










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Biológica Colombiana
versão impressa ISSN 0120-548X
Acta biol.Colomb. vol.20 no.2 Bogotá maio/ago. 2015
https://doi.org/10.15446/abc.v20n2.43639
Doi: http://dx.doi.org/10.15446/abc.v20n2.43639
Artículo de revisión
RNA-SEQ: A GLANCE AT TECHNOLOGIES AND METHODOLOGIES
RNA-Seq: un vistazo sobre las tecnologías y metodologías
Seyed Mehdi JAZAYERI1, Luz Marina MELGAREJO MUÑOZ2, Hernán Mauricio ROMERO2.
1Oil Palm Biology and Breeding Research Program. Corporación Centro de Investigación en Palma de Aceite (Cenipalma). Calle 20A nº. 43A–50. Bogotá, Colombia.
2Department of Biology, Faculty of Science, National University of Colombia (Universidad Nacional de Colombia). Av. Carrera 30, nº. 45-03, Edif. 421, Bogotá, Colombia.
For correspondence. hmromeroa@unal.edu.co
Received: 6 November 2014; Returned for revision: 10 December 2014; Accepted: 9 January 2015.
Associate Editor: Geraldo Mäder.
Citation / Citar este artículo como: Jazayeri SM, Melgarejo Muñoz LM, Romero HM. RNA-Seq: a glance at technologies and methodologies. Acta biol. Colomb. 2015;20(2):23-35. doi: http://dx.doi.org/10.15446/abc.v20n2.43639
ABSTRACT
RNA Sequencing (RNA-Seq) is a newly born tool that has revolutionized the post-genomic era. The data produced by RNA-Seq, sequencing technologies and use of bioinformatics are exploding rapidly. In recent years, RNA-Seq has been the method of choice for profiling dynamic transcriptome taking advantage of high throughput sequencing technologies. RNA-Seq studies have shown the transcriptome magnitude, notion and complexity. From 2008, as its introduction year, the relevant reports on RNA-Seq have been multiplied by more than 2822 times just in 6 years. RNA-Seq also contributes a more accurate gene expression and transcript isoform estimation than other methods. Furthermore, some of the potential applications for RNA-Seq cannot be conducted by other methods and as yet are unique to RNA-Seq. As RNA-Seq approaches increase in speed and decrease in cost, more distinct researches are applied and become more common and accurate. RNA-Seq is a cross and interdisciplinary method that interconnects biology to other scientific topics. This article describes RNA-Seq approach, technologies, methodologies, implementation, and methods done so far in characterizing and profiling transcriptomes.
Keywords: HTS High Throughput Sequencing, NGS Next Generation Sequencing, RNA-Seq, transcriptome, gene expression.
RESUMEN
En los últimos años, la técnica RNA-Seq ha tenido un desarrollo acelerado y se ha convertido en el método de elección para el estudio y la caracterización de los transcriptomas dinámicos, aprovechando las tecnologías de secuenciación de alto rendimiento. Estudios aplicando RNA-Seq han mostrado la magnitud, noción y complejidad del transcripotma. A partir de 2008, año de introducción de la técnica, los estudios con RNA-Seq se han multiplicados por más de 2822 veces sólo en 6 años. Al compararse con otros métodos, los estudios empleando RNA-Seq contribuyen a una estimación más precisa de la expresión génica y de las isoformas de los transcriptos. Además, algunas de las aplicaciones potenciales de RNA-Seq no se pueden llevar a cabo con otros métodos. El uso de RNA-Seq aumenta la velocidad de obtención de información y disminuye los costos, logrando con su uso, que investigaciones diversas se vuelvan más frecuentes y precisas. RNA-Seq es un método interdisciplinario que interconecta la biología a otros temas científicos. En este artículo se describe el planteamiento de la tecnología RNA-Seq, metodologías y los métodos realizados en la caracterización de transcriptomas.
Palabras Clave: secuenciación con alto rendimiento, próxima generación de secuenciación, RNA-Seq, transcriptoma, expresión génica.
