gel con aproximadamente 80 spots.

La electroforesis en dos dimensiones fue desarrollada en los años 70 (O'farell et al., 1975) con el objetivo de separar mezclas complejas de proteínas y como método para visualizar las diferencias en su expresión (Monteoliva et al., 2004). Su aplicación en la separación de mezclas complejas de proteínas se basa en sus propiedades, como son el punto isoeléctrico y el peso molecular. En la primera dimensión las proteínas son separadas por isoelectroenfoque de acuerdo a su punto isoeléctrico, las proteínas migran a través de un gradiente de pH inmovilizado (Monteoliva et al., 2004). En la segunda dimensión las proteínas son reducidas, alquiladas y corren en un gel de poliacrilamida SDS-PAGE, según su movilidad relativa que es proporcional a su peso molecular.
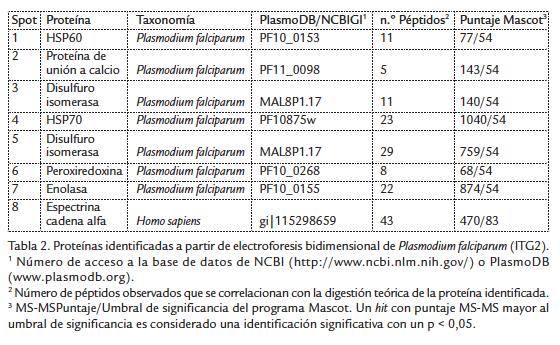
La electroforesis bidimensional es la técnica más común en estudios proteómicos en diferentes organismos y tejidos. La técnica se complementa con la identificación por espectrometría de masas (Andersen et al., 2000). Las proteínas resueltas por electroforesis en dos dimensiones deben ser digeridas mediante tripsinización para poder ser identificadas vía huella peptídica (Peptide Mass Fingerprinting) o espectrometría de masas en tándem (ion MS/MS) (Monteoliva et al., 2004), es así como a partir del perfil bidimensional de P. falciparum obtenido en nuestro grupo (Fig. 3) se llevó a cabo la identificación de algunos spots por medio de espectrometría de masas tipo MALDI-TOF/TOF, en la tabla 2 se encuentran la lista de proteínas identificadas a partir de este gel, que en su mayoría resultaron ser proteínas tipo HSP (Heat Shock Proteins) y otras involucradas en procesos como glicolisis y metabolismo redox.
La ventaja que provee la técnica de electroforesis bidimensional al dar un panorama del proteoma en un momento dado es muy útil para un organismo que tiene un ciclo de vida tan complejo como P. falciparum, además de ser una técnica útil en la identificación de posibles blancos terapéuticos y la evaluación de la expresión diferencial de proteínas bajo tratamiento o durante el ciclo de vida del parásito (Nirmalan et al., 2004; Makanga et al., 2005; Aly et al., 2007; Radfar et al., 2008; Briolant et al., 2010).
No obstante la técnica electroforética presenta algunas limitaciones como la baja resolución y poca reproducibilidad (Celis et al., 1999). El análisis del proteoma de P. falciparum por medio de electroforesis bidimensional se ha visto obstaculizado por algunas limitaciones técnicas entre las que se encuentran por ejemplo que las proteínas del parásito son notoriamente insolubles y altamente cargadas (Smit et al., 2010). El alto contenido de hemozoina de origen parasitario en los lisados proteícos impone también limitaciones en el análisis del proteoma del parásito, porque la hemozoina contiene alto contenido de hierro el cual ocasiona problemas en el isoelectroenfoque de las proteínas (Nirmalan et al., 2007). Igualmente esta técnica requiere alta calidad y cantidad de proteína inicial (lo que es difícil dadas las condiciones de cultivo del parásito), y que es prácticamente imposible separar proteínas del parásito de las del glóbulo rojo y este método a veces es insuficiente para acceder a trabajar con el proteoma de los estadios del ciclo de Plasmodium (Johnson et al., 2004). Mejoras en las técnicas de solubilización de proteínas (Rabilloud et al., 1999; Ashton et al., 2001) y la disponibilidad de gran variedad de tiras en gradiente de pH inmovilizado han incrementado el uso de la electroforesis bidimensional.
Con el fin de mejorar la electroforesis bidimensional y determinar la expresión diferencial de proteínas, se han desarrollado métodos alternativos. La técnica de electroforesis diferencial en gel conocida como DIGE por sus siglas en inglés (Differential In Gel Electrophoresis) fue desarrollada por Amershan Biosciences (Unlu et al., 1997) y permite un análisis cuantitativo de la técnica electroforética, minimiza la variabilidad suministrando un patrón de spots preciso, reproducible y facilita el análisis estadístico de los patrones de spots. Metodológicamente consiste en el marcaje de las proteínas con fluorescencia (Cy2, Cy3 o Cy5) en los residuos lisina de las proteínas previo al isoelectroenfoque (Tannu et al., 2006).
ESPECTROMETRÍA DE MASAS
La espectrometría de masas (MS) es el principal método de análisis para la identificación y función de las proteínas en diversos sistemas biológicos (Cravatt et al., 2007). La técnica permite adquirir y analizar alto contenido de información cuantitativa a partir de muestras biológicas complejas. Un espectrómetro de masas es un instrumento que analiza iones, determina la relación masa/carga (m/z) y registra las abundancias relativas en lo que se conoce como un espectro de masas (Graham et al., 2007). Hasta hace poco la espectrometría de masas estaba restringida a moléculas volátiles. El desarrollo de técnicas de "ionización suaves" en los años 80 (Fenn et al., 1989), permitió la ionización y vaporización de biomoléculas grandes, polares tales como proteínas y péptidos. La caracterización y cuantificación de proteínas se ha incrementado por el desarrollo de dos técnicas de "ionización suave" llamadas electrospray (ESI) e ionización por desorción de una matriz asistida por laser o MALDI (Matrix Assisted Laser Desorption Ionisation). Métodos de espectrometría de masas como la tecnología multidimensional para la identificación de proteínas MudPIT (por sus siglas en inglés), fueron desarrollados para identificarlas a gran escala. En un experimento típico de MudPIT una mezcla de proteínas no fraccionadas es digerida a péptidos separados por cromatografía líquida utilizando columnas de intercambio iónico, fase reversa y analizados por medio de espectrometría de masas en tándem, este enfoque puede incluir también marcaje isotópico de aminoácidos in vivo o in vitro el cual permite comparación de los patrones de expresión de proteínas (Dong et al., 2007; Fournier et al., 2007; Prieto et al., 2008).
PROTEÓMICA CUANTITATIVA
Actualmente, la cuantificación de la expresión relativa de proteínas por espectrometría de masas se hace por dos métodos: marcaje metabólico isotópico de células en cultivo (SILAC) o por el marcaje químico de extractos con Isotope Coded Affinity Tags (ICATs, por sus siglas en inglés) (Gygi et al., 1999). Las dos metodologías permiten determinar las diferencias isotópicas de un mismo compuesto químico en un espectro de masas individual donde las intensidades de los picos representan la cantidad relativa de la muestra. De esta manera se deducen las cantidades relativas de un compuesto en una mezcla (Nirmalan et al., 2004).
