










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.18 no.1 Bogotá Jan./Apr. 2013
IDENTIFICACIÓN DE ELEMENTOS cisREGULATORIOS Y PREDICCIÓN BIOINFORMÁTICA DE FACTORES DE TRANSCRIPCIÓN INVOLUCRADOS EN LA REGULACIÓN DE miARNs EN PLANTAS
Identification of cisregulatory Elements and Bioinformatic Prediction of Transcriptional Factors Involved in Regulation of miRNAs in Plants
ÁLVARO PÉREZ-QUINTERO1,2*, Biólogo, M.Sc. (c); CAMILO LÓPEZ1, Ph. D.
1 Grupo Manihot biotec, Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia. 2 Horticulture & Crop Science Dpt, Ohio State University, Columbus, OH, USA. Autor de correspondencia: Camilo López, celopezc@unal.edu.co . Universidad Nacional de Colombia, Departamento de Biología, calle 45 con carrera 30. Oficina 222. Bogotá D. C., Colombia.
Presentado el 24 de enero de 2013, aceptado el 27 de febrero de 2013, correcciones el 4 de marzo de 2013.
RESUMEN
Los microARNs (miARNs) son pequeños ARN no codificantes que juegan un papel importante en el control de la expresión génica a través de la degradación de ARNm complementarios a su secuencia. La expresión de los miARNs es dependiente de la ARN polimerasa II como la mayoría de genes que codifican para proteínas. La regulación de la expresión de miARNs está bajo el control coordinado y combinatorio de factores de transcripción (FT). En este trabajo, se realizó una aproximación bioinformática para identificar sitios de unión de FT, TFBS (del inglés Transcription Factors Binding Sites) en regiones promotoras de genes miRNAs en 17 especies vegetales y se analizó el papel de algunos FT en defensa contra bacterias. Se encontró que nueve de las plantas analizadas presentaban diferencias significativas entre la distribución de TFBS presentes en los promotores de los miRNAs cuando se compara con los presentes en los genes codificantes de proteínas. En varios de los promotores de los miRNAs de yuca que son inducidos en respuesta a la infección por la bacteria Xanthomonas axonopodis pv. manihotis se identificaron elementos de unión como CCA1, T-box y SORLREP3, los cuales se presentan también en los genes que codifican proteínas implicadas en respuestas al ciclo circadiano y a la luz, sugiriendo que estos procesos y las respuestas inmunes en plantas pueden ser coordinados. En conjunto este trabajo aporta luces sobre los posibles mecanismos transcripcionales del control de la expresión génica de los miARNs.
Palabras clave: expresión génica, factores de transcripción, microARN, promotor.
ABSTRACT
MicroRNAs (miRNAs) are a group of small non coding RNAs involved in the control of gene expression through the degradation of mRNAs in a sequence specific manner. miRNAs expression is dependent on RNA polymerase II as most of the coding protein genes. The regulation of miRNAs expression is under the coordinated and combinatorial control of transcription factors (TFs). A bionformatic approach was carried out to identify transcription factor binding sites (TFBS) in the promoter of miRNAs genes in 17 different plant species and the possible involvement of TF in antibacterial response was analyzed. In nine of the plants studied significant differences in TFBS distribution in the promoter of miRNAs were observed when compare to the promoter of protein coding genes. TFBS as CCA1, T-box y SORLREP3 were present on the promoters of the cassava miRNAs induced in response to the infection by the bacteria Xanthomonas axonopodis pv. manihotis. These TFBS are also present in the promoter of genes coding for proteins involved in circadian rhythm and light responses, suggesting a crosstalk between these process and immune plant responses. Taken together, the results here described give insight about the transcriptional mechanisms involved in the expression of miRNAs.
Keywords: gene expression, miRNA, promoter, transcription factor.
INTRODUCCIÓN
En eucariotas la regulación transcripcional está controlada fundamentalmente por el reclutamiento de factores de transcripción (FTs) a elementos reguladores en la secuencia promotora de los genes. Los FTs interactúan con elementos específicos del ADN, con otros FTs y con la maquinaria de transcripción basal para regular la expresión de los genes a los que se unen. En plantas existen más de 1500 FTs, cada uno controlando la expresión de decenas o miles de genes en complejas redes de señalización. Los elementos cis o sitios de unión de factores de transcripción, TFBS (del inglés Transcription Factor Binding Sites) son elementos de ADN funcional que influyen la actividad transcripcional (Riechmann et al., 2000; Priest et al., 2009). En algunas ocasiones múltiples TFBS constituyen un módulo de regulación cis que integra las señales de múltiples FTs, resultando en control combinatorio y en patrones altamente específicos de expresión de genes. La conservación de estos elementos de ADN en distintas plantas hace relativamente fácil su identificación y la inferencia de patrones de regulación de genes a partir de la presencia o combinación de estos (Riechmann et al., 2000; Priest et al., 2009).
