Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.18 no.2 Bogotá May/Aug. 2013
COMPARACIÓN ENTRE EL POTENCIAL DE LAS REGIONES VARIABLES DEL 16s RDNA PARA LA IDENTIFICACIÓN DE Lactobacillus spp. (LACTOBACILLIACEAE)
COMPARING THE POTENTIAL FOR IDENTIFICATION OF Lactobacillus spp. OF 16s RDNA VARIABLE REGIONS
LAURA N. GONZÁLEZ GARCÍA1 , Microbióloga; MARÍA C. VANEGAS LÓPEZ2 , M.Sc.; DIEGO M. RIAÑO PACHÓN3,4 , Dr. rer. nat.
1Grupo de Biología Computacional y Evolutiva y Laboratorio de Ecología Microbiana y de Alimentos, Departamento de Ciencias Biológicas, Universidad de los Andes, Bogotá, Colombia. ln.gonzalez138@uniandes.edu.co. 2Laboratorio de Ecología Microbiana y de Alimentos, Departamento de Ciencias Biológicas, Universidad de los Andes, Bogotá, Colombia. mvanegas@uniandes.edu.co. 3Grupo de Biología Computacional y Evolutiva, Departamento de Ciencias Biológicas, Universidad de los Andes, Bogotá, Colombia. 4Afiliación actual: Grupo de Biología Computacional e Evolutiva. Laboratório Nacional de Ciência e Tecnologia do Bioetanol, Centro Nacional de Pesquisa em Energia e Materiais, Campinas, Brasil. diego.riano@bioetanol.org.br Autor de correspondencia: Diego M. Riaño Pachón, diego.riano@bioetanol.org.br.
Presentado el 3 de diciembre de 2012, aceptado el 28 de junio de 2013, correcciones el 16 de julio de 2013.
RESUMEN
El 16s rDNA es utilizado para la identificación bacteriana dada su tasa de variación entre especies. Algunas de las regiones variables de la subunidad ribosomal son más informativas que otras por lo cual en este estudio se evalúa el potencial de identificación aportado por cada región y combinaciones entre ellas. Se extrajeron las regiones variables V1 a la V8 del 16s rDNA de diferentes cepas y especies de Lactobacillus y se analizaron mediante los paquetes de STAP (ss-RNA Taxonomy Assigning Pipeline) y RDP (Ribosomal Database Project) multiclassifier. Adicionalmente se evaluaron árboles filogenéticos de máxima verosimilitud. Nuestros resultados muestran que la mayoría de regiones variables logran dar una correcta clasificación hasta género, sin embargo no son suficientes para clasificar hasta especie usando STAP. La región que presenta el mayor número de amplímeros es V5V6, sin embargo es la que presenta la mayor cantidad de falsos negativos. La que presenta el mayor número de verdaderos positivos es V1V3 (especie) para STAP y V5V8(género) para RDP. Las filogenias evaluadas mostraron que la topología de referencia se puede obtener con diferentes combinaciones de regiones variables e.g., V1V3 y V1V8. El estudio experimental de las cepas contenidas en un tampón comercial mostró que el amplicón V1V8 y el V1V3 dan una misma clasificación correcta. Proponemos la región V1V3 como la región mínima para clasificación correcta de Lactobacillus spp.. En conclusión, la región mínima para clasificar especies del género Lactobacillus es la V1V3, la cual es útil para estudios metagenómicos de muestras de probióticos.
Palabras clave: 16s rDNA, estructura secundaria, inferencia filogenética, Lactobacillus, regiones variables.
ABSTRACT
16s rDNA is used for bacterial identification because its variation rate between species allows differentiation. The gene for this ribosomal subunit has 9 variable regions and some of them give more information than others. We were interested in evaluating the potential for species identification of each region and their combinations. We extracted the V1 to V8 regions of 16s rDNA from different strains and species of Lactobacillus and analyzed them using STAP (ss-RNA Taxonomy Assigning Pipeline) and RDP (Ribosomal Database Project) multiclassifier packages. Phylogenetic trees obtained by maximum likelihood analyses were compared. Classification results show that many regions give the correct genus classification using RDP and STAP, however they are not enough to classify up to the level of species. V5V6 region presents the highest quantity of informative fragments but also present the highest rate of false negatives. V1V3 region presents the highest rate of true positives (species) using STAP and the region V5V8 in RDP (genus).The phylogenetic result shows that the reference topology could be obtained using different combination of regions as V1V3 and V1V8.The experimental validation was done using commercial strains from a probiotic tampon. Sequencing analysis show that the V1V3 region gives the same information and result as the complete 16s rDNA; the three isolated strains correspond to the strains indicated in the product. We conclude that the V1V3 region is the minimum required region to classify Lactobacillus spp. in the correct way and this region is useful in metagenomics to analyze probiotics samples.
Keywords: 16s rDNA, Lactobacillus, phylogenetic inference, secondary structure, variable regions.
INTRODUCCIÓN
Los estudios metagenómicos con técnicas de secuenciación de segunda generación han permitido estudiar la diversidad de organismos en diversos ambientes independientemente de cultivos in vitro, sin embargo, presentan el gran reto de identificar las regiones mínimas que permitan distinguir entre bacterias, dado que las lecturas generadas no cubren el gen completamente (Petrosino et al., 2009). En el cuerpo humano, la diversidad microbiana es muy alta, encontrando Lactobacillus en varios tejidos, los cuales tienen gran valor médico e industrial por su uso como probióticos.