Uno de los problemas de ICAT en el estudio del proteoma de P. falciparum es la dependencia de la presencia de residuos de cisteína, los cuales son de baja abundancia en el proteoma de P. falciparum. Igualmente la posibilidad de reacciones no controladas de esos residuos es alta, por lo que se hace necesario una considerable optimización incrementando los costos (Smolka et al., 2001; Nirmalan et al., 2004). En contraste SILAC (Stable Isotope Labeling by/with aminoacids in cell culture) permite detectar las diferencias en abundancia relativa de proteínas entre muestras usando marcaje isotópico no radioactivo. Posteriormente la espectrometría de masas identifica péptidos químicamente idénticos pero isotópicamente diferentes. La relación de las intensidades de los picos en el espectro entre los péptidos químicamente similares pero isotópicamente diferentes refleja su abundancia relativa (Ong et al., 2002). El marcaje metabólico en estudios cuantitativos ha sido explorado en mamíferos (Ong et al., 2002), levaduras (Berger et al., 2002), e invertebrados como Caenorhabditis elegans (Krijgsveld et al., 2003) y Drosophilla melanogaster (Krijgsveld et al., 2003). Otro método de marcaje denominado iTRAQ (Isobaric Tag for Relative and Absolute Quantitation), permite cuantificar proteínas a partir del marcaje con isótopos covalentemente ligados al extremo N-terminal y aminas laterales de péptidos. Las muestras de proteínas marcada y no marcada son agrupadas, fraccionadas por medio de nanocromatografía líquida y detectadas por espectrometría de masas en tándem (MS/MS) (Aggarwal et al., 2006).
PROTEÓMICA DE P. falciparum Y ANTIMALÁRICOS
Debido a las limitaciones de los geles bidimensionales la técnica de elección es LC- MS/MS complementada con marcación isotópica o metabólica para el estudio del efecto de antimaláricos en el parásito. Nirmalan et al., 2004, reportaron el marcaje metabólico para el análisis cuantitativo de proteínas en P. falciparum. Esta técnica de marcaje isotópico tiene la ventaja de eliminar la variabilidad existente en las etapas de lisis celular, extracción de proteínas, geles bidimensionales y cromatografía líquida, porque después de marcados, el control y la muestra, pueden ser mezclados y manipulados simultáneamente (Nirmalan et al., 2004). El enfoque se fundamentó en la tecnología SILAC (Stable Isotope Labelling by Aminoacids in Cell Culture) (Ong et al., 2002). La abundancia de isoleucina en las proteínas de P. falciparum conduce a que esta estrategia de marcaje sea aplicable esencialmente a cada una de las 5300 proteínas del parásito. Nirmalan et al., 2004, utilizaron la cepa HB3, compararon la expresión proteíca de trofozoítos jóvenes, maduros y esquizontes, además compararon los perfiles proteícos de cultivos tratados con tetraciclina y pirimetamina (PYR), contra los controles no tratados utilizando espectrometría de masas y electroforesis bidimensional. Se encontró que los niveles de enolasa, fosfoetanolamina N-metiltransferasa y actina fueron regulados negativamente por la presencia de PYR, mientras que la proteína lactato deshidrogenasa fue afectada en menor grado. En cuanto a la tetraciclina se observó que los niveles de las cuatro proteínas anteriormente mencionadas fue elevado lo cual es un patrón diferente al inducido por PYR (Nirmalan et al., 2004). Este trabajo fue el primero en utilizar la electroforesis bidimensional para estudiar el efecto de antimaláricos en la expresión de proteínas en el parásito.
Makanga et al., 2005, evaluaron los antimaláricos artemeter (ARM) y lumefrantina (LUM) en cultivos de P. falciparum in vitro cepa K1. Se evidenció sobreregulación de 22 y 41 proteínas en ARM y LUM respectivamente. Estas drogas indujeron cambios en el proteoma del parásito, sin embargo los patrones fueron específicos para cada droga. Bajo tratamiento con ARM se redujo hasta tres veces la expresión de enzimas glicolíticas como enolasa, fosfoglicerato kinasa, fructosa bifosfato aldolasa y gliceraldehído-3- fosfato deshidrogenasa. La expresión de esas mismas enzimas estuvo sobre expresada más de tres veces bajo tratamiento con LUM (Makanga et al., 2005).
Un número de proteínas sobre expresadas bajo tratamiento con ARM, incluyeron proteínas de unión a calcio asociadas a membrana, una proteinasa aspártica putativa, proteínas de choque térmico HSP60, HSP70,HSP90 y también dos proteínas involucradas en funciones relacionadas con metabolismo de proteínas (Makanga et al., 2005). Aly et al., 2007, utilizaron la cepa FCR-3 originalmente susceptible a la droga experimental N-89, la cual fue sometida a crecientes concentraciones del endoperóxido N-89 durante dos años. La presión con la droga generó la resistencia a N-89. Los clones resistentes fueron utilizados para llevar a cabo las electroforesis bidimensionales y comparadas contra cepas normales sin resistencia. Los análisis de los geles bidimensionales mostraron 12 proteínas sobre expresadas en las cepas resistentes entre las que se encuentran fructosa bifosfato aldolasa y 14 proteínas reguladas negativamente como la gliceraldehído-3-fosfato deshidrogenasa (GAPDH), siendo estas dos proteínas parte de la ruta de la glicolisis, sugiriendo la glicolisis como un posible blanco terapéutico de N-89 (Aly et al., 2007). Otras proteínas sobre expresadas e identificadas fueron: la proteína de superficie de merozoito 7 (MSP-7), proteína de unión a calcio (PfERC), proteína con dominio DNAJ, fructosa bifosfato aldolasa (FBP), y proteínas de choque térmico (HSPs). En contraste, las proteínas reguladas negativamente identificadas fueron: actina, proteína homologa 14-3-3, fosfoetanolamina N metil transferasa (PfPEMT), enolasa, ornitin aminotransferasa (OAT), s-adenosilmetionina sintetasa (SAMS), gliceraldehído-3-fosfato deshidrogenasa (GAPDH), chaperonina 60 y uridina fosforilasa (Aly et al., 2007).