INTRODUCTION
Transcriptomics is the study of the transcriptome of a living organism. The transcriptome is the complete set of transcripts and its amount for a specific developmental stage or physiological state and can be described as a complete list of all classes of RNA molecules i.e. coding or non-coding when expressed in a particular cell, tissue or organ. Transcriptome perceiving and apprehending is essential for genomic functional element performance to reveal molecular components of cells and tissues, their biological functions and effects, to decipher environmental cues and their relation with and effects on functional genome and transcriptome, to comprehend development and disease, to understand gene expression and coexpression, to disclose biological networks, systems biology and expression networking, to perceive epigenetic events, and to know more on living organizations from molecule and cell to biome and biosphere.
Key objectives of transcriptomics are to catalog all species of transcripts including mRNA, antisense RNA and small RNA; to determine the structure of gene transcription depending on the starting points, 5' and 3' ends, splicing patterns and other post-translational and transcriptional modifications and to quantify levels of each transcript expression change during development under different conditions (Wang et al., 2009).
In parallel to transcriptomics there are other special transcriptome approaches called metatranscriptomics and bacterial transcriptomics that generally talk about virus, microbes and bacteria in environment, where can be any place that microorganisms live. The extraction and analysis of metagenomic mRNA or metatranscriptome provide information on structure, function, regulation and expression profiles of complex communities of prokaryotes, bacteria, virus and microbes in milieu. Metatranscriptomics offers the opportunity to reach beyond the community's genomic potential as assessed in DNA-based methods, towards its in situ activity by disclosing metatranscriptome. Also it can be used as a catalog of organisms found in under study ambience and as an evolutionary tool.
After 1964, the year of the first published RNA sequencing report and also after introduction of Northern Blot and RT-PCR as two other methods of RNA and transcriptome study, the sequencing world has seen a revolution and passed different steps to reach to RNA-Seq (Table 1). Several technologies have been developed to derive and quantify the transcriptome, including hybridization and sequence-based approaches.

Hybridization based methods mainly known as microarrays are based on fluorescently labeled cDNA and hybridization of unknown target samples with previously known transcripts and are grouped in custom-made or commercial high-density oligo microarrays. Also it is possible to create specialized and customized microarrays according to the research aims. To determine different splicing isoforms, arrays with probes spanning exon junctions can be designed. They need previous information about the transcripts whose expression is desired to reveal and it is not possible to detect unknown genes and novel transcripts. Unlike hybridization-based, sequence-based approaches detect directly cDNA sequence.
Originally, cDNA or EST libraries were sequenced by Sanger sequencing approach but it did not continue as this approach is costly and just qualitative not quantitative and lacks relatively for desired performance. Methods based on tags (Tag-based) including Serial Analysis of Gene Expression (SAGE), Cap Analysis of Gene Expression (CAGE) and Massively Parallel Signature Sequencing (MPSS) were evolved to overwhelm the mentioned limitations.
The automated method of Sanger is referred as the "first generation" technology, and new methods possessing different applications in genomics, metagenomics, epigenomics, functional genomics, transcriptomics and single-cell sequencing are known as Next-Generation Sequencing (NGS) or High-Throughput Sequencing (HTS). From the first time in 2005 presented to the market by "Roche" as the sequencing method of "454 Pyrosequencing", NGS technologies has had a tremendous influence on genomics and transcriptomics field (Nyrén, 2007).
Next-generation sequencing technologies have been used for sequencing as standard application such as genome sequencing and re-sequencing, and also for new applications previously unexplored by Sanger sequencing method such as DNA-Seq (DNA sequencing), RNA-Seq (RNA sequencing), ChIP–Seq (Chromatin Immunoprecipitation sequencing), ChIP coupled to DNA microarray (ChIP-chip), Methyl-Seq (Methylation sequencing), MethylCap-Seq, Methyl-C-Seq or BS-Seq (Bisulfite sequencing) and other NGS based approaches for epigenomics.
According to Lister et al., (2009) these technologies have been used to study the genome sequence variations, ancient DNA, methylation of DNA cytosine, DNA-protein interactions, mRNA expression, alternative splicing, small RNA populations, posttranscriptional, posttranslational and regulation events of mRNA. The advantages of NGS platforms such as money saving and low cost, data analysis ability by software and bioinformatics tools allow obtaining massive sequence data more simply and affordably and have opened new horizons to understand global processes regulating gene expression.