Los probióticos son microorganismos vivos que al ser administrados en una cantidad adecuada confieren un beneficio en la salud del hospedero (Reid et al., 2003). El estudio de la evolución de los probióticos es fundamental para el desarrollo de alimentos funcionales que los contengan dada la variación fenotípica que pueden presentar (Guerzoni, 2010). Los alimentos funcionales fueron descritos en Japón y se consideran aquellos que, además de hacer un aporte nutricional, ejercen efectos beneficiosos sobre otras funciones del organismo, fomentando la salud y/o reduciendo el riesgo de enfermedad (Sarmiento, 2008).
Son utilizados como probióticos la mayoría de cepas de Lactobacillus spp. pertenecientes al phylum Firmicutes, clase Bacilli, orden Lactobacillales (Berger et al., 2007). En el cuerpo humano pueden encontrarse en el intestino, cavidad oral, leche y vagina. En el intestino, superando de diez a veinte veces las células de todos los tejidos del cuerpo (Suau et al., 1999). En el tracto vaginal superan el 70 % de la microbiota actuando como protectoras de la mucosa mediante adherencia al epitelio, producción de agentes antimicrobianos y coagregación con patógenos, se han aislado cepas de Lactobacillus acidophilus, L. crispatus, L. gasseri, L. gallinarum, L. amylivorus, L. johnsonii, L. jensenii y L. iners (Anukam et al., 2005; Martin et al., 2008). En la leche pueden encontrarse confiriendo propiedades antiinfecciosas a neonatos y niños lactantes menores de un año, de las cuales han sido aisladas cepas de L. gasseri, L. rhamnosus, L. plantarumy, L. fermentum (Olivares et al., 2006; Martin et al., 2007).
Sin embargo, a pesar de la alta frecuencia de Lactobacillus spp., en humanos, las técnicas clásicas de detección no son suficientes para establecer la biodiversidad total en el cuerpo humano, esto se debe a que algunas especies no se pueden cultivar en el laboratorio y en aquellas que son cultivables, las técnicas bioquímicas no llegan a distinguir cepas de una misma especie, por lo cual se han desarrollado técnicas moleculares para su identificación y/o clasificación (Sarmiento, 2008). Se han realizado estudios filogenéticos comparando diferentes especies comerciales utilizando genes como 16s rDNA, groEL, rpoA, que codifican para una subunidad ribosomal, una chaperona y subunidad de la RNA polimerasa, respectivamente, con lo cual se ha comprobado que este es un grupo monofilético. También se han genotipado lactobacilos por análisis de ERIC (Secuencias repetitivas consenso de enterobacterias, por sus siglas en inglés) dado que cada una presenta una marca de repeticiones similar a los STR en humanos, i.e., altamente discriminantes (Berger et al., 2007). Además, se han hecho estudios utilizando la técnica MLST (Multilocus Sequence Typing), la cual consiste en secuenciar regiones de genes house-keeping de aproximadamente 450 pb asignando números arbitrarios para identificar cada secuencia única, de este modo se determina un perfil alélico para cada cepa con el cual se pueden establecer dendogramas (Maiden et al., 1998); así se han logrado diferenciar cepas de L. casei con base en los genes fusA, ileS, lepA, leuS, pyrG y recG (Diancourt et al., 2007) y L. paraplantarum con base en los genes pgm, ddl, gyrB, purK1, gdh, mutS y tkt4 (de las Rivas et al., 2006) mostrando su poder altamente discriminatorio.
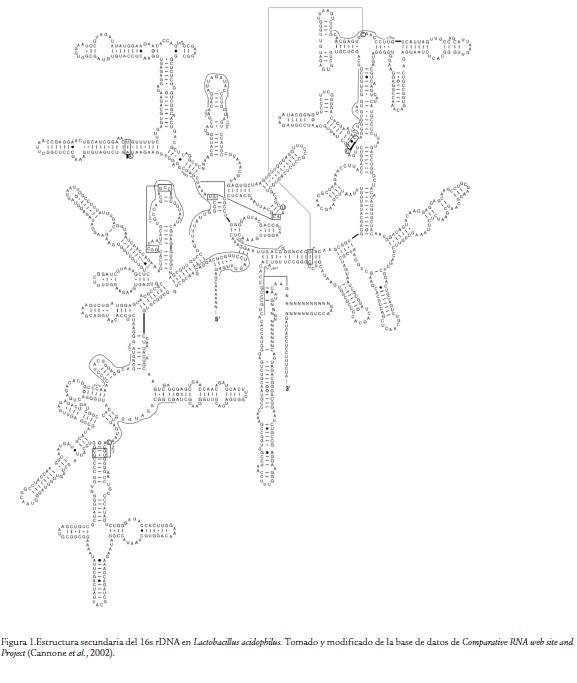
La subunidad ribosomal 16s se utiliza comúnmente para análisis filogenéticos y clasificación porque es fundamental para el desarrollo de los microorganismos y la tasa de cambios evolutivos permite la diferenciación a nivel de especies; sin embargo, se ha encontrado que pueden existir de una a 15 copias de los operones rrn en un genoma microbiano de las cuales aproximadamente solo el 40 % tienen una copia idéntica (Acinas et al., 2004). Por otro lado, el RNA ribosomal se caracteriza por la conservación de su estructura secundaria (Fig. 1), porque esta determina interacciones a corto y largo alcance que organizan la molécula en dominios y son responsables de su papel biológico (Macke et al., 2011). Todo esto permite agrupar las secuencias nucleotídicas basándose en la estructura secundaria que resulta más conservada que la estructura primaria, este análisis se puede realizar usando programas que emplean algoritmos que incluyen modelos de covarianza de las estructuras como Infernal (Inference of RNA alignments) (Nawrocki et al., 2009). Algunas herramientas computacionales desarrolladas incluyen análisis de secuencias de 16s rDNA teniendo en cuenta la estructura secundaria: RDP (Ribosomal Database Project Classifier), que utiliza el algoritmo de naÏve Bayesian classifier permitiendo asignar hasta nivel de género de la secuencia de interés con un porcentaje de confianza del 98 % (Cole et al., 2009); y STAP (ss-RNA TaxonomyAssigning Pipeline) cuyo algoritmo funciona generando alineamientos y rutas evolutivas tomadas por el gen a lo largo del tiempo (Wu et al., 2008).