Los microARNs (miARNs) son un grupo particular de moléculas de ARN implicadas en el control de la expresión génica. Los miARNs son moléculas de aproximadamente 21 nt que se originan a partir de genes nucleares. Un gen nuclear miARN es primero transcrito por la ARN polimerasa II en un miARN primario (primi-ARN), el cual es procesado a un ARN intermediario llamado precursor de miARN (premi-ARN) por la proteína Dicer-like 1 (DCL1) en plantas y Drosha en animales (Bartel, 2004; Tang et al., 2008; Zhu, 2008). Los premi-ARNs tienen una estructura secundaria característica y estable, con una alta y negativa energía libre de plegamiento (Meyers et al., 2008). Los premi-ARNs son procesados a un dúplex miARN:miARN* por DCL1 en plantas y Dicer en animales. El dúplex miARN/miARN* o el miARN de cadena sencilla es exportado al citoplasma donde solo la hebra activa o miARN maduro es incorporada al complejo de silenciamiento inducido por ARN, RISC (del inglés RNA Induced Silencing Complex), mientras que la hebra pasajera (miARN*) es degradada. El complejo RISC, al cual se asocia la proteína ARGONAUTE1 (AGO1), es el encargado de guiar el silenciamiento de ARNs mensajeros complementarios al miARN maduro conocidos como targets o blancos (Bartel, 2004; Tang et al., 2008; Zhu, 2008). Los miARNs normalmente se encuentran representados por varios loci en el genoma y son funcionalmente redundantes. Los miARNs se agrupan en familias que incluyen moléculas con secuencias de más de un 90 % de identidad (Meyers et al., 2008).
Existen algunas diferencias en la biogénesis y los mecanismos de acción de miARNs en plantas y animales, notablemente los miARNs de plantas requieren una casi perfecta complementariedad con los ARNms para producir silenciamiento, mientras que en animales la complementariedad con la región 5' del miARN es suficiente para producir silenciamiento (Bartel, 2004). Otra diferencia importante consiste en que mientras que los miARNs de animales comúnmente se originan a partir de regiones intrónicas u otras regiones no traducidas de ARNs codificantes, los genes miARN en plantas son se encuentran comúnmente en regiones intergénicas y son transcritos independientemente (Bartel, 2004). Al ser transcritos por la ARN polimerasa II al igual que la mayoría de los genes codificantes de proteínas, los genes miARNs están sometidos al control por factores de transcripción. En la mayoría de los genes de miARNs analizados se han encontrado elementos comunes de promotores tipo Pol-II como la caja TATA en proporciones similares a genes codificantes (Zhou et al., 2007). Se sabe además que la región promotora de los genes miARNs se encuentra comúnmente a menos de 1000 pb curso arriba del inicio del premi-ARN, especialmente en el caso de miARNs de regiones intergénicas, no policistrónicas (Megraw et al., 2006; Zhou et al., 2007; Cui et al., 2009). Esto es de vital importancia para la predicción de TFBS puesto que en muchos casos el sitio de inicio de la transcripción (TSS) no está caracterizado para miARNs.
Varios trabajos han caracterizado TFBS en genes miARNs en diferentes plantas. Se ha encontrado que algunos TFBS parecen estar sobrerepresentados en genes miARNs con respecto a genes codificantes para proteínas, por ejemplo los motivos AtMYC2, ARF, SORLREP3 y LFY así como la caja TATA fueron encontrados sobrerepresentados en genes miARNs en Arabidopsis (Megraw et al., 2006). Se han encontrado también diferencias en frecuencias de TFBS entre distintas especies de plantas (Zhou et al., 2007) y entre miARNs conservados y especieespecíficos (Cui et al., 2009). De igual manera se han encontrado TFBS sobrerepresentados en grupos de miARNs involucrados en varios procesos incluyendo respuestas a estrés oxidativo (Petrov et al., 2012), UVB (Zhou et al., 2007; Jia et al., 2009), salinidad (Ding et al., 2009), sequía (Zhou et al., 2010), baja temperatura (Zhou et al., 2008), así como en procesos de organogénesis y desarrollo (Joung y Fei, 2009).
Estos trabajos demuestran la importancia del estudio de los procesos de regulación transcripcional de genes miARNs para entender su papel en procesos fisiológicos. Adicionalmente, el hecho de que la mayoría de los targets de los miARNs son ARNm que codifican para FTs (Croft et al., 2012) revela la importancia del estudio del control de la expresión génica de los miARNs y pone en evidencia la presencia de un mecanismo de regulación recíproca entre FT y miARNs, el cual hace parte crucial de las redes de re gulación génica en plantas (Mejia-Guerra et al., 2012). En este trabajo se buscó comprender las diferencias de regulación de genes miARNs ha ciendo un análisis exhaustivo de las frecuencias de TFBS en distintas plantas. Así mismo, se buscó identificar TFBS determinantes para la regulación transcripcional de miARNs en respuesta a bacterias para el caso particular del patosistema yuca-Xanthomonas axonopodis pv. manihotis (Xam), encontrando que factores de transcripción involucrados en control de desarrollo floral y en el control del ritmo circadiano pueden estar regu lando la respuesta a bacterias mediada por miARNs.
