










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.17 no.3 Bogotá Sept./Dec. 2012
FILOGENIA DE ESPECIES DEL SUBGENERO Parides (LEPIDOPTERA: PAPILIONIDAE) BASADA EN SECUENCIAS DEL GEN CITOCROMO OXIDASA I
Species phylogeny of the Subgenus Parides (Lepidoptera: Papilionidae) Based in Sequences of Citochrome Oxidase I Gene
INGRID MARCELA GUTIÉRREZ R.1, M.Sc. (c); GIOVANNY FAGUA2, M.Sc.
1Maestría en Biotecnología y Biología Molecular Médica. Universidad de Buenos Aires. Buenos Aires, Argentina. ingriddna@yahoo.com.
2Laboratorio de Entomología. Grupo de Sistemática Molecular. Pontificia Universidad Javeriana, carrera 7 # 43-82, edificio Jesús Emilio Ramírez (54). Departamento de Biología. Bogotá, Colombia.Tel.: 57 1 320 83 20 ext. 4081. Fax: 57 1 320 83 20 ext. 4057. fagua@javeriana.edu.co.
Correspondencia: Giovanny Fagua, fagua@javeriana.edu.co.
Presentado el 19 de abril de 2012, aceptado el 6 de junio de 2012, correcciones el 21 de octubre de 2012.
RESUMEN
Parides Hübner es el taxón terminal de Troidini, un grupo de mariposas aposemáticas diversificado en el trópico y subtrópico, y modelos de varios complejos miméticos batesianos y mullerianos. Varias de las especies americanas de Parides son simpátricas e involucran poblaciones con variaciones intraespecíficas en los patrones de coloración, lo que genera confusiones en la definición del estatus taxonómico, especialmente en Colombia, punto de convergencia de las biotas de Norte y Suramérica. Este trabajo genera una aproximación a la filogenia de este grupo de mariposas y establece una definición más robusta de algunos de los taxones. Para ello se analizaron ejemplares pertenecientes a 15 taxones del subgénero americano Parides ( Parides ) como grupo interno y se utilizó como grupo externo especies de otros dos géneros estrechamente relacionados de Troidini. Para la extracción del ADN se utilizó el protocolo de Pascual et al. (1997) y DNeasy Kit. Se amplificó el fragmento final del gen Citocromo Oxidasa I (COI) de 476 pb. Para obtener una hipótesis filogenética se realizaron análisis de máxima parsimonia y se evaluó el soporte de cada nodo mediante Jackknife y soporte absoluto de Bremer. También se realizó un análisis bayesiano. La hipótesis resultante sugiere que el subgénero Parides es un grupo parafilético. Molecularmente se hicieron también válidas una especie y cinco subespecies. Los ejemplares analizados de Parides se dividieron en tres grupos principales coincidentes con los grupos Lysander (grupo 1) y Aeneas (grupos 2 y 3) de Rothschild y Jordan (1906).
Palabras clave: ADN mitocondrial, distribución geográfica, filogenia molecular, mariposas, neotrópico.
ABSTRACT
Parides Hübner is a terminal taxon of Troidini, an aposematic butterfly group that is diverse in the tropics and subtropics, and a model of mullerian and batesian mimetic complexes. Several American species of Parides are sympatric and include populations with intraspecific variation in color pattern, thus creating confusion on their taxonomic status, mainly in Colombia where the biota of North and South America converge. This work presents a phylogenetic hypothesis of these butterflies and proposes a more robust definition of some taxa. For this, 15 taxa of the subgenus Parides were analyzed as ingroup; species of other two genera of Troidini, closer to Parides , were used as out-group. DNA was extracted using the Pascual et al. (1997) protocol and Quiagen DNAeasy kit. A terminal fragment of Cytochrome Oxidase I gen (476 bp) were amplified. We obtained a phylogenetic approximation using maximum parsimony and evaluated the branch support with Jackknife and absolute Bremer support. We also conducted a bayesian analysis. The resulting phylogenetic hypothesis suggested that Parides is a paraphyletic group; the molecular evidence support one species and five subspecies. The analyzed taxa were divided in three principal groups coincident with the Lysander (group 1) and Aeneas (groups 1 and 2) groups proposed by Rothschild and Jordan (1906).
Keywords: Butterflies, geographical distribution, mitocondrial DNA, molecular phylogeny, neotropic.
INTRODUCCIÓN
Las mariposas del subgénero Parides Hübner, 1819 presentan generalmente alas negras con manchas amarillas, rojas o verdes, el cuerpo es negro con manchas rojas, las uñas tarsales son asimétricas y son dimórficas, siendo siempre más conspicuo el macho. Sus larvas se alimentan de plantas tóxicas de la familia Aristolochiaceae (DeVries, 1987). Las hembras presentan un ducto de la bursae corto y ancho, que corresponde con el edeagus corto y grueso de los machos (Fagua, 1997). Las hembras son muy similares entre especies y forman parte de un importante complejo mimético Mülleriano asociado a otro complejo mimético Batesiano (DeVries, 1987).
Parides sensu lato está diversificado en el trópico y el subtrópico y se encuentra dividido en tres subgéneros (sensu Miller, 1987): Panosmia y Atrophaneura del viejo mundo (zona tropical asiática, los Himalayas, el sureste de China y sur de Japón) con 26 especies; y el subgénero Parides en el Neotrópico, con 34 especies y 107 subespecies (sensu Tyler et al., 1994). En Colombia este subgénero está diversificado en los bosques húmedos de la cuenca Amazónica, la Costa Pacífica y el Magdalena Medio (Fagua, 1997). Fagua (1997) registró para Colombia 17 especies y aceptó 28 subespecies, catalogando como formas otras 20 subespecies debido a su completa simpatría, mientras que Le Crom et al. (2002) registran 16 especies y aceptan 38 subespecies, varias de ellas simpátricas. Existe una gran confusión en la identificación de los taxones subespecíficos del subgénero, al utilizar características morfológicas y de coloración, debido a que las especies y subespecies son generalmente simpátricas e incluyen poblaciones con variaciones intraespecíficas en los patrones de coloración. Esto ha generado confusiones en la definición del estatus taxonómico, especialmente en Colombia, donde convergen especies y poblaciones de Norte y Suramérica (Fagua, 1997).