En este trabajo se estudian las regiones variables del gen 16s rDNA para postular la región necesaria que permita hacer una clasificación correcta de género y/o especie de Lactobacillus spp., utilizando herramientas bioinformáticas como RDP, STAP e inferencia filogenética por máxima verosimilitud utilizando los paquetes INFERNAL y RAxML (Stamatakis, 2006). También se busca implementar herramientas de clasificación eficiente en aplicaciones web de fácil acceso.
MATERIALES Y MÉTODOS
Interfaz web de programas de clasificación
Se desarrolló una interfaz web en lenguaje XML explotando la plataforma Mobyle@Pasteur (http://mobyle.pasteur.fr/ y http://projets.pasteur.fr/) anclada a la página web del Grupo de Biología Computacional y Evolutiva en el servidor de la Universidad de los Andes (http://bce.uniandes.edu.co/) y se implementaron para los programas RDP multiclassifier y STAP disponibles para correr localmente en http://rdp.cme.msu. edu/misc/resources.jsp y http://bobcat.genomecenter.ucdavis.edu/mediawiki/index.php/STAP_download, respectivamente.
Selección de datos
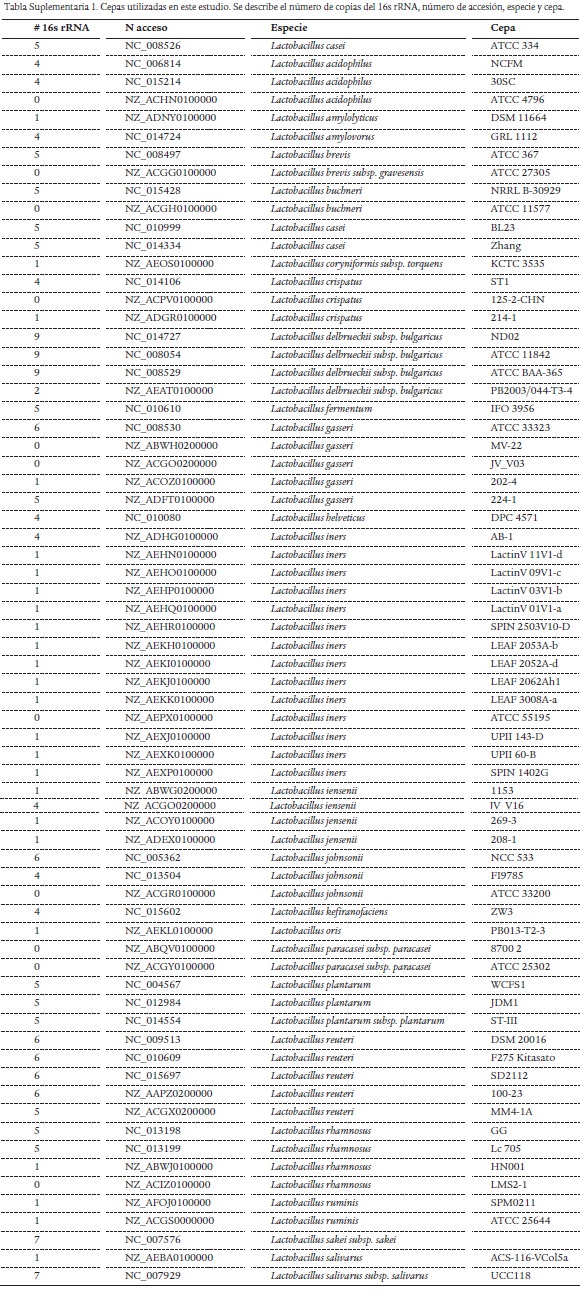
Se descargaron los genomas completos clasificados como Lactobacillus de NCBI Genomes (http://www.ncbi.nlm.nih.gov/sites/genome), los cuales tenían un tamaño entre 2 y 3 Mpb. Estos genomas pertenecen a las especies Lactobacillus iners, L. acidophilus, L. amylovorus, L. amylolyticus, L. buchneri, L. plantarum, L. ruminis, L. reuteri, L. casei, L. sakei, L. salivarus, L. coryniformis, L. crispatus, L. delbrueckii, L. brevis, L. rhamnosus, L. fermentum, L. gasseri, L. helveticus, L. jensenii, L. johnsoni, L. rhamnosus. Los números de acceso, cepas y número de copias del 16s rDNA se describen en la tabla suplementaria 1.