MATERIALES Y MÉTODOS
PREDICCIÓN DE TFBS
Para llevar a cabo la identificación de TFBS, se extrajeron inicialmente las regiones promotoras de genes miARNs y genes codificantes para proteínas en distintas plantas. Para esto se extrajeron las secuencias de los premi-ARNs de las plantas Arabidopsis lyrata (aly), Arabidopsis thaliana (ath), Brachypodium distachyon (bdi), Chlamydomonas reinhardtii (cre), Citrus sinensis (csi), Glycine max (gma), Malus domestica (mdm), Manihot esculenta (mes), Medicago truncatula (mtr), Oryza sativa (osa), Physcomitrella patens (ppt), Populus trichocarpa (ptc), Ricinus communis (rco), Sorghum bicolor (sbi), Selaginella moellendorffii (smo), Vitis vinifera (vvi) y Zea mays (zma). Los premi-ARNs se mapearon contra los genomas respectivos disponibles en phytozome (http://www.phytozome.net/ v. 8.0), utilizando Blastn, (parametros: e-value <0.0001, ungapped, word size = 4, -max target seqs 1best hit score edge 0.05 -best hit overhang 0.25). Se extrajo la región de mil bases curso arriba del inicio (5') del premi-ARN utilizando fastacmd, se ha encontrado que esta región contiene normalmente la región promotora del gen miARN en plantas (Megraw et al., 2006; Zhou et al., 2007; Cui et al., 2009). Se conservaron solo las secuencias de promotores de más de 500 nt de longitud puesto que en algunos genomas esta secuencia se encontraba incompleta. Las regiones promotoras de los genes anotados se extrajeron utilizando la herramienta biomart de phytozome (http://www.phytozome.net). Como control negativo las secuencias de promotores de genes miARN y de genes codificantes fueron aleatorizadas utilizando shuffleseq de la suite EMBOSS (Rice et al., 2000).
Para la identificación de los TFBS se utilizaron matrices de peso posicional (PMWs, del inglés Positional Weight Matrices) y los scripts del programa PlantTFBS descrito en Megraw y Hatzigeorgiou (2010). Las secuencias correspondientes a PMWs de los 99 FTs fueron confirmadas en la última versión de Agris (Yilmaz et al., 2011). PlantTFBS utiliza una función de log-likelihood para asignar puntajes a los posibles sitios de unión. Para esto primero se calcula la composición de nucleótidos del set de secuencias a escanear, se genera un set de secuencias posibles de unión para cada TFBS y se calcula el menor puntaje posible para un sitio de unión aceptado. La función del puntaje calcula la tasa de probabilidad de que una base se encuentre en una posición de un sitio de unión de acuerdo a la PMW sobre la probabilidad de esta base dada la distribución de nucleótidos, se hace la sumatoria (normalizada por longitud) de estas probabilidades y se reportan los sitios de unión con un puntaje mayor al mínimo puntaje posible para cada PMW (Megraw y Hatzigeorgiou, 2010). En la búsqueda se asignó un número de pseudocounts (probabilidad base para cualquier nucleótido en cualquier posición) de 0,02.
Una representación de la metodología utilizada para la identificación de TFBS se muestra en la figura suplementaria 1.

ANÁLISIS ESTADÍSTICO
Las frecuencias de TFBS para cada planta y para cada grupo de genes se calculó como el porcentaje de genes que contenían al menos un sitio de unión identificado para el FT respectivo. Las pruebas de diferencias en distribución de frecuencias se realizaron mediante la prueba Kolmogorov-Smirnov. Esta prueba es ideal para detectar diferencias entre distribuciones empíricas y ha sido utilizada en análisis similares de regiones promotoras (Zhang et al., 2006). Las diferencias en frecuencias de TFBS particulares entre grupos se analizaron mediante pruebas de Fisher, las cuales han sido extensivamente utilizadas para la identificación de elementos sobrerepresentados en regiones promotoras (Frith et al., 2004; Ho et al., 2004; Spangler et al., 2012). Para calcular distancias entre plantas basadas en la distribución de frecuencias se calculó una matriz de correlación entre las distribuciones utilizando el método de Pearson, esta matriz se utilizó para calcular distancias euclidianas y realizar un cluster jerárquico con hclust. Todos los análisis se llevaron a cabo con paquetes de Bioconductor en R (Reimers y Carey, 2006).