Una subespecie es una población fenotípicamente diferenciable cuyo rango de distribución no se solapa con el de otras subespecies (Fox, 1955; Frost et al., 1991; Mayr y Ashlock, 2001; Patten y Unitt, 2002) según esto, muchas de las formas descritas como subespecies para Colombia y América no se ajustan a dicha definición. Además, debido a la amplia capacidad de desplazamiento de los papiliónidos, es frecuente la generación de híbridos o la distribución clinal de los caracteres empleados como diagnósticos en algunas formas con categoría de subespecie (Sperling y Harrison, 1994). Por ello se hace necesario el análisis de caracteres moleculares que permiten una definición más robusta del estatus taxonómico de los taxones del subgénero Parides a partir de diferencias en secuencias de ADN mitocondrial (ADNmt), para el caso, generando una hipótesis filogenética que incluyó el mayor número de taxones del grupo Aeneas de Rothschild y Jordan (1906), justamente el de mayor número de taxones problemáticos. El uso de regiones del ADN mitocondrial como marcadores moleculares han permitido la obtención de filogenias robustas, la delimitación de especies objetivo y la estimación del flujo de genes (Moritz et al., 1987; Harrison, 1989; Hillis, 1996). Las regiones de los genes mitocondriales COI y COII han sido bien estudiadas en Lepidoptera y existe una base de datos importante de estas secuencias. El gen COI se considera una región que está evolucionando relativamente rápido a nivel nucleotídico (Caterino et al., 2001), por lo que se asume como una excelente herramienta de trabajo para resolver relaciones a nivel de especies y grupos de especies entre Papilionidae (Caterino y Sperling, 1999).
MÉTODOS
ESPÉCIMENES
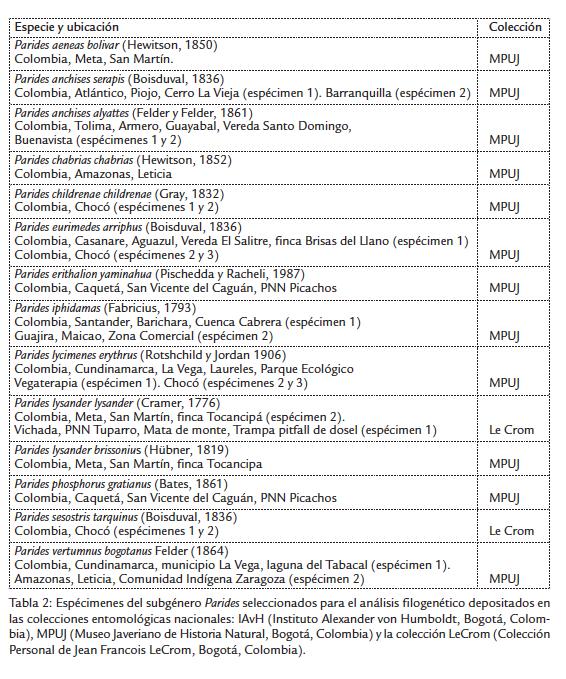
Se utilizaron espécimenes depositados en las colecciones entomológicas del IAvH (Instituto Alexander von Humboldt, Bogotá, Colombia), MPUJ (Museo Javeriano de Historia Natural, Bogotá, Colombia) y la CP LeCrom (Colección Personal de Jean Francois LeCrom, Bogotá, Colombia). Los adultos del subgénero Parides se identificaron a nivel de especies y subespecies mediante las claves morfológicas e ilustraciones de Rothschild y Jordan (1906), Tyler et al.(1994), Fagua (1997) y Le Crom et al.(2002), para posteriormente confrontar estas determinaciones con secuencias del gen mitocondrial COI, de cada espécimen se obtuvo material fotográfico.
El análisis filogenético incluyó 30 espécimenes (Tabla 1 y Tabla 2); el grupo interno lo conformaron 15 taxones de Parides de los que se obtuvieron secuencias, 12 de Colombia (25 espécimenes de los cuales se extrajo ADN y se pudo amplificar exitosamente el fragmento mitocondrial de comparación, (Tabla 2), y tres de Brasil (P. bunichus, P. neophilus y P. panthonus jaguarae) cuyas secuencias se obtuvieron del GenBank (Silva-Brandão et al., 2005). Las secuencias de las especies del grupo externo, Atrophaneura alcinous y Pachliopta neptunus, también se obtuvieron del GenBank (Fig. 1; Tablas 1 y 2).
Para la obtención de las secuencias se extrajeron las tres patas de un costado de cada espécimen o cualquier pata(s) si no las presentaban. En ejemplares antiguos (más de diez años de colecta) se realizó extracción del tórax. Patas o tórax se depositaron por espécimen en tubos estériles de 1,5 ml debidamente marcados y se almacenaron a -20 °C para evitar la degradación del ADN.


EXTRACCIÓN DE ADN Y AMPLIFICACIÓN
La extracción de ADN de los espécimenes se realizó utilizando DNeassy Tissue Kit (Qiagen QIAamp) para los ejemplares con más de un año de colecta y el protocolo de Pascual et al.(1997) para los ejemplares menores a un año de colectado. Al protocolo de Pascual et al.(1997) se le incluyó una fase previa de maceración de los tejidos con nitrógeno líquido.
El ADN obtenido se preservó a -4 °C para luego realizar las amplificaciones de los fragmentos definidos por PCR. Se amplificó el fragmento final de 476 pb del gen COI, utilizando los primers Jerry (CAACATTTATTTTGATTTTTTGG) y Mila (GCTAATCCAGTGAAT AATGG), Caterino y Sperling, 1999: Caterino et al., 2001, por presentar una mayor tasa de variación (Lumpley y Sperling, 2010). Las amplificaciones se realizaron en un termociclador empleando calor de arranque; se inició con una denaturación a 95 °C por 5 min; seguido por 35 ciclos de 30 seg a 94 °C, 30 seg a 47 °C, 1 min 30 segs a 72 °C y una extensión final 10 min a 72 °C. Una vez amplificadas todas las muestras, se almacenaron a -4 °C, hasta su posterior traslado a la fase de purificación y secuenciación (efectuada directamente en ambos sentidos de la doble cadena) realizada por MACROGEN Ltda.
ANÁLISIS FILOGENÉTICO
Las secuencias nucleotídicas se alinearon manualmente con el programa BioEdit versión 7.0.5 (Hall, 2005), utilizando como referencia a la secuencia de COI de Drosophila yakuba, por estar la molécula de ADN mitocondrial de esta especie completamente secuenciada (Clary y Wolstenholme, 1985); se compararon ambos sentidos de la doble cadena para cada muestra (espécimen) y todas las secuencias se analizaron por medio de POY 3.0.11 (Wheeler et al., 2003). Los gaps no se observaron como entidades, pero se tomaron como indels (eventos de inserción/deleción) para optimizar el alineamiento (Wheeler, 1996).