Extracción de regiones variables de 16s rDNA
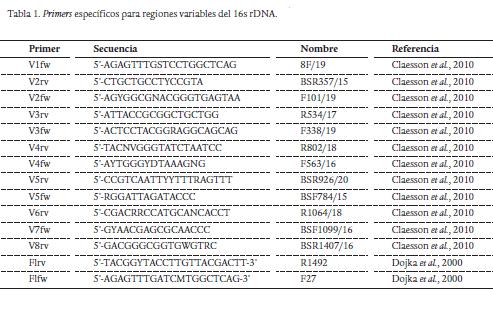
Se desarrolló un script en Perl (Disponible en http://bce.uniandes.edu.co/node/65) utilizando BioPerl con el cual se extrajeron los fragmentos de 16s rDNA de cada una de las cepas pertenecientes a diferentes combinaciones de regiones variables. Los primers utilizados para la amplificación in silico fueron tomados de un estudio de secuenciación de nueva generación, y anillan para la mayoría de genomas bacterianos (Claesson et al., 2010). Los primers empleados se describen en la Tabla 1.
Análisis de regiones variables
Los fragmentos de mayor tamaño (incluyendo las regiones V1 hasta V8), se analizaron en CD-HIT-EST (Li y Godzik 2006), con el fin de crear grupo de 100 % de identidad. Posteriormente todos los fragmentos de las regiones variables fueron clasificados taxonómicamente mediante RDP para el 16s rDNA. Se clasificaron como Verdaderos positivos (TP) aquellos que asignaban cada uno de los niveles taxonómicos correctamente, falsos negativos (FN), aquellos que clasificaban incorrectamente asumiendo como correcta la clasificación del NCBI de las secuencias; y NA aquellas regiones que no tuvieron amplímeros in silico para los pares de primers especificados o que no daban información de todos los niveles taxonómicos.
Alineamientos
Los alineamientos de las diferentes regiones se hicieron utilizando el software Infernal el cual utiliza un modelo de covarianza de estructuras secundarias. Se eliminaron las copias de 16s rDNA de cada cepa que presentaban 100 % de identidad y se seleccionó aleatoriamente una copia de cada cepa para hacer el alineamiento, y esta fue la misma para todas las regiones variables. El modelo de covarianza se generó con el modulo cmaln de Infernal a partir del alineamiento disponible en la base de datos de RDP (http://rdp.cme.msu.edu/), el cual incluía 508 especies bacterianas. Se tuvo en cuenta que el alineamiento solo tomara las posiciones comunes entre las secuencias y el modelo sin incluir aquellas presentes únicamente en el modelo de covarianza, de igual manera se especificó que las regiones eran truncadas. El formato de salida por defecto es Stockholm por lo cual se utilizaron scripts disponibles en la web (http://blog.mckuhn.de/2010/08/convert-stockholm-sequence-format-to.html) para convertirlo a Phylip relajado.
Análisis filogenético
Se realizó inferencia filogenética usando métodos de máxima verosimilitud implementados en RAxML, con 1000 réplicas de bootstrap y se tomó como modelo de substitución el S7A según el manual de Phase (Gowry-Shankar y Jow, 2006) que incluye 21 parámetros y permite hacer una regresión acertada en el tiempo.
Validación experimental
En el trabajo de laboratorio se estandarizó PCR para regiones del gen 16s rDNA con cepas de un tampón comercial.
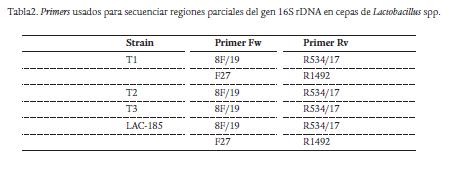
Muestras. Se usó un tampón comercial con probióticos de marca Ellen para determinar que cepas estaban presentes. Se tomó el tampón y se sembró en caldo MRS por dos días a 30 °C. Se numeraron las colonias de morfología característica (colonias blancas cremosas) y se plaqueó en agar MRS, después de dos días se evaluaron las colonias. Las colonias fueron numeradas como T1, T2 y T3. Se describieron morfológicamente las colecciones por microscopia de luz (tinción de Gram) y observación de las colonias en MRS, se caracterizaron de acuerdo a la prueba bioquímica de catalasa.
Extracción de ADN y PCR.
Se sembró cada una de las cepas en 2 ml de MRS caldo y se incubó a 33 °C Over Night. Se hizo extracción de ADN por boiling. Brevemente, se centrifugaron los tubos 5 min a 1500 rpm y se descartó el sobrenadante. Se agregó 1 ml de agua desionizada estéril y se resuspendió el pellet. Se pasó a un tubo de microcentrífuga de 1,5 ml y se microcentrifugó a 2000 rpm durante cinco minutos. Se descartó el sobrenadante y se repitió el lavado dos veces. Se resuspendió el pellet en agua desionizada estéril en un volumen de 100 µL. Se siguió el mismo procedimiento para una cepa aislada previamente en nuestro laboratorio, LAC-185 (Cepario del Laboratorio de Ecología Microbiana y de Alimentos - LEMA - Universidad de los Andes).