Algunas de las proteínas que resultaron afectadas en su regulación hacían parte de la ruta glicolítica del parásito, dado que la producción de energía de los estadios sanguíneos del parásito proviene enteramente de la ruta glicolítica. Así las enzimas de la ruta glicolítica parecen ser promisorios candidatos a drogas (Aly et al., 2007). Sin embargo, la resistencia es multifactorial lo que se refleja por el efecto en más de una proteína y enzimas de diversos procesos y localizaciones celulares (Aly et al., 2007). Koncarevic et al., 2007, utilizando la tecnología SELDI-TOF Surface Enhanced Laser Desorption/Ionisation (SELDI) Time Of Flight Mass Spectrometry (TOF-MS) describieron perfiles de expresión de proteínas de P. falciparum en las cepas K1 y Dd2 (resistentes a cloroquina) HB3 y 3D7 (sensibles a cloroquina). Con esta técnica las proteínas son selectivamente absorbidas a arreglos de superficie cromatográfica antes de la adición a la matriz. La selectividad de los arreglos de superficie para retener porciones del proteoma permite el análisis directo de mezclas biológicas crudas de proteínas tales como suero, orina o lisados celulares (Koncarevic et al., 2007). SELDI-TOF-MS ha sido usado satisfactoriamente para estudiar perfiles de expresión comparativos de organismos como Yersinia pestis (Thulasiraman et al., 2001), Helicobacter pylori (Hynes et al., 2003) y Trypanosoma brucei (Papadopoulos et al., 2004 ), además ha sido aplicado para el descubrimiento de biomarcadores diagnósticos en cáncer (Engwegen et al., 2006). Koncarevic et al., 2007, encontraron cambios en la expresión de proteínas bajo tratamiento con cloroquina en las cuatro cepas resistentes a cloroquina (CQR) y sensibles (CQS) estudiadas. Se reportaron diez proteínas reguladas por la droga y tres de esos productos fueron enriquecidos, purificados e identificados. Los productos regulados fueron identificados como la proteína plasmodial EXP-1, también conocida como antígeno relacionado al circumsporozoito y las cadenas alfa, beta y delta de la hemoglobina humana. La expresión de la proteína EXP-1 fue modulada bajo presión con cloroquina, esta proteína fue regulada positivamente en dos cepas resistentes y negativamente en cepas sensibles, no obstante hasta ahora no ha sido posible predecir la función fisiológica de EXP-1 o si esta directamente involucrada en la resistencia a cloroquina. SELDI- TOF proveyó una aproximación rápida para comparar perfiles de expresión de proteínas de bajo peso molecular en cepas de P. falciparum CQS (3D7 y HB3) CQR (Dd2 y K1).
En otro estudio, Radfar et al., 2008 compararon las proteínas oxidadas a través del ciclo intraeritrocítico de los parásitos en cultivos tratados y no tratados con cloroquina en la cepa Dd2 resistente a cloroquina. Aunque se ha sugerido que el estrés oxidativo cumple un papel clave en la patogénesis de la malaria (Becker et al., 2004), se sabe que el parásito de la malaria es vulnerable al estrés oxidativo (Muller et al., 2004) por lo que se ha planteado el metabolismo redox como blanco promisorio antimalárico. Los autores del estudio reportan que el tratamiento con cloroquina incrementó el número de proteínas oxidadas en el estadio de esquizonte de los parásitos (Radfar et al., 2008). Se encontró que las proteínas funcionales plasmodiales sufren los mayores daños oxidativos, entre estas se encuentran proteínas involucradas en plegamiento (HSPs), proteólisis, metabolismo energético (fosfoglicerato kinasa, fructosa bifosfato aldolasa, gliceraldehido-3-fosfato deshidrogenas y enolasa), señalización y patogénesis. Los hallazgos de este trabajo proveen indicios de la respuesta al tratamiento con cloroquina. La proteómica redox, la cual se encarga de estudiar las proteínas en su estado de oxidoreducción, es una herramienta apropiada para identificar respuesta a antimaláricos y proveer más herramientas para enfrentar la resistencia a la cloroquina (Radfar et al., 2008).
Prieto et al., 2008, utilizó marcaje isotópico (SILAC) y la tecnología multidimensional para la identificación de proteínas por sus siglas en inglés MudPIT, determinando que bajo tratamiento con artemisinina y cloroquina, 41 y 38 proteínas fueron reguladas positivamente mientras que 14 y ocho proteínas lo fueron negativamente respectivamente. Ninguna de estas proteínas se asoció con proteínas de estrés (HSPs) o funciones de glicólisis como en los trabajos con geles bidimensionales. Esta técnica basada en marcaje metabólico con isoleucina mostró que es eficiente en la identificación de marcadores de resistencia a antimaláricos en el parásito.
La metodología SILAC se complementó con el uso de cromatografía de intercambio aniónico para disminuir la complejidad de la mezcla peptídica. Esta separación de péptidos seguida de cromatografía en fase reversa e intercambio catiónico y posteriormente LC-MS/MS permite la identificación y cuantificación de proteínas. Los autores describen que con su metodología, 100 ug de proteína son necesarios para el análisis LC-MS, este método puede ser aplicado en otros apicomplexa para los cuales el rendimiento de extractos celulares es limitado. Este estudio describe proteínas sobrexpresadas a nivel del núcleo, así como proteínas involucradas en procesos postraduccionales. No obstante el análisis de los resultados se ve afectado por el alto número de proteínas hipotéticas diferencialmente expresadas bajo tratamiento con los antimaláricos, por lo que es posible que sea factible hacer anotación de genomas a partir del proteoma en organismos como P. falciparum con un contenido alto de AT en su genoma.
Los resultados proteómicos de Prieto et al., 2008, estuvieron en concordancia con los resultados transcriptómicos de Gunasekera et al., 2003 y Gunasekera et al., 2007, quienes observaron cambios en la expresión de mRNA en parásitos tratados con cloroquina. Los cambios afectaron particularmente proteínas ribosomales, moléculas de señalización, procesamiento de proteínas (HSPs, subunidades de proteasoma), también como metabolismo de RNA (factores de transcripción). Una comparación directa de los resultados del transcriptoma y el proteoma tiene sus limitaciones dada la variación en la sincronicidad del parásito, las dosis de los antimaláricos y el tiempo de exposición.
En cuanto a los efectos de la artemisinina se mostró regulación negativa de la expresión de la ATP sintasa vacuolar (PF13_0130), mientras que proteínas involucradas en el metabolismo de ácidos nucléicos, transporte y secreción mostraron una ligera regulación positiva. Una de las observaciones más importantes fue la sobreregulación de la proteína de resistencia a múltiples drogas de P. falciparum (pfmdr1) bajo tratamiento con cloroquina y artemisinina. La pfmdr1 es una proteína que se conoce regula resistencia a la cloroquina en el parásito (Duraisingh et al., 2005).
Dado que el 60 % de las proteínas conocidas en P. falciparum son hipotéticas, es difícil asignarles funciones, sin embargo el trabajo de Prieto et al., 2008, ha permitido establecer que algunos de estos productos hipotéticos pueden verse afectados por los antimaláricos, además de ilustrar la complejidad de la biología del parásito y contribuir en la identificación y cuantificación de la expresión de proteínas en respuesta al tratamiento con cloroquina y artemisinina. De esta manera el trabajo de Prieto et al., 2008, se convierte en referencia para estudios cuantitativos de los cambios inducidos por el tratamiento con antimaláricos.