RNA-Seq can help us find expression levels of transcripts and genes during ontogeny of an organism and understand more about functional genome and transcriptome. From 2008, the year of the first RNA-Seq publication to 2014, an increasing growth from 74 to 208,892 runs in Sequence Read Archive (SRA) database (http://cell-innovation.nig.ac.jp/cgi-bin/pub_stat/pub_stat_all.cgi, data accessed and taken in Aug. 2014) has been reported, i.e. more than 2,822 times more reports in just about 6 years (Figure 1). This shows how RNA-Seq has been important and contributed on transcriptomics and RNA studies as some of its applications are considered newly presented utilizations which are not applicable in other methods.

In this article our attention will be on NGS technologies, platforms, methods and tools, in functional genomics and transcriptomics research, focusing on RNA-Seq. RNA-Seq approach is discussed in details based on a general workflow and some of its applications are presented. The article continues with RNA-Seq challenges and suggested solutions.
RNA-SEQ APPROACH
"RNA-Seq" term applies to any of several different methods of HTS or NGS used for obtaining whole transcriptome profiles of RNA both in terms of type and quantity that can be studied in cell, tissue, organ, whole organism, community, ecosystem or biome scale. RNA-Seq ability to discover previously uncharacterized mRNA isoforms and new classes of non-coding RNAs represents why this fast developing technology is used excessively, assuming a key role in the analysis of RNA.
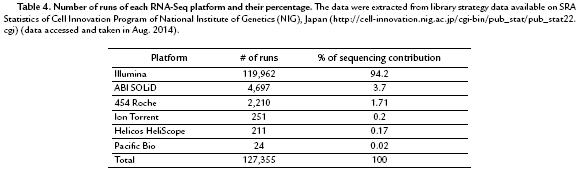
Each of NGS or HTS technologies can be used as RNA-Seq methodology. Illumina/Solexa, Applied Biosystems ABI SOLiD, 454 Pyrosequencing Roche Genome Sequencer, PacificBio or combination of them have been applied as different RNA-Seq platforms and are being reported tremendously. Although they are based on a same principle, i.e. to generate RNA sequencing data, they are different regarding to price, throughput, read length and generation and error rate (Loman et al., 2012) .
Hereinafter, we present a suggested workflow that can be considered and employed in each RNA-Seq study as a basic starting point. There are three important basic steps in RNA-Seq including "wet-lab" part, "in equipo" part, and finally "in silico" part, qua shown in details in Figure 2 as an RNA-Seq overall workflow.

"Wet-lab" as laboratory section includes experiment design according to the expected aims of research and then relevant subsequent procedures toward obtaining a pure and without contamination RNA extraction in order to detect the fragments that give better and more precise reads in downstream steps.
"In equipo" as sequencing section in almost all cases is done following the protocols and procedures and using kits recommended by each platform technology. This part also can include the last part of wet-lab part that is cDNA library preparation to run in the sequencing machine depending on library construction strategies of the used platform.
"In silico" as bioinformatics section is how to analyze sequencing data precisely and correctly according to project objectives, requirements, tools and available programs to have more reliable results. However the programs are enough reliable in most cases as increasing publications comparing distinct programs used for each part of analyses like assembly (Schliesky et al., 2012) and differential expression (Soneson and Delorenzi, 2013) mostly have explained that the results of a program can confirm and validate the results of other programs and regarding to researcher design and aims, most of them work well and are reliable enough to use in replace of each other or in parallel.
Each steps of an RNA-Seq project can face some drawbacks those can be problematic if not alleviated. In wet-lab step RNA quality, quantity and purity, RNA degradation and ribosomal RNA elimination can be mentioned those affect the results of RNA-Seq. RNA extraction quality is crucial as all analyses are done based on RNA input. However some techniques have been provided by which RNA can be processed even in low-input cases to ameliorate RNA extraction quality, quantity and purity.
In equipo drawbacks can be listed as not enough economical prices of either sequencing machines or services offered by sequencer companies to allow RNA-Seq available to all research teams those have limit budget, technology limits like read length, read accuracy and sequencing time. To solve such in equipo problem, multiplexing methods are available to handle many samples in one same sequencing run by different barcodes in order to decrease sequencing expenses. However library construction is the most expensive part of sequencing that cannot be solved by multiplexing as each sample needs to have its own library.
Required high performance computation unit, lacking available suitable software and hardware to handle the data completely and adequately are some impediments for in silico. The solutions for the mentioned shortcomings will be discussed later.