Para la lisis de la célula y separación del material genético se calentó por 20 min. en baño de agua a 100 °C. Se tomó como templado para la reacción de PCR una alícuota de 4,5 -7,5 µL. Los primers usados para cada cepa aparecen en la Tabla 2. Los ciclos de PCR fueron: denaturación a 94 °C por 2 min, luego 35 ciclos de denaturación a 94 °C por 45 s, anillaje a 53 °C por 45 s y elongación a 72 °C por 1 min. Finalmente, se dejó un ciclo a 72 °C por 10 min para extensión final. Se obtuvo la secuencia de los amplímeros usando química de Sanger (Macrogen, Korea; http://www.macrogen.com/).
RESULTADOS
Análisis de regiones variables
El número de copias del operon rrn varió entre uno y nueve para los microorganismos en estudio. Para estas copias los primers utilizados anillaron en la mayoría de los casos, dando un número máximo de amplímeros para cada región variable de 204, con base en el cual se calculó el número de datos faltantes. De los genomas analizados se encontró que diez no presentaban región para el 16s rDNA y dos fueron clasificados como cianobacterias por lo cual se excluyeron del análisis filogenético, adicionalmente una cepa fue clasificada como Enterococcus, esta clasificación fue independiente de la región del 16 rDNA empleada. Se observaron agrupaciones diversas, al 100 % de identidad, entre copias de 16s de una misma cepa con CD-HIT-EST (Figura suplementaria 1). De igual manera se observan clusters que incluyen copias de diferentes cepas que en general pertenecen a la misma especie. Los clusters 31 y 38 (Figura suplementaria 1, tabla suplementaria 2) tienen el máximo de copias, incluyendo cepas de L. iners. La mayoría de los clusters se organizan por cepa o por especie. Las cepas reportadas como cianobacterias no presentan agrupación en ninguno de los clusters.
Programas de clasificación RDP y STAP
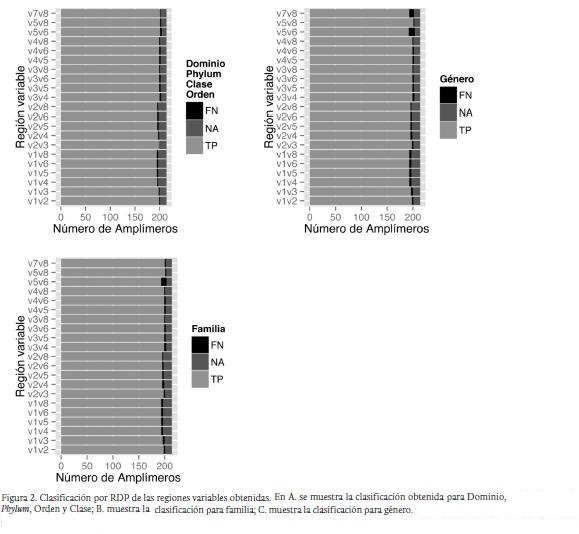
Todas las regiones variables presentan ausencia de datos para ciertas secuencias y además falsos negativos. Las figuras 2 y 3 muestran la distribución de verdaderos positivos (TP), falsos negativos (FN) y datos ausentes (NA) para los fragmentos analizados por RDP y STAP respectivamente para diferentes niveles en la jerarquía taxonómica. En la Figura 2 se observan las tasas de acierto y error de RDP. Para dominio, phylum, orden y clase, (A) el mayor número de amplímeros clasificados correctamente los presenta la región V5V6 y las regiones V1V4, V1V5, V1V6 y V1V8 presentan la menor cantidad de amplímeros clasificados; el fragmento con mayor proporción de falsos negativos comprende las regiones V2V5; para la familia y el género (B y C), V5V8 presenta el mayor número de amplímeros asignados correctamente, y la menor cantidad la tienen las mismas regiones que A.
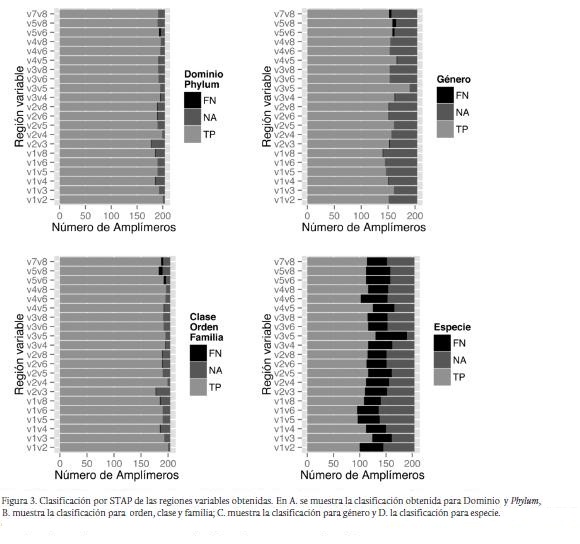
Utilizando STAP, tenemos que, como muestra la Figura 3, para dominio y phylum, la región V1V2 es la que presenta un mayor número de amplímeros clasificados correctamente con los primers seleccionados y V2V3 la que presenta el mayor número de amplímeros sin clasificación. La región con mayor número de falsos negativos es V5V6. Para orden, clase y familia se observa que V1V2continua presentando el mayor número de amplímeros clasificados correctamente, V2V3 el mayor número de amplímeros sin clasificar, pero para este caso la región con mayor error es V5V8. Para género se pierde gran cantidad de datos ya que los fragmentos no logran resolver hasta esta categoría taxonómica; se observa que V3V5 tiene el mayor número de amplímeros clasificados, y V1V8 el que presenta menor número de amplímeros clasificados. Sin embargo, aunque V3V5 tiene una gran cantidad de fragmentos clasificados, su tasa de error es aproximadamente del 50 %. Las regiones V1V5, V1V6 y V4V6 presentan las mayores tasas de error en comparación con los verdaderos positivos.