Briolant et al., 2010, analizaron los cambios en la expresión de proteínas en el estadio de esquizonte bajo tratamiento con Doxiciclina (DOX), empleando 2D-DIGE combinada con una técnica llamada iTRAQ (Isobaric Tagging Reagents for Relative and Absolute Quantification). La combinación de estas dos tecnologías permitieron identificar proteínas que presentaron cambios en su regulación en respuesta al tratamiento con DOX, estas proteínas están involucradas en procesos tales como redox, respuesta a estrés, íntesis de proteínas, síntesis de lípidos y metabolismo energético, además se planteó que DOX actúa sobre la mitocondria y el apicoplasto del parásito porque según los resultados de este trabajo de las 64 proteínas diferencialmente expresadas utilizando los dos enfoques proteómicos 21 % se localizaron en el citoplasma, 19 % en el apicoplasto, 13 % en la membrana, 13 % en el núcleo, 5 % en la mitocondria y 29 % con localización desconocida. Proteínas de membrana, citoplasma y apicoplasto estuvieron generalmente sobre expresadas, mientras las proteínas nucleares y mitocondriales fueron reguladas negativamente (Briolant et al., 2010).
En el trabajo de Briolant et al., 2010, resultaron estar sobre expresadas las proteínas asociadas a procesos redox (1-Cys peroxiredoxina, 2-Cys peroxiredoxina), las cuales han sido recientemente reportadas como potenciales blancos junto con tioredoxinas, glutaredoxinas y plasmoredoxinas (Sturm et al., 2009), esto también puede representar una respuesta no especifica al tratamiento.
PROTEÓMICA Y BIOLOGÍA DEL PARÁSITO
Los trabajos basados en proteómica relacionados con la biología de P. falciparum se han encargado de analizar los subproteomas en diferentes estadios del ciclo de vida, con el objetivo de identificar las proteínas especificas de cada uno de los estadios y encontrar la relación con los diferentes cambios metabólicos, fisiológicos y bioquímicos que sufre el parásito lo largo del ciclo.
El proteoma del parásito descrito por Florens et al., 2002, en la cepa 3D7, describe las proteínas de los estadios de esporozoíto, merozoito, trofozoíto y gametocito, utilizando separación por cromatografía líquida de alta resolución, acoplada a un espectrómetro de masas MS/MS para su identificación. Se encontró que 6 % de las proteínas detectadas son compartidas por todos los estadios. La mayoría de estas proteínas fueron asociadas a funciones celulares básicas como proteínas ribosomales, proteínas de citoesqueleto, factores de transcripción e histonas. Las diferencias en la expresión de proteínas entre los estadios evidenció la complejidad biológica del ciclo de vida del parásito (Florens et al., 2002).
Lasonder et al., 2002, caracterizaron el proteoma de gametocitos y gametos (formas sexuales) y lo compararon con trofozoítos y esquizontes en la cepa NF54 de P. falciparum. Mediante SDS-PAGE seguido de separación cromatográfica y análisis de masas, demostraron la capacidad de la espectrometría de masas en la anotación del genoma de P. falciparum. Posteriormente, los mismos autores caracterizaron el proteoma del esporozoíto, el cual es un estadio presente en el mosquito. Este trabajo reportó la primera comparación proteómica de diferentes estadios de la forma transmitida por el mosquito (Anopheles), el esporozoíto cumple un papel esencial en la primera fase de la infección por malaria, el entendimiento de su biología será de gran importancia con el fin de desarrollar métodos de intervención contra la infección inicial (Lasonder et al., 2008). Los autores caracterizaron el proteoma de oocistos que contienen esporozoítos tempranos y tardíos y esporozoítos infectivos maduro, no obstante los esporozoítos obtenidos a partir de oocitos difieren de aquellos provenientes de glándulas salivares. A pesar de las similitudes morfológicas entre esporozoítos de intestino y glándulas salivares, sus proteomas son diferentes, de acuerdo con el incremento en la infectividad del hepatocito. Los distintos proteomas de esporozoítos contienen un gran número de proteínas específicas de estadio involucradas en maduración del esporozoíto, movilidad, infección del hospedero humano y metabolismo. En general el análisis del proteoma de los esporozoítos de P. falciparum muestra que en oocitos derivados de esporozoítos y esporozoítos de glándulas salivares se identificaron 728 proteínas de las cuales 250 son específicas del estadio de oocito/ esporozoíto. La identificación de las proteínas y sus distribuciones relativas dentro de los diferentes proteomas sugieren adaptaciones metabólicas específicas y otras funciones biológicas del esporozoíto maduro.
Una alternativa para mejorar la sensibilidad de las técnicas en gel es el marcaje metabólico, que saca ventaja de la poca capacidad catabólica del eritrocito humano comparada con la del parásito. Wu et al., 2006, utilizando marcaje metabólico con (35S) metionina, lograron identificar proteínas del parásito presentes en la membrana de eritrocitos infectados que median interacciones con receptores del hospedero. Los autores identificaron proteínas del parásito, presentes en la membrana de eritrocitos infectados. Usando la técnica de marcaje metabólico combinado con electroforesis bidimensional se logra evidenciar cambios en los perfiles de proteínas de P. falciparum en cepas con diferentes fenotipos adhesivos. Los cambios encontrados fueron alteraciones en el punto isoeléctrico, lo que sugiere activación vía fosforilación, un pequeño grupo de proteínas pudieron ser identificados usando espectrometría de masas, igualmente usando técnicas libres de geles tales como LC/MS/MS mejoran la sensibilidad, aunque las proteínas de membrana y algunas proteínas de la superficie del eritrocito infectado de interés en estudios de adherencia no pudieron ser identificadas (Wu et al., 2006).
Wu et al., 2009, con el objeto de obtener los perfiles de fosforilación a través del ciclo de vida eritrocítico examinaron los cambios en el fosfoproteoma de glóbulos rojos parasitados, a partir de electroforesis bidimensional de diferentes cepas del parásito en combinación con inmunoblot usando anticuerpos monoclonales específicos a serina/treonina y tirosina fosforiladas.
Se utilizó cromatografía de afinidad para purificar proteínas fosforiladas. Posteriormente se separaron usando 1D SDS-PAGE y se identificaron con LC/MS/MS, 34 bandas fueron cortadas y las proteínas identificadas representaron un fosfoproteoma "shotgun" de trofozoítos de la cepa ITG. Entre las proteínas fosforiladas en el parásito se encuentran proteínas asociadas a citoadherencia como las pertenecientes a la familia CLAG (Cytoadherence Linked Asexual Proteins), otras proteínas de interés fosforiladas fueron la proteína DNAJ, la cual es similar a RESA y se ha predicho que es exportada al exterior del parásito y contiene el motivo PEXEL, una proteína que participa en el remodelamiento de la cromatina e involucrada en expresión génica y un gran número de proteínas de choque térmico y con actividad chaperona.