Para el análisis filogenético se analizó primero la matriz de datos completa mediante PAUP 4.0q (Swofford, 1999). Por medio del programa MODELTESTS 3.06 (Posada y Crandall, 1998), se determinó el modelo de evolución al que mejor se ajustaba al gen, la proporción de transición/transversión, la proporción de sitios invariables y el modelo de substitución que mejor se ajustaba a los datos obtenidos. Se utilizó máxima parsimonia (MP) utilizando caracteres moleculares. Los análisis de MP se ejecutaron por medio de Tree Analysis using New Technology; TNT 1.1 (Goloboff et al.2008), empleando los siguientes parámetros: búsqueda heurística, TBR branch-swapping para obtener los árboles más parsimoniosos, adición de 500 réplicas y un ratchet de 200 iteraciones; los caracteres multiestado fueron tratados como polimorfismos y todos con pesos diferentes, los gaps fueron contados como datos faltantes. El índice de consistencia (IC) y el índice de retención (IR) fueron calculados en Winclada 1.00.08 (Nixon, 1999-2002). De igual manera, con TNT 1.1 (Goloboff et al., 2008) se determinó el grado de soporte o la robustez de las ramas empleando: soporte por análisis de Jackknife (Felsenstein, 2004) y el soporte absoluto de Bremer (Felsenstein, 2004). Los valores del soporte se definieron como: "soporte débil" valores de Bremer de 1-6 (correspondiendo a valores de Jackknife 3 % - 40 %), "soporte moderado" con valores entre 6-16 (valores de Jackknife 41 % -70 %), "soporte bueno" con valores entre 17-22 (valores de Jackknife 71 % - 88 %) y "soporte fuerte" con valores > 22 (valores de Jackknife 89 % - 100 %). Por medio de PAUP 4.0q (Swofford 1999), se determinó el número de sitios constantes, sitios variables no informativos y el número de sitios informativos de parsimonia.
Se realizaron análisis Bayesianos con el programa MrBayes 3.1 (Ronquist et al., 2005), utilizando el modelo de evolución obtenido para el gen; se corrieron simulaciones de seis cadenas simultáneas para 1,0 x 106 generaciones, el muestreo de los árboles se realizó sobre 250 ciclos. Se descartaron los primeros 1000 árboles como "calientes". Los valores de probabilidad posterior (PP) se definieron como: "débil" valores entre 53 % - 61 %, "bueno" con valores entre 65 % - 95 % y "fuerte" con valores > 97 %.
RESULTADOS
REVISIÓN Y ANÁLISIS MOLECULAR DE LOS ESPÉCIMENES
Se revisaron 403 ejemplares del subgénero Parides en las colecciones visitadas. Desafortunadamente, la mayoría fueron colectados hace más de 20 años, constituyéndose en material antiguo para conservar ADN en buen estado. Para la extracción se seleccionaron 137 espécimenes colectados después de 1996; de estos se logró obtener ADN de 130, pero solo en 80 se pudo amplificar el fragmento de estudio. Una vez alineadas las 80 muestras, se decidió incluir en el análisis solo a 25, dado que las 55 restantes presentaron un bajo porcentaje de coincidencia nucleotídica (< 80 %) al alinearlas respecto a la secuencia de referencia (COI de Drosophila yakuba, Clary y Wolstenholme, 1985), evento consecuencia del mal estado de conservación de su ADN.
Como se comentó, al método de extracción de ADN de Pascual et al.,1997, se adicionó la maceración de los tejidos en nitrógeno líquido, previo a su paso al buffer de lisis, evento que facilitó la rotura adecuada de los tejidos, en muy poco tiempo, y que aumentó la cantidad obtenida de ADN. Se evidenció que el fragmento amplifica dependiendo de la especie. Es decir, se presentaron especies con una alta cantidad de ADN en donde el marcador molecular funcionó perfectamente, mientras que en otros casos, como en Parides neophilus, la muestra no amplificó, a pesar de realizar diferentes pruebas de amplificación (cambios en la temperatura de anillamiento, en el volumen del buffer de la polimerasa, el cloruro de magnesio, la concentración de la polimerasa y el volumen final de la reacción). En otros casos las señales de amplificación fueron nulas o bajas (P. sesostris, P. childrenae y P. eurimedes), lo que puede corresponder a que el ADN de algunas especies de Parides podría presentar una mutación en la zona de complementariedad con los primers (alelo-específicos), ocasionando una disminución o perdida de especificidad de los primers hacia la secuencia complementaria.
ANÁLISIS FILOGENÉTICO
Se analizaron las bases y posiciones de 695 pb del gen COI (695 caracteres), incluyendo nucleótidos y gaps; 161 caracteres fueron constantes, 56 caracteres variables no informativos de parsimonia y 476 caracteres informativos de parsimonia. Los análisis filogenéticos se determinaron bajo el modelo de evolución simple de Jukes-Cantor, GTR + G (General Time-Reversible model; Rodríguez et al., 1990), con distribución gamma y sin proporción de sitios invariables. El promedio de A+T fue de 56,16 % (26,10 % A, 30,06 % T) mayor que el promedio de G+C de 43,83 % (19,89 % G, 23,94 % C). La proporción de transición/transversión fue de 0,816, lo que corresponde a una saturación de transición en las secuencias analizadas; se observó que la proporción de sustitución de C-T fue sustancialmente más alta que otro tipo de sustitución.
MÁXIMA PARSIMONIA
El análisis de máxima parsimonia produjo dos árboles igualmente parsimoniosos con una longitud de 3236 (IC = 0,5, IR = 0,31), cuyo consenso estricto dio un árbol con una longitud de 3240 y los mismos índices de consistencia y retención (Fig. 2). En los dos árboles y el consenso se encontró una baja homoplasia (IC = 0.5) y la parafilia del subgénero Parides . El árbol de consenso estricto mostró menor resolución por colapsar el clado en donde se encuentran las subespecies P. anchises alyattes, P. anchises serapis y P. erithalion yaminahua, mostrando relaciones inciertas entre estas (Fig. 2). Asimismo, el árbol reveló que los nodos de las ramas básales están pobremente resueltos, presentando soporte de Jackknife débil y moderado (3 % - 47 %) y soporte de Bremen débil (1-6); por el contrario, los nodos de las ramas terminales estuvieron bien soportados por Jackknife (71 % - 100 %) y Bremen (17-32).