RESULTADOS
IDENTIFICACIÓN DE SITIOS DE UNIÓN DE FACTORES DE TRANSCRIPCIÓN (TFBS) EN PROMOTORES DE GENES miARNs EN PLANTAS
Con el fin de obtener una perspectiva amplia de los posibles mecanismos de regulación asociados a miARNs, se analizaron las regiones promotoras de los premi-ARNs (1000 nucleótidos curso arriba del extremo 5' del premi-ARN) identificados en 17 especies de plantas para las que se cuenta con un genoma secuenciado y al menos 50 premi-ARNs depositados en miRBase (Griffith-Jones et al., 2008). Adicionalmente, se analizaron también las regiones promotoras de genes codificantes de proteínas en estas 17 especies. Empleando el programa PlantTFBS (Megraw et al., 2010) se escanearon 3812 regiones promotoras correspondientes a miARNs y 596701 correspondientes a genes codificantes de proteínas. Se escanearon 99 TFBS, utilizando las matrices de peso posicional descritas en Megraw y Hatzigeorgiou (2010), las cuales están basadas principalmente en datos de Arabidopsis obtenidos de las bases de datos AGRIS (Yilmaz et al., 2011), PlantCare (Lescot et al., 2002) y AtProbe (http://exon.cshl.org/cgibin/atprobe/atprobe.pl). Como control y con el objetivo de confirmar que los sitios de unión identificados por el programa no se deban simplemente al azar se generaron grupos de secuencias de miARNs y genes codificantes aleatorias, manteniendo la distribución de nucleótidos de las secuencias originales. Al comparar la distribución de las frecuencias relativas entre los de sitios de unión para cada FT entre promotores de miARNs aleatorizados y no aleatorizados se encontró una diferencia estadísticamente significativa (KolmogorovSmirnov p - valor = 6,44-5). Un resultado similar se obtuvo al comparar promotores de genes codificantes aleatorizados y no aleatorizados (Kolomogorov-Smirnov p - valor = 1,56-10), indicando que efectivamente los TFBS identificados no son producto del azar. Sin embargo, algunos TFBS se encontraron con frecuencias muy altas (> 50 %) en las secuencias aleatorizadas. Estos corresponden principalmente a sitios de unión cortos (5 - 8 nt) o de baja complejidad como las cajas TATA, GATA y CAAT.
DIFERENCIAS EN FRECUENCIAS DE TFBS ENTRE TIPOS DE GENES Y ENTRE PLANTAS
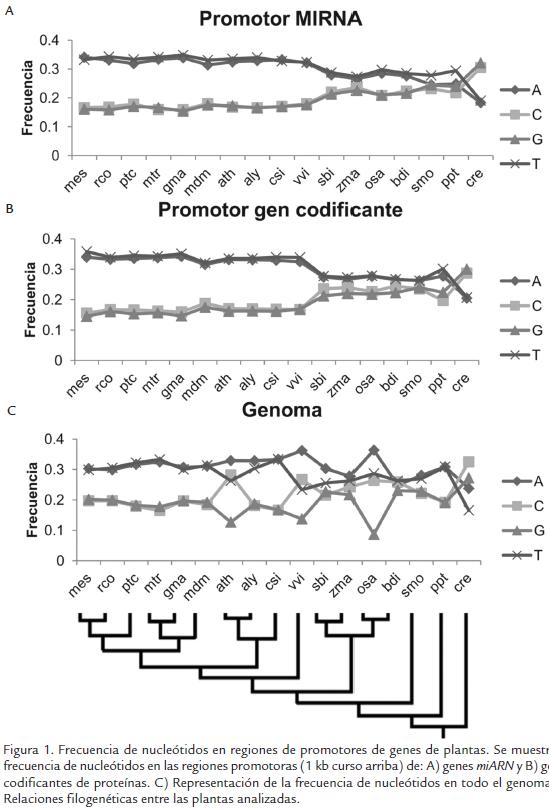
A partir de los resultados obtenidos, se procedió a comparar los TFBS presentes en los promotores de miARNs con los de los promotores en los genes codificantes. Sin embargo, antes de realizar esta comparación y con el objetivo de verificar que las diferencias encontradas no estuvieran determinadas por la distribución de nucleótidos en los promotores, para cada especie se comparó la composición de nucleótidos tanto de la región de los promotores de miARNs, la de genes codificantes y la de todo el genoma (Fig. 1). Para ninguna de las especies se encontraron diferencias significativas en la distribución de nucleótidos entre los promotores de miARNs y los promotores de genes (Kolmogorov-Smirnov p-valor > 0,05). En muchos casos la distribución de nucleótidos en promotores reflejaba la distribución de nucleótidos global de los genomas (Fig. 1). Así mismo, se pudo observar que la distribución de nucleótidos variaba ostensiblemente entre plantas, pero un patrón similar se observó para plantas cercanas filogenéticamente (Fig. 1). A partir de estos resultados se puede esperar que, de existir diferencias en TFBS de los promotores de miARNs y genes codificantes, estas se deban posiblemente a procesos de selección y no a diferencias en distribución de nucleótidos. Así mismo, estos resultados sugieren que es posible encontrar similitudes en TFBS entre plantas filogenéticamente cercanas.