Si se busca una clasificación hasta género es confiable hacerla utilizando la región V3V5 , tiene un mayor cubrimiento y tasa de error muy baja, pero si se desea llegar a una aproximación de especie o de grupo la región V1V3 presenta la menor tasa de falsos negativos aunque su cubrimiento no es el mayor. El análisis filogenético mostró que todas las regiones dan topologías diferentes, sin embargo, como se observa en la Figura 4, hay grupos que se conservan en los análisis hechos para todas las regiones y pertenecen a un mismo grupo de Lactobacillus.
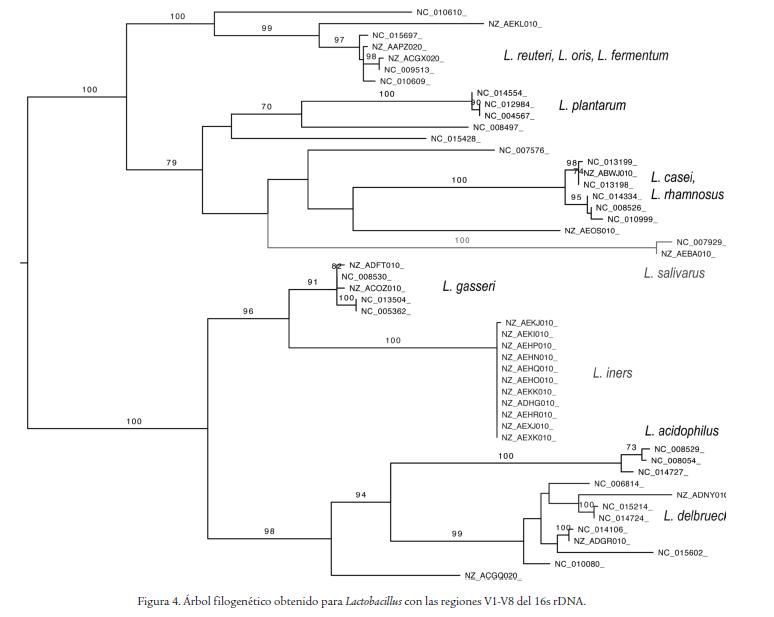
Se observan cuatro grupos definidos en todas las regiones variables, los cuales se corresponden a: primero L. oris, L. reuteri y L. fermentum; segundo L. plantarum, L. casei, L. rhamnosus y L. salivarus; tercero L. gasseri y L. iners y cuarto L. acidophilus, L. delbrueckii, L. amylovorus, L. amylolyticus, estos tienen buen soporte de clado, sin embargo, para las regiones variables V7V8, V5V8, V5V6 se pierden estos grupos totalmente. Los grupos formados presentan algunas diferencias en la topología que no se tuvieron en cuenta dado que no tenían buen soporte. Las regiones que presentan las mismas relaciones entre grupos son V1V8, V2V8, V4V8, V1V3, V2V5 y V1V5. No se pueden indicar relaciones de ancestría por falta de datos de muestreo por lo cual el árbol fue enraizado en el punto medio. Se asumió buen soporte como mayor a 70.
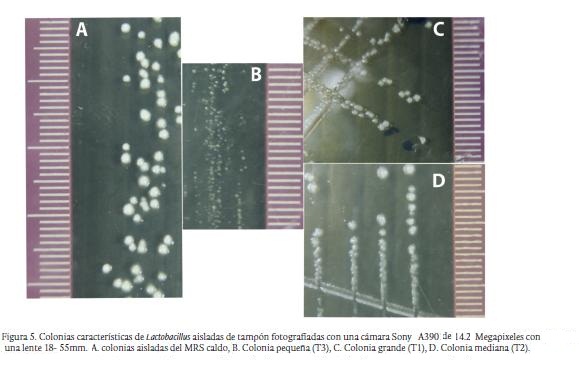
En cuanto al análisis de laboratorio, las colonias características aisladas del tampón se muestran en la Figura 5. Las tres cepas aisladas al igual que la cepa LAC-185, presentaron coloración violeta en la tinción de Gram indicando que son Gram positivas y son bacilos no esporulados. Las pruebas bioquímicas reflejaron que son catalasa negativo y presentaron crecimiento óptimo en MRS pH 6,2 a 33 °C. Las colonias aisladas presentan una morfología macroscópica redonda, cremosa, de bordes irregulares y olor característico. La cepa T3 es pequeña con un diámetro de 0,05 mm, T2 es mediana con diámetro 0,15 mm y T1 es grande con diámetro 0,2 mm. La estandarización de PCR mostró que el perfil descrito en los métodos era eficiente. Los primers anillaban correctamente y se obtenía una banda más definida y gruesa que la resultante de la amplificación con los primers universales disponibles. El amplímero tenía un tamaño de aproximadamente 550 pb (Figura Suplementaria 2) que incluía las regiones variables 1-3, y los primers universales presentaron un amplímero de aproximadamente 1200 pb que incluían una región mayor del 16s rDNA. La mayoría de secuencias obtenidas para el 16s rDNA completo no pudieron ser ensambladas dada su baja calidad (datos no mostrados). Utilizando RDP tanto para la región V1V8 como para la V1V3 todos los amplímeros clasificaron hasta el nivel de género como Lactobacillus. Con el análisis de STAP se obtuvo que la cepa T1 era L. fermentum (GenBank ACC: KC207719), la cepa T2 L. casei (GenBank ACC: KC207720) y la cepa T3 L. delbrueckii (GenBank ACC: KC207721). La cepa LAC-185 fue asignada a Lactobacillus casei (GenBank ACC: KC243415).