Cambios en la fosforilación de las proteínas de membrana y del citoesqueleto del eritrocito, son una consecuencia de la infección por P. falciparum, esto se ha examinado en las proteínas: espectrina, banda 3, banda 4.1, ankirina y glicoporinas (Wu et al., 2009). Estos cambios observados en la fosforilación de proteínas demembrana de glóbulo rojo y citoesqueleto puede ser importante en la regulación de la organización del citoesqueleto en células infectadas con P. falciparum, la fosforilación de estas proteínas está restringido a los estadios de trofozoíto y esquizonte (Wu et al., 2009).
La regulación postranscripcional cumple un papel importante en Plasmodium (Le Roch et al., 2003), se ha reportado una baja correlación entre el transcriptoma y el proteoma en cada uno de los estadios, algunos genes mostraron retrasos entre los picos de abundancia de mRNA y proteína. Mair et al., 2006, demostraron que 370 transcriptos producidos durante el estadio de gametocito se represan después de sintetizados hasta la fertilización del gameto. Este mecanismo de regulación puede explicar entonces las discrepancias entre los perfiles de abundancia de mRNA y la expresión proteica de algunas proteínas de P. falciparum (Foth et al., 2008).
La expresión de transcriptos y los niveles de abundancia de proteínas muestran según Foth et al., 2008, una situación dinámica y compleja e indican la importancia de la expresión diferencial de isoformas (Foth et al., 2008). Una ventaja de los geles bidimensionales es que permiten observar las modificaciones postraduccionales (PTMs) y resaltan la importancia de la regulación de la función de las proteínas (Jones et al., 2006; Brobey et al., 2007; De Jesus et al., 2007). Las isoformas no se pueden detectar a nivel transcripcional y son no detectables inclusive por western blot. La importancia de las isoformas, por ejemplo, fue demostrada en un estudio de resistencia de T. brucei (Foucher et al., 2006), con el uso de DIGE se mostraron diferentes niveles de expresión de las isoformas para algunas proteínas como: factor de iniciación eucariótico eIF4A, eIF5A y HSP70-2 (Foth et al., 2008). La contrastación de DIGE con los perfiles transcriptómicos reveló una posible regulación postraduccional de la expresión lo cual sugiere su importancia biológica en el ciclo de vida del parásito (Foth et al., 2008). Por otro lado, las interacciones de las cinco isoformas de los factores de iniciación eIF4A y RNA helicasa funcionan como iniciadores de la trascripción lo que puede llevar a diferencias biológicas en el parásito. La función en el desarrollo del parásito de las isoformas está por determinar.
En los tres trabajos anteriores (Wu et al., 2006; Foth et al., 2008; Wu et al., 2009) se evidencian igualmente las aplicaciones de la proteómica al estudio de las modificaciones postraduccionales, en estos artículos se demostró que los mapas proteómicos bidimensionales en P. falciparum son muy importantes para observar modificaciones postraduccionales (PTMs) además de resaltar la importancia de la detección de isoformas y la regulación de la función de las proteínas, este tipo de aproximaciones han sido igualmente aplicadas a otro tipo de parásitos (Jones et al., 2006; De Jesus et al., 2007; Brobey et al., 2007), la importancia de las isoformas por ejemplo fue demostrada en un estudio proteómico usando geles bidimensionales de T. brucei donde la ausencia de una sola isoforma de una subunidad asociada a un polipéptido se correlacionó con resistencia a drogas (Foucher et al., 2006). Los hallazgos reportados en ese estudio resaltan la importancia de las isoformas en la resistencia a ciertos medicamente en otros modelos.
Las PTMs cumplen un papel crítico en la regulación de la actividad de muchas proteínas durante el ciclo de vida del parásito, estas incluyen clivaje proteolítico (Kumar et al., 2004), glicosilación ( Kumar et al., 2004; Foucher et al., 2006 ), fosforilación (Anamika et al., 2007), acetilación (Cui et al., 2007) y ubiquitinación (Horrocks et al., 2000). Por ejemplo, en la proteína enolasa de P. falciparum enzima glicolítica esencial se han encontrado al menos cinco isoformas modificadas postraduccionalmente (Pal-Bhowmick et al., 2007). De esta manera con la proteómica se ha logrado comprender mejor que las PTMs son uno de los principales mecanismos de control para muchas funciones celulares, incluyendo la regulación de la división celular, síntesis de proteínas y transcripción (Cohen et al., 2002). Por ejemplo, la fosforilación de proteínas es un evento importante en la infección por malaria como en el proceso de citoadherencia.
La espectrometría de masas ha mostrado tener la habilidad para caracterizar las modificaciones postraduccionales de las proteínas tipo glicosilación o fosforilación a partir de muy pequeñas cantidades de material inicial (Foth et al., 2008). La glicosilación y fosforilación de proteínas en P. falciparum es crucial en la maduración del parásito y su inhibición vía vacunación puede reducir significativamente la mortalidad de los pacientes (Johnson et al., 2004; Foth et al., 2008).
Para sacar ventajas de la información visual de la electroforesis bidimensional es crítico estandarizar los métodos de preparación de la muestra y los procedimientos electroforéticos. Smit et al., 2010, optimizaron métodos de extracción, cuantificación y detección de proteínas plasmodiales a partir de los estadios de anillos y trofozoítos, bajo estas condiciones fueron capaces de visualizar 349 spots con una tasa de éxito del 95 % en la identificación por espectrometría de masas, superior a las reportadas previamente entre 50-79 % (Gelhaus et al., 2005; Makanga et al., 2005). El trabajo de Smit et al., 2010, permitió la caracterización de proteomas de los estadios de anillo y trofozoíto de P. falciparum estos mostraron que algunas proteínas están diferencialmente reguladas entre esos estadios del ciclo de vida.
EFECTO déjà vu EN PROTEÓMICA DE Plasmodium falciparum
Los estudios de expresión diferencial, empleando las técnicas de electroforesis bidimensional y espectrometría de masas, han revelado recientemente un escenario preocupante. Petrak et al., 2008, analizaron diversos trabajos de expresión diferencial basados en electroforesis bidimensional bajo distintas condiciones biológicas. El resultado indica un patrón recurrente de proteínas aparentemente expresadas diferencialmente bajo condiciones muy diferentes independiente del experimento, tejido o especie, a lo cual los autores llamaron un efecto déjà vu en proteómica (Petrak et al., 2008). Según ese trabajo la proteína identificada con mayor frecuencia fue la enzima glicolítica enolasa, esta proteína resultó estar diferencialmente expresada en casi un tercio de los experimentos analizados, las proteínas de choque térmico HSP27, HSP60, peroxiredoxinas también se encontraron diferencialmente expresadas en casi el 40 % de los experimentos. Los autores plantean que estos cambios, comúnmente observados, representan respuestas comunes al estrés celular y son un reflejo de las limitaciones técnicas de la electroforesis bidimensional.