Las relaciones de Parides con el grupo externo, Atrophaneura alcinous y Pachliopta neptunus, no fueron claramente resueltas. A. alcinous resultó externa a las Parides americanas, evento coherente con lo propuesto por Miller, 1987, mientras que las Parides estudiadas se agregaron en una rama bien soportada por Jackknife pero no por Bremer (Fig. 2). Sin embargo P. neptunus, grupo hermano de los Troidini neotropicales sensu Miller (1987), se asoció con P. lysander lysander dentro del grupo interno, con soporte de Jackknife moderado y soporte de Bremer débil. P. bunichus resultó la especie basal de las Parides americanas, con soporte de Jackknife fuerte y de Bremer bajo, coincidiendo con los resultados de Silva-Brandão et al.(2005). P. bunichus fue incluida dentro del grupo Ascanius por Rothschild y Jordan (1906).
La posición de P. neophilus como hermano de las Parides restantes (Fig. 2) no es coherente con estudios previos, aunque se resalta que su soporte de Bremer fue débil. Es importante anotar que las secuencias de los taxones obtenidas del GenBank se separaron de los taxones cuyas secuencias se obtuvieron del trabajo de laboratorio, lo anterior puede deberse a los indels introducidos en las secuencias durante el alineamiento. Sin embargo P. panthonus jaguarae, una secuencia obtenida del GenBank, se agrupó en el clado que incluyó a las Parides pero con valores de Bremer y Jackknife débiles.
Los taxones restantes de Parides se dividieron en tres grupos (1,2,3) y cuatro subgrupos (A, B, C, D; Fig. 2). El grupo 1, subgrupo A, incluyó a las especies de Parides con manchas marginales rosadas en el ala posterior, mientras que los subgrupos B, C y D incluyeron a las especies con manchas marginales blancas en el ala posterior. Este agrupamiento coincide con la propuesta por Rothschild y Jordan,1906, para quienes los taxones con manchas marginales rosadas en el ala posterior conforman el grupo Lysander, mientras que los taxones con dichas manchas blancas conforman el grupo Aeneas . En este sentido, los resultados moleculares soportan el arreglo de Rothschild y Jordan (1906), aunque con las exepciones de P. chabrias chabrias, P. vertumnus bogotanus ejemplar 2 y P. eurimedes arriphus ejemplar 3.
Todos los ejemplares de P. sesostris tarquinius (soporte de Jackknife y Bremer fuertes) y P. iphidamas (soporte de Jackknife fuerte y soporte de Bremer moderado) conformaron grupos monofiléticos (Fig. 2). Las subespecies restantes con más de un espécimen en el análisis presentaron grupos parafiléticos como P. childrenae childrenae, P. vertumnus bogotanus 1 y P. eurimedes arriphus 3, con soportes de Jackknife y Bremer fuertes. Adicionalmente, como ejemplares hermanos se encontró a las dos P. l. lysander (soportes de Jackknife y Bremer fuertes), los dos P. eurimedes arriphus (Jackknife fuerte y Bremer bueno), los ejemplares 1 y 3 de P. lycimenes erythrus (Jackknife y Bremer fuertes) y los dos P. anchises alyattes (Jackknife y Bremer fuertes).
La mayoría de las ramas terminales se encontraron bien soportadas con soportes de Jackknife y Bremer buenos y fuertes; aunque para el clado de P. anchises serapis 1 y P. erithalion yaminahua el soporte de Bremer fue débil (5), lo que se relacionó con que la rama que soporta a este clado se colapsó. Además, los nodos de las ramas de los grupos 1, 2 y 3 presentaron soportes débiles, y los nodos de las ramas de los cuatro subgrupos presentaron valores de Bremer débil moderado, por lo que las relaciones entre los grupos o subgrupos no fueron robustas.
ANÁLISIS BAYESIANO
El análisis Bayesiano obtuvo un árbol de consenso estricto (Fig. 3) de 43.228 árboles restantes; la topología del árbol al incorporar el modelo de evolución GTR+G, con distribución gamma fue heterogénea, respecto de lo obtenido según análisis de Máxima Parsimonia (MP). El promedio de probabilidad posterior (PP) para la inferencia filogenética fue de 87 %. El árbol Bayesiano presentó diez ramas colapsadas con una PP del 90 %. Ocho (de 22) nodos del grupo interno presentaron una PP de 1,0, 14 nodos y estuvieron soportados con un nivel de significancia de > 0,9 (7 nodos) y > 0,5 (7 nodos). Las relaciones del clado Parides con el grupo externo en los dos análisis presentó coincidencia con una PP fuerte (100 %) para Atrophaneura alcinous y una PP moderada (61 %) para Pachliopta neptunus. P. bunichus estuvo por fuera del grupo y P. neophilus presentó la misma ubicación en el árbol que en MP y con una PP fuerte (100 %), comparado con el soporte de Bremer débil (7) de MP, por lo que la asignación de este taxón en el árbol puede ser precisa. P. panthonus jaguarae presentó colapso de su rama con una PP indeterminada, indicando relaciones confusas con los restantes taxones de Parides. Los grupos 1 y 3 establecidos en MP se encontraron claramente definidos en este análisis; sin embargo los subgrupos presentaron colapso en algunas de sus ramas y diferente distribución de los clados y de los taxones, por lo que las relaciones entre los taxones terminales variaron. Los clados que se modificaron en análisis Bayesiano con respecto a MP, correspondieron a los clados con soportes de Bremer débil a moderado. Así mismo, los nodos de las ramas básales del árbol de MP que presentaron soportes de Jackknife y de Bremer débiles correspondieron en análisis Bayesiano con las ramas con PP débil (53-56 %) y a las ramas que se colapsaron. El análisis Bayesiano, al igual que el de MP, estableció los dos grupos principales de Parides en buena parte coincidentes con los grupos Lysander y Aeneas de Rothschild y Jordan (1906), debido a que las relaciones de los taxones terminales en gran parte no se modificaron.