Designing project run
Some items are subject to be considered as the design basics of RNA-Seq such as sequencing depth, technical and biological replication, efficient experimental design, multiplexing technical and algorithmic approaches (Robles et al., 2012). It is possible to design a RNA-Seq project in four forms: biologically unreplicated unblocked design, biologically unreplicated balanced block design, biologically replicated unblocked design, and biologically replicated balanced block design (Auer and Doerge, 2010). In these forms balanced block is considered as different sequencing in different lanes of a same plate of machine. However the results of a unreplicated unblocked design can be reliable and acceptable as in RNA-Seq published articles "no biological replication" is common (Auer and Doerge, 2010).
Required RNA samples
Basically mRNA is required for RNA-Seq to obtain transcriptome, while depending on the project aims, other types of RNA like total RNA, small RNA, microRNA and non-codifying RNA are also mentioned as RNA-Seq required material. It is common to prepare a RNA pool from different under study replicates as the accuracy of pooled RNA-Seq remains comparable with pooled genome resequencing (Konczal et al., 2014) and as it can cover and give general transcriptomics profile of all individuals if they are not very variant. The mRNA is purified from total RNA using a kit to enrich poly(A) mRNA. To purify mRNA from total RNA, there is a good method using magnetic beads to obtain mRNA according to poly (A) oligo system (Mortazavi et al., 2008) that is used in some kits for mRNA purification from total RNA or directly from tissue. When mRNA is used for sequencing, ribosomal RNA is eliminated by available kits to improve RNA extraction (Sooknanan et al., 2010) and to meliorate subsequent results.
Considering differences between Eukaryotic and Prokaryotic mRNA, the methodologies of handling RNA in Eukaryotes are different from Prokaryotes. Eukaryotic mRNA life span is longer than of prokaryotes, prokaryotic mRNA undergoes very little posttranscriptional changes and have short time interval between transcription and translation but eukaryotic mRNA are subjected to posttranscriptional modifications like Poly(A) tail, Guanylate residue capping at 5' end, heterogeneous mRNA, slicing and pertinent modifications.
For prokaryotes total RNA can be extracted using methods like salt fractionation, TRIzol reagents employing guanidium isothiocyanate and phenol solution and silica-membrane binding RNA column. Then mRNA is purified and enriched using 5'-Phosphate-Dependent Exonuclease and poly (A)-based magnetic beads to remove rRNA and mRNA from eukaryotic sources and DNase I treatment to eliminate DNA contamination.
The problem of RNA extraction is mainly degradation by RNases found everywhere from any living organism source. Also RNA degradation may occur during saving tissues in inadequate conditions. Some solutions are available to eliminate RNases and alleviate RNA degradation either in saving step (known mainly as RNAlater reagent) to conserve tissues or in extracting step (known as RNase AWAY or RNase ZAP reagents) to clean working surface and tools and to eliminate RNases. When extraction starts, the buffers used in extraction process, protect RNAs by deactivation of RNases.
Another problem is RNA purity and required amount. Purity should be considered when extracting RNA. The ratio of 260/280 can give an idea about how pure RNA extraction is as nucleic acids are measured at 260nm and proteins and other compounds at 280nm. Acceptable range of 260/280 ratio for RNA is 1.8 -2.1 with the optimum value of 2. In case of impurity, it is less than 1.8 and it may be more than 2.3 due to RNA degradation.
Depending on the platform the amount of RNA can be different to have a good coverage. In Illumina as the predominant technology, 1 µg of RNA is sufficient to have reliable data considering the number of samples those are run in one lane of each plate. Each run of Illumina HiSeq2000 machine can sequence > 2 human genomes at 30X coverage that means about 2 billion reads each ∼ 101 bp in Paired-End style in one run. Thus to have a 10X coverage of a transcriptome with about 100 Mbp (Mega base pairs), there needs 1 Gbp (Giga base pairs) data that can be generated from one hundredth of a run meaning 100 samples in each.
About more or less than 5% of genome can be considered as transcriptome (Table 2) by which it may calculate and determine reliable coverage. However in an Arabidopsis study based on Shannon entropy, it has shown that only 250000 reads or 5000 transcripts can be used to have reliable data for gene expression profiling and for each lane of Illumina up to 400 samples can be run theoretically covering 90% of transcripts to obtain trustworthy results (Kliebenstein, 2012).