Igualmente, a partir de esta revisión podemos concluir que en P. falciparum se observa un patrón de expresión diferencial de proteínas casi similar independiente de la cepa, el antimalárico o la estrategia experimental utilizada. Podemos plantear que existe un patrón recurrente en las proteínas diferencialmente expresadas entre estos estudios, de acuerdo al proceso biológico en primer lugar las proteínas asociadas a metabolismo energético (enolasa) son las que aparecen con mayor frecuencia en los trabajos de proteómica diferencial en P. falciparum, seguido por proteínas involucradas en respuesta a estrés (HSPs, chaperoninas), procesos redox (disulfuro isomerasa), proteínas de citoesqueleto (actina) y finalmente proteínas asociadas con transporte intracelular (proteína de unión a calcio). Lo anterior plantea un efecto déjà vu en los estudios de proteómica diferencial en P. falciparum efecto ya reportado por Petrak et al., 2008, en otros organismos y estudios.
El trabajo de Prieto et al., 2008, sale del patrón porque emplea las técnicas SILAC y MudPIT. En este trabajo, 40 % de las proteínas diferencialmente expresadas resultaron ser hipotéticas, un resultado nunca antes visto en trabajos de proteómica en P. falciparum, esto podría sugerir que existen grandes diferencias en el tipo y cantidad de proteínas identificadas utilizando electroforesis bidimensional y MudPIT, al parecer esta ultima técnica da un panorama más completo y profundo de los efectos de antimaláricos en la expresión de proteínas, además de lograr identificar más de 1000 proteínas en un solo experimento y más de 300 nunca antes identificadas en otros análisis proteómicos en el parásito. Con la técnica MudPIT posiblemente se podría erradicar el efecto déjà vu en los estudios del proteoma de P. falciparum.
CONCLUSIONES
En el estudio de un sistema biológico las disciplinas ómicas permiten obtener una visión integrada, en el caso del estudio de la malaria estas disciplinas han permitido este enfoque en el marco del ciclo de vida del parásito y su respuesta a los antimaláricos. De esta manera se ha visto que los datos genómicos, transcriptómicos y proteómicos son complementarios.
La electroforesis bidimensional, cromatografía líquida y la espectrometría de masas son metodologías importantes en el área de la proteómica, la electroforesis bidimensional puede ayudar a resolver y visualizar proteínas simultáneamente en un gel, además de permitir la separación y detección de proteínas modificadas postraduccionalmente, no obstante una de sus limitaciones radica en la dificultad para analizar un proteoma completo porque las proteínas visualizadas en los geles representan solo las proteínas que se expresan mayoritariamente en el proteoma por lo que solo se tiene una porción de todas las proteínas que están presentes en la muestra, el uso de otro tipo de tecnologías específicamente MudPIT logra incrementar la sensibilidad en la detección de proteínas. En general la eficacia de la electroforesis bidimensional en términos de número de proteínas observadas es siempre más baja comparada con las técnicas LC-MS/MS. En Plasmodium se predicen aproximadamente 2500 proteínas por LCMS/MS. Implementando electroforesis bidimensional, se cuentan 239 spots en geles bidimensionales utilizando tinción de plata, 345 con la técnica 2D-DIGE y 349 con la tinción fluorescente. Lo anterior sugiere que la técnica LC-MS/MS resuelve mejor el proteoma del parásito. Hacer proteómica en P. falciparum es bastante laborioso en cuanto a la obtención de la muestra a partir de cultivos continuos. De aquí que estrategias libres de geles por su mayor sensibilidad sean las técnicas de elección en proteómica para organismos en los cuales se presente la misma limitación de muestra, además de proporcionar datos precisos y cuantificables del proteoma e incrementar la habilidad para analizar más proteínas respecto a la electroforesis bidimensional en un mismo experimento.
Los estudios de expresión diferencial deben ser abordados con técnicas cuantitativas, las cuales son mucho más exhaustivas y nos pueden enmarcar mejor en el contexto de la regulación proteica del parásito bajo diferentes condiciones, a la fecha las aplicaciones de la proteómica cuantitativa en malaria son abundantes. La comparación de la expresión relativa de proteínas entre muestras tratadas con fármacos y no tratadas puede dilucidar potenciales blancos o mecanismos de acción y las comparaciones entre perfiles de expresión entre estadios del parásito pueden ayudar a comprender el ciclo de vida del parásito y a identificar las proteínas específicas de cada estadio. Sin embargo, actualmente uno de los grandes retos de la proteómica es conocer la función específica de más del 50 % de las proteínas de P. falciparum que todavía permanecen como hipotéticas. La combinación de las diferentes aproximaciones metodológicas son sin duda estrategias claves en el desarrollo de las investigaciones en proteómica de la malaria, porque han proporcionado suficiente nivel de resolución al conocimiento de la biología del parásito. La proteómica ha aportado evidencias importantes de la regulación de proteínas en P. falciparum bajo tratamiento con antimaláricos, proporcionando de esta manera una alternativa en el desarrollo de medicamentos o vacunas, y es en este sentido que se está trabajando en el Grupo Malaria de la Universidad de Antioquia con la aplicación de la proteómica al estudio de la expresión diferencial de proteínas en P. falciparum bajo tratamiento con quinina, mefloquina y el compuesto natural diosgenona.
AGRADECIMIENTOS
Los autores de este trabajo agradecen a Colciencias por la financiación del proyecto: Caracterización del Proteoma de Plasmodium falciparum bajo tratamiento con los medicamentos: quinina, mefloquina y el compuesto natural diosgenona, código: 111540820
500. Igualmente agradecen a la Universidad de Antioquia y su programa de sostenibilidad 2012-2013. A los Doctores Oscar Alzate y Cristina Osorio del System Proteomics Center, University of North Carolina-Chapel Hill, USA., por sus servicios técnicos en espectrometría de masas.