DETERMINACIÓN DE LOS TAXONES DE ESPECIE Y SUBESPECIE DE Parides EN COLOMBIA
Se analizaron 12 especies y 13 subespecies de Parides en Colombia (Tabla 2), que mostraron correspondencia respecto a la designación especifica y subespecífica morfológicamente, propuesta en principio por Rothschild y Jordan (1906) y luego por Tyler et al., (1994), Fagua (1997) y Le Crom et al., (2002). El análisis de distribución geográfica de Parides en Colombia propuesta por Tyler et al., (1994) y Fagua (1997), plantean a las subespecies de acuerdo a sus distribuciones alopátricas o simpátricas, respectivamente. Lo anterior fue un factor importante en la validación de los taxones analizados a nivel de especie y subespecie de Parides. Además, esta delimitación fue coherente con el concepto de especie filogenético de Nixon y Wheeler (1990), quienes la definen como una detectable pequeña unidad de población monofilética, por lo que la monofília que presentaron varios de los taxones con más de un individuo en el análisis de MP permitió hacer valido a una especie y cinco subespecies de Parides, para el caso: P. iphidamas, P. anchises alyattes, P. eurimedes arriphus, P. lycimenes erythrus, P. lysander lysander y P. sesostris tarquinius.
DISCUSIÓN
ANÁLISIS FILOGENÉTICO
Este análisis incluye el mayor número de taxones de Parides del grupo Aeneas de Rothschild y Jordan, 1906, del cual se analizaron 12 especies y 13 subespecies reconocidas para Colombia por Tyler et al.,1994, Fagua,1997 y Le Crom et al.,2002. Con base en los resultados de Máxima Parsimonia (MP) y análisis Bayesiano, el subgénero Parides es parafilético, siendo esto no consecuente con la propuesta de Hancock (1983) quien propone a Parides como un grupo monofilético.
Según el análisis de Máxima Parsimonia, Atrophaneura alcinous fue corroborada como parte del grupo externo del subgénero Parides , evento que apoyaría el planteamiento propuesto por Munroe,1961, y seguido por Miller,1987, en dividir a Parides en las especies del Viejo Mundo (Atrophaneura) y las del Neotrópico ( Parides ). Por su parte, Parides se dividiría en tres grupos (Fig. 2), que coinciden en buena parte con los grupos Ascanius, Lysander y Aeneas de Rothschild y Jordan (1906), quienes ubicaron a las Parides americanas en la subsección A "Aristolochia-Swallowtail" equivalente a las actuales Troidini Americanas. Estos grupos corresponderían a mariposas que presentan series submarginales de manchas rojas en el ala posterior (Hancock, 1983).
Dentro de las Parides ( Parides ) analizadas, la ubicación basal de P. bunichus corrobora lo propuesto por Rothschild y Jordan, 1906, Tyler et al., 1994 y Silva-Brandão et al.,2005, quienes ubican a esta especie en el grupo Ascanius, que retiene gran número de caracteres primitivos y tiene una distribución disyunta en América (México y Sur de Brasil). Adicionalmente, este trabajo corrobora en buena parte a la división de Parides de Rothschild y Jordan,1906, hecha con base en análisis morfológicos.
La topología general obtenida del subgénero Parides fue diferente según análisis de MP y Bayesiano, excepto para la ubicación de P. neophilus que se mantuvo estable en los dos análisis como grupo hermano del resto de las especies de Parides (Fig. 2, Fig. 3). Sin embargo las topologías coincidieron en incluir a la mayoría de los taxones estudiados dentro de los grupos Lysander y Aeneas de Rothschild y Jordan (1906). El grupo Lysander incluyó a los taxones P. lysander lysander, P. lysander brissonius, P. chabrias chabrias, P. vertumnus bogotanus 2 y P. eurimedes arriphus, que son mariposas con manchas marginales rosadas en el ala posterior y palpos labiales negros.
De estas solamente P. chabrias chabrias y P. vertumnus bogotanus 2 corresponden al grupo Aeneas de Rothschild y Jordan (1906). Esta ubicación posiblemente se deba a que chabrias pertenecen a un grupo de especies más amplio que incluye a Parides haneli, P. quadratus, P. pizarro, P. vercingetorix y P. klagesiesi, de las que no se obtuvo ADN, lo que posibilita la vinculación de P. chabrias chabrias a cualquier rama. Respecto de P. vertumnus bogotanus 2, su ubicación puede ser explicada porque la hembra analizada puede corresponder a una forma geográfica no coincidente, o a que la designación taxonómica de los espécimenes femeninos de esta subespecie, muy raras en colecciones, es incorrecta, característica que presentan algunas de las hembras de Parides (Fagua, 1997).
Las especies del grupo Aeneas pertenecerían al grupo de divergencia más reciente de Parides según análisis de MP e inferencia Bayesiana; estas incluyen a las mariposas con manchas marginales blancas en el ala posterior. No obstante, dentro de este grupo se incluyeron el ejemplar 3 de P. eurimedes arriphus y P. panthonus jaguarae, subespecies del grupo Lysander según Rothschild y Jordan (1906). La inclusión de estas dos en el grupo Aeneas puede ser simplemente una asignación azarosa dado el bajo porcentaje de coincidencia de la secuencia de P. eurimedes arriphus respecto del COI de Drosophila yakuba, y dados los soportes de Jackknife y Bremer débiles y PP débil de P. panthonus jaguarae. Este resultado contrasta con lo propuesto por Silva-Brandão et al.(2005), que designan a P. panthonus jaguarae como taxón hermano de P. lysander y relacionado con P. eurimedes, P. zacynthus y P. neophilus. En el grupo 1 (Fig. 2) la primera divergencia corresponde al clado de Pachliopta neptunus y P. lysander lysander, ubicación que se mantuvo en los análisis de MP y Bayesiano, aunque con valores de Bremer y de PP débiles, por lo que la asignación como rama de divergencia basal de Parides debe tomarse como poco precisa. De igual manera, la ubicación de Pachliopta neptunus dentro de este grupo puede ser simplemente un artefacto debido a la no inclusión en el presente análisis de espécimenes de Cressida y Euryades, taxones hermanos de Pachliopta (sensu Miller, 1987), lo que estaría respaldado por los soportes moderados de Jackknife y Bremer y la PP débil. Las relaciones entre los dos ejemplares de P. lysander lysander, fuertemente soportados, es consecuente con que, morfológicamente, corresponderían a macho y hembra de la misma subespecie.
El Grupo 2 (Fig. 2) presentó soportes débiles de Jackknife y de Bremer en su rama basal, lo que indica que las relaciones entre los subgrupos que lo integran (A y B) son imprecisas, evento evidenciado en análisis Bayesiano en donde las ramas que soportan cada subgrupo se colapsaron con PP fuertes. Referente al subgrupo A, sus ramas internas estuvieron bien soportadas y presentaron valores de Jackknife fuertes; este grupo incluyó dos clados (P. chabrias chabrias + P. lysander brissonius) y (P. vertumnus bogotanus 2+ (P. eurimedes arriphus 1 + P. eurimedes arriphus 2)).