cDNA and library preparation
mRNA must be converted to cDNA by reverse transcription reaction using random primers and/or Oligo dT primers. The advantage of using the latter is that most of cDNA should be produced from polyadenylated mRNA thus obtained sequence results are informative (not ribosomal) (Wilhelm and Landry, 2009). In order to construct cDNA and relevant libraries, special kits are specifically used for each platform according to its technology and recommendation. When library preparation is done and cDNA fragments are created, keeping information of mRNA is the most important factor to have clean and intact results and data.
Also FRT-Seq approach has been presented that uses Poly A+ RNA as the template rather than cDNA, is strand-specific, amplification-free, compatible with paired-end sequencing, and reverse transcription takes place on the flowcell surface (Mamanova et al., 2010). Random oligonucleotide primer approach is available to synthesize cDNA from any prokaryotic source.
SEQUENCING AND IN-USE PLATFORMS
All platforms share the same basic principle of total RNA isolation, mRNA purification, cDNA construction and attachment to a solid matrix of a single piece of cDNA by limiting dilution, followed by amplification of this molecule through a specialized emulsion PCR (emPCR) in SOLiD, 454 Pyrosequencing, Ion Torrent, or a connector based on the bridging reaction (Illumina), while Pacbio or Pacificbio technology does not need to amplify cDNA and performs sequencing directly from the target cDNA.
The cDNA transcripts of identical RNA molecules can be sequenced in parallel, either by measuring the incorporation of fluorescent nucleotides (Illumina), fluorescent short linkers (SOLiD), by the release of the by-products derived from the incorporation of normal nucleotides (454), fluorescence emissions or by measuring pH change (Ion Torrent). In Table 3 different platforms are compared with more details regarding to their features, specifications and technologies.
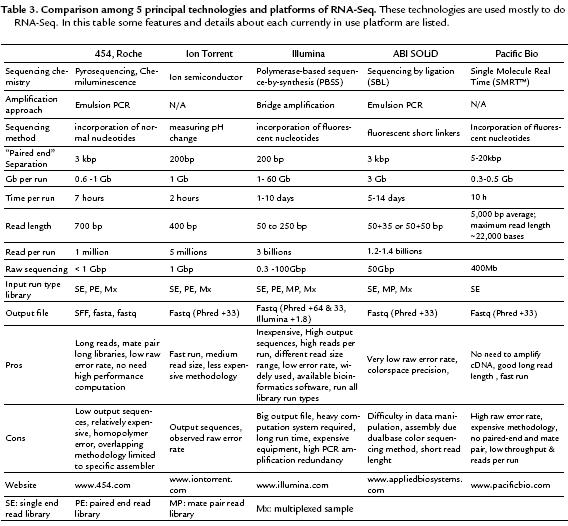
Regarding to available information on RNA-Seq library strategy runs for different platforms in SRA, Illumina is located at the top with more than 94.2% runs followed by ABI SOLiD, 454 Roche, Ion Torrent, Helicos HeliScope, and Pacific Bio, respectively (Table 4).
Illumina/Solexa
This technology has been developed based on modifying dideoxynucleotide terminator used in Sanger sequencing method. Illumina has designed a reversible version of termination known as cyclic reversible termination (CRT) (Metzker, 2010). During sequencing, each modified dNTP is bound to a fluorophore specific base that becomes fluorescent when incorporated into the DNA fragment. The emission is recorded by a high resolution camera. This process is repeated in each cycle as occurring incorporation of one labeled dNTP followed by taking a picture of fluorescent, and removing the terminator.
In this method, fragments are amplified by bridge PCR that is amplification in constructed bridge among fragments. The fragments hybridize to a set of forward and reverse immobilized primers in the substrate corresponding to the adapters used to prepare the library. Several million clusters can accumulate in each of the independent set channels existing in the flow cell, where sequencing reactions occur.
SOLID
The initial technology was described in 2005 and the first machine was released by Applied Biosystems in 2007. The DNA or cDNA fragments are denatured and fixed on magnetic beads, following the strategy; one fragment for each bead. The library is amplified by emulsion PCR and each bead having a set of amplified template is covalently bound to the surface of a glass slide, which is inserted into a flow cell. Each point in the matrix is called a "polony" as an analogy with the bacterial colonies on a plate or PCR colonies. SOLiD sequencing strategy is based on oligonucleotide ligation hence its name, SOLiD, stands for Sequencing by Oligo Ligation and Detection (http://www.lifetechnologies.com/). It is known as "two-base encoding" or dual base encoding method as each base is read and defined twice by fluorochrome on separate primer rounds with different color codes for the di-base and then the signal is registered for each two-base encoding fragment.