DISCUSIÓN
Los resultados de este estudio muestran la importancia de conocer las regiones más variables e informativas del 16s rDNA para la identificación de cepas bacterianas dado que no siempre una región más grande va a representar un resultado mejor, y para todas las especies no se va a dar el mismo patrón de variación. Los resultados de CD-HIT-EST en la Figura suplementaria 1, evidenciaron que no siempre los fragmentos de la misma especie se agrupan, se observaron casos en que fragmentos de diferentes cepas formaban grupos. En estudios anteriores fue reportado que alrededor del 40 % de las secuencias bacterianas tiene al menos una copia idéntica en el genoma y en el 38 % de los genomas todas las copias del operón son idénticas entre sí (Acinas et al., 2004).
La clasificación por STAP mostró diferentes regiones candidatas para clasificación dependiendo del grado de discriminación al que se pretenda llegar con el estudio. Anteriormente se había determinado que las regiones V1V3 dan una mejor resolución al identificar especies (Petrosino et al., 2009; Wu et al., 2010), lo cual tiene relación con el resultado de este ensayo al tomar los datos de clasificación de especie. Para RDP, se observó un patrón diferente para la clasificación, las regiones de V1V4-V8 presentan el mayor error y por ende pueden presentar bastante ruido en la identificación de cepas al igual que la región V5V6 que antes había sido reportada como la mejor región para identificación de bacterias (Claesson et al., 2010). Fue reportado anteriormente que el menor error se daba en bacterias en la clasificación hecha con las regiones V2V4, dando el mejor resultado (88,7 %) para regiones superiores a 400 pb (Cole et al., 2009).
Los árboles filogenéticos debieron ser comparados entre sí para cada combinación posible de copias del 16s rDNA, con lo cual se tendría un resultado más acertado que por la escogencia al azar dado que pudieron haberse elegido las copias más similares entre cepas. Además, se debería evaluar en el laboratorio cual de las copias esta siendo expresada o si todas se expresan al mismo tiempo, lo cual podría explicar problemas de clasificación, dado que los sitios más variables se perderían como errores de secuenciación. Sin embargo, se obtuvo una organización en grupos similar a la obtenida por Canchaya et al., (2006) quienes hicieron la filogenia con el 16s rDNA de 111 cepas disponibles en la base de datos de RDP. Han sido descritos cinco grandes grupos de Lactobacillus los cuales son L. acidophilus que incluye a L. acidophilus, L. amylovorus y L. amylolyticus; L. johnsonii que incluye a L. gasseri y L. iners; L. sakei, L. plantarum y L. salivarus. Estos mismos grupos a excepción de L. sakei se encuentran conservados.
Por medio de la validación experimental se obtuvo que las cepas aisladas del tampón probiótico correspondieron a las indicadas por la casa comercial (Ellen AB; http://www.ellenab.se/en/) aunque no fue posible obtener su clasificación hasta el nivel de subespecie. Las cepas indicadas por la casa comercial son L. gasseri LN40 el cual puede corresponder a la cepa T3 aislada que clasificó como L. delbrueckii ya que son muy cercanos filogenéticamente, L. fermentum LN99 que corresponde a la cepa T1 y L. rhamnosus LN113 el cual se aisló como la cepa T2 y se clasificó hasta dentro de L. casei del cual L. rhamnosuses una subespecie (Petricevic et al., 2008). En el caso de LAC-185 y T1 la clasificación obtenida fue idéntica al usar tanto el gen 16s rDNA completo como la región V1V3. Esto nos permite sugerir que esta región mínima obtenida in silico mediante el análisis en STAP también es útil para la identificación de muestras ex silico.
Este flujo de trabajo puede ser útil para cualquier tipo de procariotas para definir las regiones mínimas para su clasificación las cuales pueden ser empleadas en diferentes tecnologías de secuenciación y para análisis de metagenómica. Sin embargo, para una clasificación más específica pueden realizarse otros tipos de análisis entre los cuales se incluyan más genes y caracteres fenotípicos relevantes en cada grupo.
AGRADECIMIENTOS
DMRP reconoce el apoyo de la Facultad de Ciencias de la Universidad de los Andes mediante el programa "Proyecto de Profesor Asistente". LNGG agradece la beca otorgada por la "International Society for Computational Biology - ISCB", para presentar resultados preliminares de este trabajo en la "International Societyfor Computational Biology Latin American regional meeting (ISCB-LatinAmerica)" en Santiago, Chile. Los autores agradecen a Dra. Flavia Vischi Winck por su colaboración en la realización de este proyecto y la ayuda en la obtención del tampón probiotico.