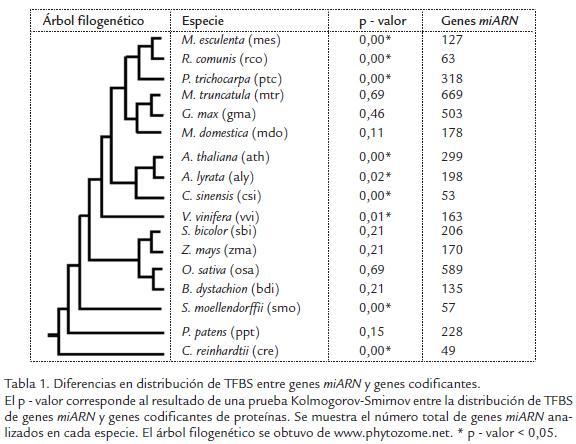
La distribución de frecuencias de TFBS entre promotores de miARNs y de genes fue diferente para nueve de las 17 plantas evaluadas (Tabla 1). Notablemente no se encontraron diferencias en la distribución de frecuencias de TFBS entre promotores de miARNs y genes codificantes para dos grupos de plantas: gramíneas (B. dystachyon, O. sativa, S. bicolor, Z. mays) y plantas fijadoras de nitrógeno (G. max, M. domestica, M. truncatula) (Tabla 1). Es posible que estos resultados estén influenciados por el número de genes miARNs reportados y depositados en la base de datos, dado que entre las plantas en las que no se presentaron diferencias significativas se encontraban cinco con el mayor número de miARNs (> 200 genes) descritos.
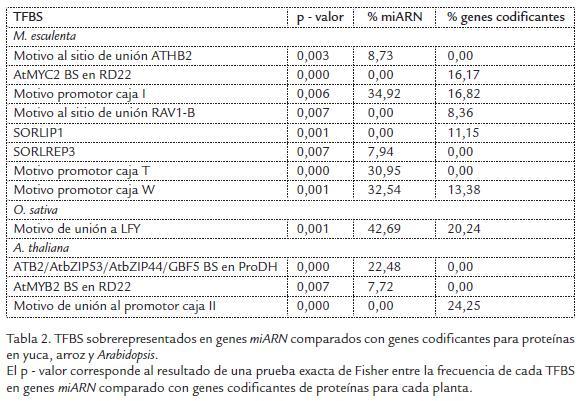
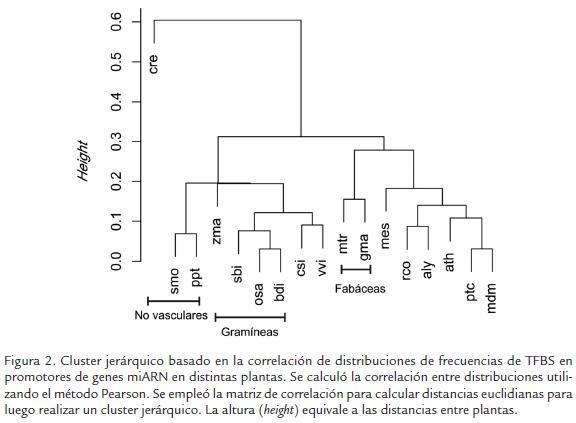
Con el fin de identificar TFBS sobrerepresentados en los promotores de los miARNs con respecto a los presentes en los de los genes codificantes se realizó una comparación en tres plantas: yuca, arroz y Arabidopsis. En yuca se encontraron cinco TFBS sobrerepresentados en promotores de miARNs, en Arabidopsis dos y en arroz uno (Tabla 2). En ninguno de los casos se encontró un FT común entre las tres plantas, sugiriendo regulación especie-especifica de miARNs. Para determinar si las diferencias entre TFBS presentes en los promotores de miARNs de distintas plantas presentaban alguna relación con la filogenia de ellas, se utilizaron las frecuencias de TFBS de cada planta para calcular una matriz de correlación a partir de la cual se construyó una matriz de distancias y se realizó un clúster jerárquico (Fig. 2). A partir de este análisis se pudo observar que las frecuencias de TFBS reflejan parcialmente relaciones filogenéticas. Así por ejemplo, algunas especies pertenecientes a las mismas familias (Gramineas y Fabaceas) se agrupan en este clúster, lo cual sugiere que pueden existir patrones de regulación transcripcional similares entre plantas filogenéticamente cercanas.