El subgrupo B integrado por las subespecies P. phosphorus gratianus, P. lycimenes erythrus, P. childrenae childrenae, P. vertumnus bogotanus 1 y P. eurimedes arriphus 3 presentó diferente topología en los análisis de MP e inferencia Bayesiana, por lo que las relaciones de sus taxones terminales pueden ser ambiguas. Esto posiblemente se deba a que la rama interna que soporta este subgrupo mostró un valor de Jackknife moderado y soporte de Bremer débil; además, en análisis Bayesiano los clados (P. phosphorus gratianus + P. lycimenes erythrus) y (P. childrenae childrenae 1 + (P. childrenae childrenae 2 + (P. vertumnus bogotanus + P. eurimedes mycale))) se encontraron separados debido a que sus ramas se colapsaron con una PP de 99 %.
En el grupo 3 los subgrupos C y D incluyeron a los taxones P. anchises alyattes, P. anchises serapis, P. erithalion yaminahua, P. lycimenes erythrus, P. iphidamas, P. aeneas bolivar y P. sesostris tarquinius. Según MP este grupo presentó un soporte de Bremer débil y en análisis Bayesiano cuatro de sus ramas se colapsaron, por lo que las relaciones filogenéticas de la mayor parte de los taxones variaron, lo que demuestra confusas las relaciones entre sus taxones. El grupo C incluyó a los ejemplares de P. anchises alyattes, P. anchises serapis, P. erithalion yaminahua. Las relaciones filogenéticas de P. anchises serapis y P. erithalion yaminahua no se encontraron totalmente resueltas y en el análisis de MP las ramas que soportan a estas subespecies se colapsaron. Posiblemente la relación entre estas subespecies puede estar relacionada con la secuencia de P. erithalion yaminahua, que presentó un alto porcentaje de coincidencia nucleotídica con las secuencias de P. anchises serapis, lo que puede mostrar una afinidad genética entre estas subespecies a pesar de que los patrones de genitalia, la coloración y la morfología son diferentes (Fagua, 1997). No obstante, hay que destacar que solo se analizó un espécimen de P. erithalion yaminahua y que esta subespecie está restringida al Perú (Tyler et al., 1994), por lo que puede corresponder a una forma o aberración de alguna especie de P. erithalion presente en Colombia, lo que debe ser ratificado mediante el análisis de más material de esta subespecie. Referente a los dos espécimenes analizados (dos machos) de P. anchises serapis, a pesar de provenir de localidades distantes, mostraron igual fenotipo y una fuerte relación filogenética al ubicarse en el árbol dentro de un mismo clado, lo que podría corroborar lo planteado por Fagua (1997), quien la define como una subespecie muy distintiva y poco variable.
En el subgrupo D, que incluye a las subespecies P. lycimenes erythrus, P. iphidamas, P. aeneas bolivar y P. sesostris tarquinius, las relaciones entre los taxones no son claras y la rama pre- sentó un valor de Bremer moderado en análisis de MP y en análisis Bayesiano, donde dos de sus clados se colapsaron. Finalmente, el análisis de MP reveló que el grupo 3, parte del Aeneas de Rothschild y Jordan (1906), fue el grupo divergencia más temprana dentro de las Parides estudiadas.
Varios autores han argumentado que tanto el número de caracteres como el número de taxones en ser muestreados son importantes en la buena estimación de los árboles filogenéticos (Swofford, 1999). En análisis de datos moleculares adicionar taxones a un grupo de genes puede a veces mejorar las inferencias del árbol filogenético, sobre todo en los casos donde el interés es el largo de las ramas (Hillis, 1996); además incrementar los taxones puede mejorar la reconstrucción del estado ancestral de los caracteres, estimar la proporción y los modelos de evolución de las secuencias, y generalmente proporcionan un mejor resumen de la historia evolutiva de un clado (Sanderson et al., 2003).
El alto número de caracteres informativos que presentaron las secuencias, permiten entender porque los dos análisis mostraron topologías semejantes frente a la filogenia de Paride. Por consiguiente, se plantea que la hipótesis filogenética obtenida para Parides es robusta y con altos valores de Jackknife , Bremer y PP de los grupos de taxones terminales, aunque es importante aumentar el número de caracteres a analizar, lo que permitiría incrementar la exactitud (Hillis, 1998) y el soporte (Felsenstein, 2004; Sanderson et al., 2003) de la hipótesis filogenética generada.
El grado de saturación encontrado en COI, correspondió a 0,816, siendo un valor alto, lo que puede deberse a que la gran mayoría de caracteres informativos presentaron cambios rápidos (sinónimos) en la tercera posición de un codón. El resultado de esta saturación se evidenció en las altas probabilidades que se obtuvieron en el análisis Bayesiano, estos modelos altos de probabilidad pueden ser el mejor ajuste de estos caracteres para generar una topología (Monteiro y Pierce, 2000).
DETERMINACIÓN Y DISTRIBUCIÓN GEOGRÁFICA DE LOS TAXONES DE ESPECIE Y SUBESPECIE DE Parides EN COLOMBIA
De los 25 espécimenes analizados de Parides en este estudio y comparado con un mayor número de individuos analizados en los trabajos de Tyler et al.(1994), Le Crom et al.(2002) y especialmente el de Fagua (1997), se ratifica que la validación de varios de los taxones de Parides es poco robusta, a pesar de la coincidencia con estudios previos. Lo anterior se complementaría con el análisis genético de un mayor número de espécimenes, primordialmente para la definición de subespecies, porque la mayoría de las subespecies de Parides descritas son simpátricas e involucran poblaciones en las cuales se han reconocidos híbridos (Fagua, 1997).