BIBLIOGRAFÍA
AGGARWAL K, CHOE LH, LEE KH. Shotgun proteomics using the iTRAQ isobaric tags. Brief Funct Genomic Proteomic. 2006;5(2):112-120. [ Links ]
ALY NS, HIRAMOTO A, SANAI H, HIRAOKA O, HIRAMOTO K, KATAOKA H, et al. Proteome analysis of new antimalarial endoperoxide against Plasmodium falciparum. Parasitol Res. 2007;100(5):1119-1124. [ Links ]
ANAMIKA K, SRINIVASAN N. Comparative kinomics of Plasmodium organisms: unity in diversity. Protein Pept Lett. 2007;14(6):509-517. [ Links ]
ANDERSEN JS, MANN M. Functional genomics by mass spectrometry. FEBS Lett. 2000;480(1):25-31. [ Links ]
ASHTON PD, CURWEN RS, WILSON RA. Linking proteome and genome: how to identify parasite proteins. Trends Parasitol. 2001;17(4):198-202. [ Links ]
BECKER K, TILLEY L, VENNERSTROM JL, ROBERTS D, ROGERSON S, GINSBURG H. Oxidative estrés in malaria parasite-infected erythrocytes: host-parasite interactions. Int J Parasitol. 2004;34(2):163-189. [ Links ]
BERGER SJ, LEE SW, ANDERSON GA, PASA-TOLIC L, TOLIC N, SHEN Y, et al. High-throughput global peptide proteomic analysis by combining stable isotope amino acid labeling and data-dependent multiplexed-MS/MS. Anal Chem. 2002;74(19):4994-5000. [ Links ]
BOZDECH Z, LLINAS M, PULLIAM BL, WONG ED, ZHU J, DERISI JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1(1):85-100. [ Links ]
BRIOLANT S, ALMERAS L, BELGHAZI M, BOUCOMONT-CHAPEAUBLANC E, WURTZ N, FONTAINE A, et al. Plasmodium falciparum proteome changes in response to doxycycline treatment. Malar J. 2010;9(9):141-154. [ Links ]
BROBEY RK, SOONG L. Establishing a liquid-phase IEF in combination with 2DE for the analysis of Leishmania proteins. Proteomics. 2007;7(1):116-120. [ Links ]
CELIS JE, GROMOV P. 2D protein electrophoresis: can it be perfected? Curr Opin Biotechnol. 1999;10(1):16-21. [ Links ]
CIACH M, ZONG K, KAIN KC, CRANDALL I. Reversal of mefloquine and quinine resistance in Plasmodium falciparum with NP30. Antimicrob Agents Chemother. 2003;47(8):2393-2396. [ Links ]
COHEN P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4(5):127-130. [ Links ]
CUI L, MIAO J, FURUYA T, LI X, SU XZ. PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot Cell. 2007;6(7):1219-1227. [ Links ]
CRAVATT BF, SIMON GM, YATES J III. The biological impact of mass- spectrometry-based proteomics. Nature. 2007;450(7172):991-1000. [ Links ]
DATE SV, STOECKERT CJ Jr Computational modeling of the Plasmodium falciparum interactome reveals protein function on a genome-wide scale. Genome Res. 2006;16(4):542-549. [ Links ]
DE JESUS JB, CUERVO P, JUNQUEIRA M, BRITTO C, SILVA-FILHO FC, SABOIAVAHIA L, et al. Application of two-dimensional electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for proteomic analysis of the sexually transmitted parasite Trichomonas vaginalis. J Mass Spectrom. 2007;42(11):1463-1473. [ Links ]
DONG MQ, VENABLE JD, AU N, XU T, PARK SK, COCIORVA D, et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007;317(5838):660-663. [ Links ]
DURAISINGH MT, COWMAN AF. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005;94(3):181-190. [ Links ]
ENGWEGEN JY, GAST MC, SCHELLENS JH, BEIJNEN JH. Clinical proteomics: searching for better tumour markers with SELDI-TOF mass spectrometry. Trends Pharmacol Sci. 2006;27(5):251-259. [ Links ]
FENN JB, MANN M, MENG CK, WONG SF, WHITEHOUSE CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64-71. [ Links ]
FIDOCK DA, ROSENTHAL PJ, CROFT SL, BRUN R, NWAKA S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509-520. [ Links ]
FOTH BJ, ZHANG N, MOK S, PREISER PR, BOZDECH Z. Quantitative protein expression profiling reveals extensive post-transcriptional regulation and post-translational modifications in schizont-stage malaria parasites. Genome Biol. 2008;9(12):177-195. [ Links ]
FOUCHER AL, MCINTOSH A, DOUCE G, WASTLING J, TAIT A, TURNER CM. A proteomic analysis of arsenical drug resistance in Trypanosoma brucei. Proteomics. 2006;6(9):2726-2732. [ Links ]
FOURNIER ML, GILMORE JM, MARTIN-BROWN SA, WASHBURN MP. Multidimensional separations-based shotgun proteomics. Chem Rev. 2007;107(8):3654-3686. [ Links ]
FLORENS L, WASHBURN MP, RAINE JD, ANTHONY RM, GRAINGER M, HAYNES JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419(6906):520-526. [ Links ]
GARDNER MJ, HALL N, FUNG E, WHITE O, BERRIMAN M, HYMAN RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002; 419(6906):498-511. [ Links ]
GARDNER MJ, SHALLOM SJ, CARLTON JM, SALZBERG SL, NENE V, SHOAIBIA, et al. Sequence of Plasmodium falciparum chromosomes 2, 10, 11 and 14. Nature. 2002;419(6906):531-534. [ Links ]
GELHAUS C, FRITSCH J, KRAUSE E, LEIPPE M. Fractionation and identification of proteins by 2-DE and MS: towards a proteomic analysis of Plasmodium falciparum. Proteomics. 2005;5(16):4213-4222. [ Links ]
GILSON PR, NEBL T, VUKCEVIC D, MORITZ RL, SARGEANT T, SPEED TP, et al. Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2006;5(7):1286-1299. [ Links ]
GOWDA DC, GUPTA P, DAVIDSON EA. Glycosylphosphatidylinositol anchors represent the major carbohydrate modification in proteins of intraerythrocytic stage Plasmodium falciparum. J Biol Chem. 1997;272(10):6428-6439. [ Links ]
GUNASEKERA AM, PATANKAR S, SCHUG J, EISEN G, WIRTH DF. Drug-induced alterations in gene expression of the asexual blood forms of Plasmodium falciparum. Mol Microbiol. 2003;50(4):1229-1239. [ Links ]
GUNASEKERA AM, WICKRAMARACHCHI T, NEAFSEY DE, GANGULI I, PERERA L, PREMARATNE PH, et al. Genetic diversity and selection at the Plasmodium vivax apical membrane antigen-1 (PvAMA-1) locus in a Sri Lankan population. Mol Biol Evol. 2007;24(4):939-947. [ Links ]
GRAHAM R, GRAHAM C, MCMULLAN G. Microbial proteomics: a mass spectrometry primer for biologists. Microb Cell Fact. 2007;6:26. [ Links ]
GRAVES PR, KWIEK JJ, FADDEN P, RAY R, HARDEMAN K, COLEY AM, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol. 2002;62(6):1364-1372. [ Links ]
GYGI SP, RIST B, GERBER SA, TURECEK F, GELB MH, AEBERSOLD R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17(10):994-999. [ Links ]
HORROCKS P, NEWBOLD CI. Intraerythrocytic polyubiquitin expression in Plasmodium falciparum is subjected to developmental and heat-shock control. Mol Biochem Parasitol. 2000;105(1):115-125. [ Links ]
HYNES SO, MCGUIRE J, WADSTROM T. Potential for proteomic profiling of Helicobacter pylori and other Helicobacter spp. using a ProteinChip array. FEMS Immunol Med Microbiol. 2003;36(3):151-158. [ Links ]