TFBS SOBREREPRESENTADOS EN FAMILIAS DE MIARNS INVOLUCRADAS EN RESPUESTA A BACTERIAS EN YUCA
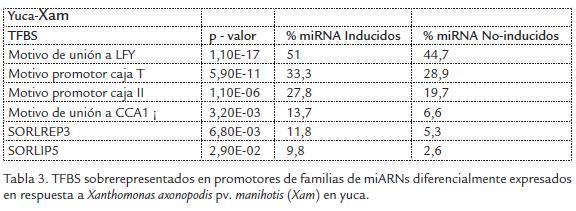
En un trabajo previo, se identificaron miARNs en yuca involucrados en respuestas a la bacteria Xanthomonas axonopodis pv. manihotis (Xam) (Pérez-Quintero et al., 2012). Con el fin de identificar TFBS relacionados con esta respuesta se realizó una prueba de Fisher entre las frecuencias de los distintos TFBS en promotores de genes miARNs de familias cuyo perfil de expresión fue modificado en respuesta a la bacteria (inducidos o reprimidos) y se comparó con las frecuencias de presencia de estos sitios en las familias de miARNs cuyo perfil de expresión no fue modificado. Se encontraron seis TFBS sobrerepresentados en las familias de miARNs inducidas y ninguno en las familias reprimidas (Tabla 3).
DISCUSIÓN
En este trabajo se analizó la presencia de TFBS en regiones promotoras de genes miARNs. Se encontró que en muchas plantas no existen diferencias significativas en la distribución de TFBS en genes miARNs comparado con genes codificantes para pro teínas, especialmente en plantas para las que un gran número de miARNs ha sido reportado (por ejemplo arroz y Medicago). Curiosamente, en este trabajo no se identificaron los mismos TFBS de Arabidopsis que se encontraron sobrerepresentados en genes miARNs respecto a genes codificantes obtenido en un trabajo previo que empleó una estrategia similar (Megraw et al., 2006). Una posible razón para explicar estas diferencias es considerando el hecho de que actualmente existen muchos más genes miARNs identificados en Arabidopsis (299 genes) respecto a los utilizados en el estudio anterior (59 genes) (Megraw et al., 2006). De esta manera se resalta la importancia del número de loci estudiados para estos estudios. Desafortunadamente se desconoce si la identificación de estos genes en algunas plantas ha alcanzado el punto de saturación.
Por otro lado se encontró que tanto la distribución de nucleótidos y la distribución de TFBS en regiones promotoras de genes miARNs es muy variable, siendo más similar entre plantas filogenéticamente relacionadas. Esto concuerda con patrones de regulación de miARNs especie-específicos comúnmente encontrados al comparar por ejemplo arroz y Arabidopsis (Zhou et al., 2007) y con la multiplicidad de funciones observadas para algunas familias de miARNs en distintas plantas (Lopez y Pérez-Quintero, 2012; Kruszka et al., 2012). Así mismo, se resalta la importancia de estudiar plantas de distintos grupos taxonómicos para trabajos como estos en los que se busca identificar la función de miARNs en distintos procesos celulares.
Entre los TFBS sobrerepresentados en yuca se encontró el de unión para factores de transcripción LFY, el cual está involucrado principalmente en procesos de desarrollo floral mediante la activación de genes tipo AGAMOUS (AG), APETALA (1, 2 y 3) (Moyroud et al., 2011). Este motivo fue abundante en miembros de la familia 172, la cual es inducida en respuesta a Xam y tiene entre sus targets principalmente genes tipo AP2 involucrados en desarrollo floral (Frazier et al., 2011; Jung et al., 2011; Zhu et al., 2011). Esto sugiere la presencia de un bucle de regulación entre factores de transcripción y miARNs en desarrollo floral. Es posible que en respuesta a bacterias se favorezca la acción de los miARNs para frenar el desarrollo floral.
Este estudio también permitió identificar una sobrerepresentación de elementos relacionados con el supresión del ciclo circadiano y las respuestas a luz en miARNs involucrados en respuesta a Xam: CCA1, T-box y SORLREP3 (Tabla 3) (Wang y Tobin, 1998; Hudson y Quail, 2003). Previos estudios han mostrado que existe una estrecha conexión entre procesos de defensa, ritmos circadianos y respuesta a luz (Hudson y Quail, 2003). Por ejemplo, los procesos normalmente controlados por el reloj circadiano como la apertura de estomas o por la luz como la síntesis de ácido salicílico son modificados durante los procesos de defensa (Hudson y Quail, 2003). No es sorprendente, por tanto, que la inducción de miARNs pueda estar regulada por factores similares (Roden y Ingle, 2009). Por otro lado, se esperaba encontrar sobrerepresentación de motivos de unión de factores de respuesta auxinas (ARF) puesto que los ARF son directamente regulados por miARNs como parte de las respuestas de defensa contra bacterias (Navarro et al., 2006; Pérez-Quintero et al., 2012) y se sabe que las familias involucradas en señalización de auxinas normalmente participan en bucles regulatorios en los que los miARNs son a la vez regulados por sus targets (Megraw et al., 2006; Meng et al., 2009). Los motivos de unión para ARF fueron abundantes únicamente en la familia miR160 y no en otras familias inducidas. Esta familia efectivamente está implicada en la regulación de señalización de auxinas (Pérez-Quintero et al., 2012), de modo que se puede decir que existe un bucle de regulación entre miRNAs y ARF en yuca, al menos en el caso de la familia miR160.