Asimismo, el análisis molecular mostró que en la validación de las especies y subespecies de Parides es útil emplear machos y hembras, porque los dos conforman grupos monofiléticos dentro del árbol filogenético según análisis de MP e inferencia Bayesiana, como ocurre con los taxones de P. lysander lysander, P. eurimedes arriphus, P. anchises alyattes y P. sesostris tarquinius. Sin embargo, que en P. lycimenes erythrus una hembra se haya separado de los restantes ejemplares, puede estar relacionado con lo propuesto por Fagua (1997), quien menciona que varias de las hembras de las subespecies de Parides se encuentran taxonómicamente mal asignadas o pueden corresponder a formas geográficas no coincidentes o, incluso, podrían corresponder a híbridos interespecíficos ocasionales. La hipótesis filogenética plantea al subgénero Parides como un grupo parafilético y las Parides analizadas se dividieron en tres grupos principales coincidentes con los grupos Lysander y Aeneas de Rothschild y Jordan (1906). Por su parte varias de las relaciones filogenéticas que se describieron como incongruencias, se justifican por el bajo porcentaje de coincidencia nucleotídica que presentaron algunos espécimenes respecto a la secuencia de COI de Drosophila yakuba. Aunque también podría ser explicado por el bajo número de espécimenes analizados dentro de cada taxón. Se encontró coincidencia frente al análisis morfológico de otros autores y el desarrollado en este trabajo en relación a la designación de taxones de especie y subespecie para el subgénero Parides , lo que permite establecer que los dos análisis se complementan, lo que integrado con la distribución geográfica, permite validar los estatus taxonómico de especies y subespecies para este subgénero en Colombia. Con los datos obtenidos los ejemplares soportaron, a nivel molecular, una especie y cinco subespecies, infortunadamente existe muy poco material fresco en colecciones para poder incrementar la muestra y la robustez de un análisis filogenético.
CONCLUSIONES
El subgénero Parides fue recuperado como un grupo parafilético. Las Parides analizadas se dividieron en tres grupos principales, coincidentes con los grupos Ascanius, Lysander y Aeneas de Rothschild y Jordan (1906). La ubicación basal de P. bunichus, especie del grupo Ascanius, corrobora lo propuesto por Tyler et al.(1994) y Silva-Brandão et al.(2005). El resto de los taxones se distribuyeron en dos grupos y cuatro subgrupos. En el grupo 1, la primera rama divergente incluyó a Pachliopta neptunus, un macho y una hembra de P. l. lysander. La ubicación de P. neptunus dentro de este grupo puede ser un artefacto debido a la no inclusión de espécimenes de Cressida y Euryades, taxones hermanos de Pachliopta. La estrecha asociación de macho y hembra de P. l. lysander es consecuente con su asignación subespecífica hecha a nivel morfológico.
El subgrupo A del grupo 2 correspondió principalmente al grupo Lysander de Rothschild y Jordan (1906) e incluyó a P. l. lysander, P. lysander brissonius y P. eurimedes arriphus. P. chabrias y P. vertumnus bogotanus 2 que también fueron incluidas en este grupo pero con soportes muy débiles, lo que es coherente con que estos taxones hayan sido incluidos dentro del grupo Aeneas por Rothschild y Jordan (1906). El subgrupo B del grupo 2 incluyó a las subespecies P. phosphorus gratianus, P. lycimenes erythrus, P. childrenae childrenae, P. vertumnus bogotanus 1 y P. eurimedes arriphus 3. Este presentó diferente topología en los análisis de MP e inferencia Bayesiana, por lo que las relaciones de sus taxones terminales pueden ser ambiguas. En este grupo una hembra de P. lycimenes erythrus se vinculó a un macho de P. phosphorus gratianus, lo que puede estar relacionado con una mala asignación taxonómica de la forma femenina, que podría corresponder a formas geográficas no coincidentes o a algún híbrido interespecífico.
En el grupo 3 los subgrupos C y D incluyeron a los taxones P. anchises alyattes, P. anchises serapis, P. erithalion yaminahua, P. lycimenes erythrus, P. iphidamas, P. aeneas bolivar y P. sesostris tarquinius. Estas especies son parte del grupo Aeneas de Rothschild y Jordan (1906), y fue el grupo divergencia más temprana dentro de las Parides estudiadas.
Un aspecto relevante de este estudio es dejar en claro que se está presentando una filogenia basada en la evidencia de cambio de un gen mitocondrial, por tanto de transferencia materna dentro de parte de las especies que conforman el subgénero. En este sentido, los alcances deben ser proporcionados y hacen parte de la resolución final de la filogenia de este muy interesante grupo de mariposas. No deja de ser interesante, sin embargo, ver como los resultados coinciden en términos generales con los trabajos morfológicos taxonómicos realizados a principios del siglo XX.
Es conveniente comentar que la filogenia estará definida por el gen que presente una tasa de cambio adecuada al nivel taxonómico que se desea dilucidar. En este caso se analizaron cambios relativamente recientes desde el nivel intragenérico al subespecífico, y en este nivel, una de las mejores herramientas es el gen de la COI, como lo resaltan Caterino y Sperling (1999) y lo corroboran posteriormente Wahlberg y Wheat (2008).
AGRADECIMIENTOS
A la Vicerrectoría Académica de la Pontificia Universidad Javeriana (proyecto 03099) y a la Sociedad Colombiana de Entomología (SOCOLEN), programa de becas a estudiantes en Entomología, por su apoyo financiero. Este trabajo se basó en la información geográfica y taxonómica obtenida en el proyecto "El género Parides Hübner (Lepidoptera Papilionidae) en Colombia" financiado por la Fundación para la Promoción de la Ciencia y la Tecnología del Banco de la República. A Mónica Higuera y DianaÁlvarez por su asesoría y orientación durante la realización del proyecto. Al laboratorio de Genética de Poblaciones-Biología Evolutiva y al Grupo Elitros de trabajo en Entomología por su ayuda permanente. Un especial agradecimiento al Dr. Olaf Mielke, a Jean Françoise Le Crom y a Julián Salazar por su significativo apoyo con los espécimenes.