BIBLIOGRAFÍA
Acinas S, Marcelino LA, Klepac-Ceraj V, Polz MF. Divergence and Redundancy of 16s rDNA Sequences in Genomes with Multiple rrn Operons. J Bacteriol 2004;186(9):2629-2635. [ Links ]
Anukam K C, Osazuwa E O, Ahonkhal I, Raid G. 16s rDNA gene sequence and phylogenetic tree of Lactobacillus species from vagina of healthy Nigerian women. Afr J Biotechnol. 2005;4(11):1222-1227. [ Links ]
Berger B, Pridmore RD, Barretto C, Delmas-Julien F, Schreiber K, Arigoni F, et al. Similarity and differences in the Lactobacillus acidophilus group identified by polyphasic analysis and comparative genomics. J Bacteriol. 2007;189(4):1311-1321. [ Links ]
Canchaya C, Claesson M, Fitzgerald G, Van Sinderen D, O'oole PW. Diversity of the genus Lactobacillus revealed by comparative genomics of five species. Microbiol. 2006;152(11):3185-3196. [ Links ]
Cannone JJ, Subramanian S, Schnare MN, Collett Jr, D'souza LM, Du Y, et al. The Comparative RNA Web (CRW) Site: An Online Database of Comparative Sequence and Structure Information for Ribosomal, Intron, and Other RNAs.BMC Bioinformatics 2002;3:2. [Correction: BMC Bioinformatics3:15. [ Links ]
Claesson M J, Wang Q, O'sullivan O, Greene-Diniz R, Cole J R, Ross P, O'toole P W. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16s rDNA gene regions. Nucl Ac Res. 2010;38(22):e200 [ Links ]
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. The Ribosomal Database Project: improved alignments and new tools for rDNA analysis. Nucl Ac Res. 2009;37 (Database issue):D141-145. [ Links ]
De Las Rivas B, Marcobal A, Munoz R. Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiol. 2006;152(1):85-93. [ Links ]
Diancourt L, Passet V, Chervaux C, Garault P, Smokvina T, Brisse S. Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl Environ Microbiol. 2007;73(20):6601-6611 [ Links ]
Dojka MA, Harris JK, Pace NR. Expanding the Known Diversity and Environmental Distribution of an Uncultured Phylogenetic Division of Bacteria. Appl Environ Microbiol. 2000;66(4):1617-1621. [ Links ]
Gowri-Shankar V, Jow H. PHASE: a Software Package for Phylogenetics And Sequence Evolution. University of Manchester, 2006. [ Links ]
Guerzoni ME. Human food chain and microorganisms: a case of co-evolution. Front Microbiol. 2010;1:106. doi: 10.3389/fmicb.2010.00106 [ Links ]
Kohavi R. Wrappers for performance enhancement and obvious decision graphs [Tesis doctoral. Standford: Computer Science Department, Stanford University, 1995. [ Links ]
Li W, Godzok A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658-1659. [ Links ]
Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNA Motif, an RNA secondary structure def inition and search algorithm. Nucl Ac Res. 2011;29(22):4724-4735. [ Links ]
Maiden M, Bygraves J, Feil E, Morelli G, Russell J, Urwin R et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998;95(6):3140-3145. [ Links ]
Martín R, Heilig GH, Zoetendal EG, Smidt H, Rodriguez JM. Diversity of the Lactobacillus group in breast milk and vagina of healthy women and potential role in the colonization of the infant gut. J Appl Microbiol. 2007;103(6):2638-2644. [ Links ]
Martín R, Soberón N, Vázquez F, Suárez JE. La microbiota vaginal: composición, papel protector, patología asociada y perspectivas terapéuticas. Enferm Infecc Microbiol Clin. 2008;26(3):160-167. [ Links ]
Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: Inference of RNA alignments. Bioinformatics. 2009;25(10):1335-1337. [ Links ]
Olivares M, Diaz-Ropero MP, Martin R, Rodriguez JM, Xaus J. Antimicrobial potential of four Lactobacillus strains isolated from breast milk. J Applied Microbiol. 2006;101(1):72-79. [ Links ]
Petricevic L, Witt A. The role of Lactobacillus caseirhamnosus Lcr35 in restoring the normal vaginal flora after antibiotic treatment of bacterial vaginosis. BJOG. 2008;115(11):1369-1374. [ Links ]
Petrosino JF, Highlander S, Ann R, Gibbs RA, Versalovic J. Metagenomic Pyrosequencing and Microbial Identification. Clin Chem. 2009;55(5):856-866. [ Links ]
Reid G, Jass J, Sebulsky MT, Mccormick JK.Potential uses of probiotics in clinical practice. Clin Microbiol Rev. 2003;16(4):658-672. [ Links ]
Sarmiento LA. Influencia del consumo de sorbitol en la microbiota intestinal de un modelo animal [Tesis doctoral. Valencia: Universidad Politécnica de Valencia, 2008. [ Links ]
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16s rDNA from comple x communities reveals many novel molecular species within the human gut. Appl Environ. 1999;65(11):4799-4807. [ Links ]
Stamatakis A. RAxML-VI-HPC: Maximum Likelihood-based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics. 2006;22(21):2688-2690. [ Links ]
Wu D, Hartman A, Ward N, Eisen JA. An automated phylogenetic tree-based small subunit rDNA taxonomy and alignment pipeline (STAP).PLoS ONE. 2008;3(7):e2566. [ Links ]
Wu GD, Lewis JD, Hoffman C, Chen Y, Knight R, Bittinger K, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16s sequence tags. BMC Microbiology. 2010;10:206. [ Links ]