Aunque estos resultados aportan algunas luces sobre los procesos de transcripción de miARNs, la aproximación utilizada tiende a simplificar el efecto de los factores de transcripción dado que no considera patrones complejos de interacción entre distintos factores o la posibilidad de transcripción no independiente en miARNs policistrónicos o miARNs asociados a genes. De igual manera, en este trabajo asume que los sitios de unión y los factores de transcripción analizados son conservados entre plantas, porque los modelos utilizados provienen principalmente de Arabidopsis, lo cual puede no ser cierto. Estas consideraciones aun no se pueden incorporar a un análisis masivo de regulación entre distintas especies principalmente por ausencia de suficiente información para modelarlos adecuadamente en otras especies de plantas.
AGRADECIMIENTOS
Los autores agradecen a los integrantes del grupo Manihot biotec por sus discusiones que enriquecieron la elaboración del presente trabajo.
BIBLIOGRAFÍA
BARTEL DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281-297. [ Links ]
CROFT L, SZKLARCZYK D, JENSEN LJ, GORODKIN J. Multiple independent analyses reveal only transcription factors as an enriched functional class associated with microRNAs. BMC Syst Biol. 2012;6:90. [ Links ]
CUI X, XU SM, MU DS, YANG ZM. Genomic analysis of rice microRNA promoters and clusters. Gene. 2009;431(1-2):61-66. [ Links ]
FRAZIER TP, SUN G, BURKLEW CE, ZHANG B. Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol Biotechnol. 2011;49(2):159-165. [ Links ]
FRITH MC, FU Y, YU L, CHEN JF, HANSEN U, WENG Z. Detection of functional DNA motifs via statistical overrepresentation. Nucleic Acids Res. 2004;32(4):1372-1381. [ Links ]
GRIFFITHS-JONES S, SAINI HK, VAN DONGEN S, ENRIGHT AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154-158. [ Links ]
HO SUI SJ, MORTIMER JR, ARENILLAS DJ, BRUMM J, WALSH CJ, KENNEDY BP, WASSERMAN WW. oPOSSUM: identification of overrepresented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33(10):3154-3164. [ Links ]
HUDSON ME, QUAIL PH. Identification of promoter motifs involved in the network of phytochrome A-regulated gene expression by combined analysis of genomic sequence and microarray data. Plant Physiol. 2003;133(4):1605-1616. [ Links ]
JIA X, REN L, CHEN QJ, LI R, TANG G. UV-B-responsive microRNAs in Populus tremula. J Plant Physiol. 2009;166(18):2046-2057. [ Links ]
JOUNG JG, FEI Z. Identification of microRNA regulatory modules in Arabidopsis via a probabilistic graphical model. Bioinformatics. 2009;25(3):387-393. [ Links ]
JUNG JH, SEO PJ, KANG SK, PARK CM. miR172 signals are incorporated into the miR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental transitions. Plant Mol Biol. 2011;76(1-2):35-45. [ Links ]
KRUSZKA K, PIECZYNSKI M, WINDELS D, BIELEWICZ D, JARMOLOWSKI A, SZWEYKOWSKA-KULINSKA Z, VAZQUEZ F. Role of microRNAs and other sRNAs of plants in their changing environments. J Plant Physiol. 2012;169(16):1664-1672. [ Links ]
LESCOT M, DEHAIS P, THIJS G, MARCHAL K, MOREAU Y, VAN DE PEER Y, ROUZE P, ROMBAUTS S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325-327. [ Links ]
LÓPEZ C, PÉREZ-QUINTERO Á. The Micromics Revolution: MicroRNA-Mediated Approaches to Develop Stress-Resistant Crops. Improving Crop Resistance to Abiotic Stress. 1 ed. WillerBlackwell; 2012. p. 559-590. [ Links ]
MEGRAW M, BAEV V, RUSINOV V, JENSEN ST, KALANTIDIS K, HATZIGEORGIOU AG. MicroRNA promoter element discovery in Arabidopsis. RNA 2006;12(9):1612-1619. [ Links ]
MEGRAW M, HATZIGEORGIOU AG. MicroRNA promoter analysis. Methods Mol Biol. 2010;592:149-161. [ Links ]
MEJIA-GUERRA MK, POMERANZ M, MOROHASHI K, GROTEWOLD E. From plant gene regulatory grids to network dynamics. Biochim Biophys Acta. 2012;1819(5):454-465. [ Links ]
MENG Y, HUANG F, SHI Q, CAO J, CHEN D, ZHANG J, NI J, WU P, CHEN M. Genome-wide survey of rice microRNAs and microRNA-target pairs in the root of a novel auxin-resistant mutant. Planta. 2009;230(5):883-898. [ Links ]