BIBLIOGRAFÍA
CATERINO MS, SPERLING FAH. Papilio Phylogeny Based on Mitochondrial Cytochrome Oxidase I and II Genes. Mol Phylogenet Evol. 1999;11(1):122-137. [ Links ]
CATERINO MS, REED RD, KUO MM, SPERLING FAH. A Partitioned Likelihood Analysis of Swallowtail Butterfly Phylogeny (Lepidoptera: Papilionidae). Syst Biol. 2001;50(1):106-127. [ Links ]
CLARY DO, WOLSTENHOLME DR. The mitochondrial-DNA molecule of Drosophila yakuba nucleotide-sequence, gene organization, and genetic-code. J Mol Evol. 1985;22(3):252-271. [ Links ]
DEVRIES PHJ. The butterflies of Costa Rica and their natural history. Papilionidae, Pieridae, Nymphalidae. Princeton University Press. Princeton; 1987. p. 456. [ Links ]
FAGUA G. El género Parides Hübner, 1819 (Lepidoptera:Papilionidae) En Colombia. Tesis de Maestría en Biología Sistemática. Facultad de Ciencias. Universidad Nacional de Colombia. Informe final presentado a la Fundación para la Promoción de la Investigación y la Tecnología. Banco de la República. Bogotá; 1997. p. 146. [ Links ]
FELSENSTEIN J. Inferring phylogenies. Sinauer Associates, Inc. Sunderland, Massachusetts; 2004. p. 580. [ Links ]
FOX EM. On subspecies. Syst Zool. 1955;4(2):93-95. [ Links ]
FROST D, KLUGE AG, HILLIS DM. Species in contemporary herpetology: comments on phylogenetic inference and taxonomy. Herpetol Rev. 1992;23:46-54. [ Links ]
GOLOBOFF P, FARRIS J, NIXON K. TNT: a free program for phylogenetic analysis. Cladistics. 2008;24(5):774-786. [ Links ]
HALL T. BIOEDIT. Biological sequence alignment editor [programa de ordenador]. version 7.0.5. Computer program; 2005. [ Links ]
HANCOCK DL. Classification of the Papilionidae: a phylogenetic approach. Smitheria. 1983;2(1):1-48. [ Links ]
HARRINSON RG. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol Evol. 1989;4(1):6-11. [ Links ]
HILLIS DM. Inferring complex phylogenies. Nature. 1996;383(6596):130-131. [ Links ]
HILLIS DM. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst Biol. 1998;47(1):3-8. [ Links ]
LE CROM JF, CONSTANTINO LM, SALAZAR JA. Mariposas de Colombia. Tomo I: Papilionidae. Carlec Ltda, Bogotá; 2002. p. 108. [ Links ]
LUMLEY LM, SPERLING FAH. Integrating morphology and mitochondrial DNA for species delimitation within the spruce budworm (Choristoneura fumiferana) cryptic species complex (Lepidoptera: Tortricidae). Syst Entomol. 2010;35(3):416-428. [ Links ]
LUNT DH, ZHANG D-X, SZYMURA, HEWITT GM. The insect cytochrome oxidase I gene: Evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol. 1996;5(3):153-165. [ Links ]
MAYR E, ASHLOCK, PD. Principles of systematic biology. New York, USA: McGraw-Hill; 1991. p. 475. [ Links ]
MILLER JS. Phylogenetic studies in the Papilionidae (Lepidoptera: Papilionidae). Bull Am Mus Nat Hist. 1987;186(4):365-512. [ Links ]
MITCHELL SE, COCKBURN AF, SEAWRIGHT JA. The mitochondrial genome of Anopheles quadrimaculatus species A: Complete nucleotide sequence and gene organization. Genome. 1993;36(6):1058-1073. [ Links ]
MONTEIRO A, PIERCE N. Phylogeny of Bycilus (Lepidoptera: Nymphalidae) inferred from COI, COII, and EF-1a gene sequences. Mol Phylogenet Evol. 2001;18(2):264-281. [ Links ]
MORITZ C, DOWLING TE, BROWN WM. Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Evol Syst. 1987;18(1):269-292. [ Links ]
MUNROE E. The classification of the Papilionidae (Lepidoptera). Can Entomol. Supplement. 1961;17(1):1-51. [ Links ]
NIXON KC. Winclada (BETA). [programa de ordenador] version 0.9.9. Published by the author. Ithaca, NY. 1999. [ Links ]
NIXON KC. WinClada. [programa de ordenador]. ver. 1.0000 Published by the author. Ithaca, NY. 2002. [ Links ]
NIXON KC, WHEELER QD. An amplification of the phylogenetic species concept. Cladistics. 1990;6(3):211-223. [ Links ]
PATTEN MA, UNITT P. Diagnosability versus Mean Differences of Sage Sparrow Subspecies. Auk. 2002;119(1):26-35. [ Links ]
PASCUAL MJ, BALANYÁ A, TORRE LA, SERRA L. Analysis of the variability of Drosophila azteca and Drosophila thabasca populations revealed by random amplified polymorphic DNA. J Zoolog Syst Evol Res. 1997;35(4):159-164. [ Links ]
POSADA D, CRANDALL K. MODELTEST: Testing the model of DNA substitution. Bioinformatics. 1998;14(9):817-818. [ Links ]
RONQUIST F., HUELSENBECK JP, VAN DER MARK P. MrBayes [programa de ordenador]. version 3.1. Computer program. 2005. [ Links ]
ROTHSCHILD W, JORDAN K. A revision of the American Papilios. Novitates Zoology. 1906;13: 412-752. [ Links ]
SANDERSON MJ, DRISKELL AC, REE RH, EULENSTEIN O, LANGLEY S. Obtaining maximal concatenated phylogenetic data sets from large sequence databases. Mol Biol Evol. 2003;20(7):1036-1042. [ Links ]
SILVA-BRANDÃO KL, FREITAS AVL, BROWER AVZ, SOLFERINI VN. Phylogenetic relationships of the New World Troidini swallowtails (Lepidoptera: Papilionidae) based on COI, COII, and EF-1a genes. Mol Phylogenet Evol. 2005;36(3):468-483. [ Links ]
SPERLING FAH, HARRISON RG. Mitochondrial DNA variation within and between species of the Papilio machaon group of of swallowtail butterflies. Evolution. 1994;48 (2):408-422. [ Links ]
SWOFFORD DL. PAUP: Phylogenetic analysis using parsimony [programa de ordenador]. version 4.0. MODELTEST, version 3.06. Computer program distributed by Sinauer, Sunderland, Massachusetts; 1999. [ Links ]
TYLER HA, BROWN Jr KS, WILSON KH. Swallowtail butterflies of the Americas. Scientific Publishers, Inc. Gainesville; 1994. p. 376. [ Links ]
WAHLBERG N, WHEAT CW. Genomic outposts serve the phylogenomic pioneers: designing novel nuclear markers for genomic DNA extractions of Lepidoptera. Syst Biol. 2008;57(2):231-242. [ Links ]
WHEELER WC. Optimization alignment: The end of multiple sequence alignment in phylogenetics?. Cladistics. 1996;12(1):1-9. [ Links ]
WHEELER W, GLADSTEIN D, DE LAET J. POY: Phylogeny Reconstruction via Optimization of DNA [programa de ordenador]. Version 3.0.11. Computer program. 2003. [ Links ]