JOHNSON JR, FLORENS L, CARUCCI DJ, YATES JR III. Proteomics in malaria. J Proteome Res. 2004;3(2):296-306. [ Links ]
JONES A, FALDAS A, FOUCHER A, HUNT E, TAIT A, WASTLING JM, et al. Visualisation and analysis of proteomic data from the procyclic form of Trypanosoma brucei. Proteomics. 2006;6(1):259-267. [ Links ]
KONCAREVIC S, BOGUMIL R, BECKER K. SELDI-TOF-MS analysis of chloroquine resistant and sensitive Plasmodium falciparum strains. Proteomics. 2007;7(5):711-721. [ Links ]
KRIJGSVELD J, KETTING RF, MAHMOUDI T, JOHANSEN J, ARTAL-SANZ M, VERRIJZER CP, et al. Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics. Nat Biotechnol. 2003;21(8):927-931. [ Links ]
KUMAR R, MUSIYENKO A, OLDENBURG A, ADAMS B, BARIK S. Post-translational generation of constitutively active cores from larger phosphatases in the malaria parasite, Plasmodium falciparum: implications for proteomics. BMC Mol Biol. 2004;1:5-6. [ Links ]
LASONDER E, ISHIHAMA Y, ANDERSEN JS, VERMUNT AM, PAIN A, SAUERWEIN RW, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419(6906):537-542. [ Links ]
LASONDER E, JANSE CJ, VAN GEMERT GJ, MAIR GR, VERMUNT AM, DOURADINHA BG, et al. Proteomic profiling of Plasmodium sporozoite maturation identifies new proteins essential for parasite development and infectivity. PLoS Pathog. 2008;4(10):e1000195. [ Links ]
LE ROCH KG, ZHOU Y, BLAIR PL, GRAINGER M, MOCH JK, HAYNES JD, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301(5639):1503-1508. [ Links ]
LIST EO, BERRYMAN DE, BOWER B, SACKMANN-SALA L, GOSNEY E, DING J, et al. The use of proteomics to study infectious diseases. Infect Disord Drug Targets. 2008;8(1):31-45. [ Links ]
MAIR GR, BRAKS JA, GARVER LS, WIEGANT JC, HALL N, DIRKS RW, et al. Regulation of sexual development of Plasmodium by translational repression. Science. 2006;313(5787):667-669. [ Links ]
MAKANGA M, BRAY PG, HORROCKS P, WARD SA. Towards a proteomic definition of CoArtem action in Plasmodium falciparum malaria. Proteomics. 2005; 5(7):1849-1858. [ Links ]
MONTEOLIVA L, ALBAR JP. Differential proteomics: an overview of gel and non-gel based approaches. Brief Funct Genomic Proteomic. 2004;3(3):220-239. [ Links ]
MULLER S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol Microbiol. 2004;53:1291-1305. [ Links ]
NIRMALAN N, SIMS PF, HYDE JE. Quantitative proteomics of the human malaria parasite Plasmodium falciparum and its application to studies of development and inhibition. Mol Microbiol. 2004;52(4):1187-1199. [ Links ]
ONG SE, BLAGOEV B, KRATCHMAROVA I, KRISTENSEN DB, STEEN H, PANDEY A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376-386. [ Links ]
PAL-BHOWMICK I, SADAGOPAN K, VORA HK, SEHGAL A, SHARMA S, JARORI GK. Cloning, over-expression, purification and characterization of Plasmodium falciparum enolase. Eur J Biochem. 2004;271(23):4845-4854. [ Links ]
PAL-BHOWMICK I, VORA HK, JARORI GK. Sub-cellular localization and post-translational modifications of the Plasmodium yoelii enolase suggest moonlighting functions. Malar J. 2007;6:45. [ Links ]
PAPADOPOULOS MC, ABEL PM, AGRANOFF D, STICH A, TARELLI E, BELL BA, et al. A novel and accurate diagnostic test for human African trypanosomiasis. Lancet. 2004;363(9418):1358-1363. [ Links ]
PETRAK J, IVANEK R, TOMAN O, CMEJLA R, CMEJLOVA J, VYORAL D, et al. Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics. 2008;8(9):1744-1749. [ Links ]
PRIETO JH, KONCAREVIC S, PARK SK, YATES JR III, BECKER K. Large-scale differential proteome analysis in Plasmodium falciparum under drug treatment. PLoS One. 2008;3(12):e4098. [ Links ]
RABILLOUD T, BLISNICK T, HELLER M, LUCHE S, AEBERSOLD R, LUNARDI J, et al. Analysis of membrane proteins by two-dimensional electrophoresis: comparison of the proteins extracted from normal or Plasmodium falciparum-infected erythrocyte ghosts. Electrophoresis. 1999;20(18):3603-3610. [ Links ]
RADFAR A, DIEZ A, BAUTISTA JM. Chloroquine mediates specific proteome oxidative damage across the erythrocytic cycle of resistant Plasmodium falciparum. Free Radic Biol Med. 2008;44(12):2034-2042. [ Links ]
SIMS PF, HYDE JE. Proteomics of the human malaria parasite Plasmodium falciparum. Expert Rev Proteomics. 2006;3(1):87-95. [ Links ]
SMIT S, STOYCHEV S, LOUW AI, BIRKHOLTZ LM. Proteomic profiling of Plasmodium falciparum through improved, semiquantitative two-dimensional gel electrophoresis. J Proteome Res. 2010;9(5):2170-2181. [ Links ]
SMOLKA MB, ZHOU H, PURKAYASTHA S, AEBERSOLD R. Optimization of the isotope-coded affinity tag-labeling procedure for quantitative proteome analysis. Anal Biochem. 2001;297(1):25-31. [ Links ]
STURM N, JORTZIK E, MAILU BM, KONCAREVIC S, DEPONTE M, FORCHHAMMER K, et al. Identification of proteins targeted by the thioredoxin superfamily in Plasmodium falciparum. PLoS Pathog. 2009;5(4):e1000383. [ Links ]
TANNU NS, HEMBY SE. Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat Protoc. 2006;1(14):1732-1742. [ Links ]
THULASIRAMAN V, MCCUTCHEN-MALONEY SL, MOTIN VL, GARCIA E. Detection and identification of virulence factors in Yersinia pestis using SELDI Protein Chip system. Biotechniques. 2001;30(2):428-432. [ Links ]
UNLU M, MORGAN ME, MINDEN JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071-2077. [ Links ]
VIEIRA PP, FERREIRA MU, ALECRIM MG, ALECRIM WD, DA SILVA LH, SIHUINCHA MM, et al. pfcrt Polymorphism and the spread of chloroquine resistance in Plasmodium falciparum populations across the Amazon Basin. J Infect Dis. 2004; 190(2):417-424. [ Links ]
WU Y, CRAIG A. Comparative proteomic analysis of metabolically labelled proteins from Plasmodium falciparum isolates with different adhesion properties. Malar J. 2006;(5):67. [ Links ]
WU Y, NELSON MM, QUAILE A, XIA D, WASTLING JM, CRAIG A. Identification of phosphorylated proteins in erythrocytes infected by the human malaria parasite Plasmodium falciparum. Malar J. 2009;(8):105. [ Links ]
WHO. World Health Organization. World Malaria Report. Geneva, Switzerland. 2010. [ Links ]
YEH I, HANEKAMP T, TSOKA S, KARP PD, ALTMAN RB. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res. 2004;14(5):917-924. [ Links ]
ZHU H, PAN S, GU S, BRADBURY EM, CHEN X. Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun Mass Spectrom. 2002;16(22):2115-2125. [ Links ]