MEYERS BC, AXTELL MJ, BARTEL B, BARTEL DP, BAULCOMBE D, BOWMAN JL, CAO X, CARRINGTON JC, CHEN X, GREEN PJ, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20(12):3186-3190. [ Links ]
MOYROUD E, MINGUET EG, OTT F, YANT L, POSE D, MONNIAUX M, BLANCHET S, BASTIEN O, THEVENON E, WEIGEL D, et al. Prediction of regulatory interactions from genome sequences using a biophysical model for the Arabidopsis LEAFY transcription factor. Plant Cell. 2011;23(4):1293-1306. [ Links ]
NAVARRO L, DUNOYER P, JAY F, ARNOLD B, DHARMASIRI N, ESTELLE M, VOINNET O, JONES JD. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006;312(5772):436-439. [ Links ]
PÉREZ-QUINTERO Á, QUINTERO A, URREGO O, VANEGAS P, LÓPEZ C. Bioinformatic identification of cassava miRNAs differentially expressed in response to infection by Xanthomonas axonopodis pv. manihotis. BMC Plant Biology. 2012;12:29. [ Links ]
PETROV V, VERMEIRSSEN V, DE CLERCQ I, VAN BREUSEGEM F, MINKOV I, VANDEPOELE K, GECHEV TS. Identification of cisregulatory elements specific for different types of reactive oxygen species in Arabidopsis thaliana. Gene. 2012;499(1):52-60. [ Links ]
PRIEST HD, FILICHKIN SA, MOCKLER TC. Cis-regulatory elements in plant cell signaling. Curr Opin Plant Biol. 2009;12(5):643-649. [ Links ]
REIMERS M, CAREY VJ. Bioconductor: an open source framework for bioinformatics and computational biology. Methods Enzymol. 2006;411:119-134. [ Links ]
RICE P, LONGDEN I, BLEASBY A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276-277. [ Links ]
RIECHMANN JL, HEARD J, MARTIN G, REUBER L, JIANG C, KEDDIE J, ADAM L, PINEDA O, RATCLIFFE OJ, SAMAHA R, et al. Arabidopsis transcription factors: genomewide comparative analysis among eukaryotes. Science. 2000;290(5499):2105-2110. [ Links ]
RODEN LC, INGLE RA. Lights, rhythms, infection: the role of light and the circadian clock in determining the outcome of plant-pathogen interactions. Plant Cell. 2009;21(9):2546-2552. [ Links ]
SPANGLER JB, FICKLIN SP, LUO F, FREELING M, FELTUS FA. Conserved noncoding regulatory signatures in Arabidopsis co-expressed gene modules. PloS One. 2012;7(9):e45041. [ Links ]
TANG G, REINHART BJ, BARTEL DP, ZAMORE PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17(1):49-63. [ Links ]
WANG ZY, TOBIN EM. Constitutive expression of the CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) gene disrupts circadian rhythms and suppresses its own expression. Cell. 1998;93(7):1207-1217. [ Links ]
YILMAZ A, MEJIA-GUERRA MK, KURZ K, LIANG X, WELCH L, GROTEWOLD E. AGRIS: the Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res. 2011;39(Database issue):D1118-1122. [ Links ]
ZHANG B, PAN X, CANNON CH, COBB GP, ANDERSON TA. Conservation and divergence of plant microRNA genes. Plant J. 2006;46(2):243-259. [ Links ]
ZHANG C, XUAN Z, OTTO S, HOVER JR, MCCORKLE SR, MANDEL G, ZHANG MQ. A clustering property of highly-degenerate transcription factor binding sites in the mammalian genome. Nucleic Acids Res. 2006;34(6):2238-2246. [ Links ]
ZHOU X, WANG G, ZHANG W. UVB responsive microRNA genes in Arabidopsis thaliana. Mol Systems Biol. 2007;3:103. [ Links ]
ZHOU X, RUAN J, WANG G, ZHANG W. Characterization and identification of microRNA core promoters in four model species. PLoS Computat Biol. 2007;3(3):e37. [ Links ]
ZHOU X, WANG G, SUTOH K, ZHU JK, ZHANG W. Identification of coldinducible microRNAs in plants by transcriptome analysis. Biochim Biophys Acta. 2008;1779(11):780-788. [ Links ]
ZHOU L, LIU Y, LIUM Z, KONG D, DUAN M, LUO L. Genomewide identification and analysis of droughtresponsive microRNAs in Oryza sativa. J Exp Bot. 2010;61(15):4157-4168. [ Links ]
ZHU JK. Reconstituting plant miRNA biogenesis. Proc Natl Acad Sci USA. 2008;105(29):9851-9852. [ Links ]
ZHU QH, HELLIWELL CA. Regulation of flowering time and floral patterning by miR172. J Exp Bot. 2011;62(2):487-495. [ Links ]