454 Pyrosequencing
The principle of pyrophosphate detection as the base of the system was described in 1985 and in 1988 a new sequencing approach was released using this basic principle. However Pyrosequencing system was first introduced in 2005 (Margulies et al., 2005).
According to the official website of 454, this technology follows a system workflow as "One Fragment = One Bead = One Read" (http://454.com/products/technology.asp). The fragments are amplified using "emulsion PCR or emPCR", in which each bead is isolated within one drop of a PCR reaction mixture, i.e. in an oil emulsion. At the end of amplification, each bead contains several million copies of a single DNA fragment.
Then, the emulsion is broken, the DNA is denatured and the beads are deposited into the wells of a plate called "PicoTiterPlate". The plate contains millions of individual wells as sequencing individual reactors where sequencing reactions are catalyzed (Margulies et al., 2005). The diameter of the wells is made so that only one bead can be accepted and enters into each well.
In the reaction in each well, when a nucleotide is added to the primer by DNA polymerase, a pyrophosphate molecule is released. Pyrophosphate is converted to ATP by ATP sulfurylase enzyme, and ATP is used to produce a chemiluminescence signal by the luciferase reaction that is registered as sequencing continues.
Ion Torrent sequencing
The technology was introduced in 2010 by Ion Torrent Systems Inc. that previously had been licensed by DNA Electronics Ltd. (http://www.lifetechnologies/iontorrent). Ion Torrent works like 454 Roche following a pyrophosphate based pyrosequencing-like platform. Ion semiconductor sequencing works by determining when a hydrogen ion is released during incorporation of a dNTP to the reaction of DNA amplification and is a sequencing-by-synthesis (SBS) method. During each incorporation which is uniting all complementary nucleotides to the end of each template, the product ion is liberated and changes pH. In this manner, pH level changes designate whether incorporation occurred and, how many consecutive bases were incorporated. Then a washing cycle including washing several repetitions of a shorter sequence of nucleotides is done and each nucleotide during each flow is pre-determined (Golan and Medvedev, 2013).
The pH change is detected by an ion-sensitive field-effect transistor (ISFET) that is a facility to measure ion concentration in one solution and is used in biological sciences as DNA sensing system (Lee et al., 2009). Then the dNTP molecules are washed before the next cycle. In the new cycle, the reaction is repeated.
Pacific Bio or PacBio
The base of this technology, introduced in 2003, works with Zero-Mode Waveguides (ZMWs) that are simple nano-structure arrays in a metal film including subwave length holes (Zhu and Craighead, 2012). It offers a simple method for studying single-molecule dynamics at micromolar concentrations with microsecond temporal resolution that can be used in Real-Time DNA Sequencing from Single Polymerase Molecules. Sequencing is done in SMRT® (Single Molecule, Real-Time) cells where 150000 of ZMWs with immobilized polymerases exist. The machine can monitor all 150000 MZWs at a real time (http://www.pacificbiosciences.com).
PacBio RS II is a single molecule sequencing technology based on Real-Time DNA Sequencing System, introduced by PacBio as SMRT®. This sequencing approach produces longest read lengths of any available sequencing technology. In SMRT® technology, it is possible to observe DNA synthesis by a DNA polymerase in real time.
BIOINFORMATICS, RNA-SEQ ANALYSIS TOOL
Bioinformatics has become a strong tool to analyze the complexity of specific firms of cellular RNA and is biological analysis gadget. RNA-Seq analysis steps are employed to correct, trim and clean primer reads, low quality small reads, and other contaminants that may be entangled reads; to align and assemble reads; to map reads to transcriptome or genome; to quantify exons or genes and finally to analyze the data obtained from gene expression in order to respond to aim and hypothesis of the study.
Checking quality of sequences is the first step that is done in a RNA-Seq research. The routine program by which RNA-Seq sequences are quality controlled is FastQC used world-widely (Ramirez-Gonzalez et al., 2013). It controls the sequencing file and gives information about sequence reads (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Another QC program has been launched called NGS QC Toolkit (http://59.163.192.90:8080/ngsqctoolkit/) that can check and modify sequences generated by Illumina and Roche 454.
To perform better downstream processes, the sequencing files need to be ameliorated as they can contain ribosomal RNA, barcodes, primers, contamination and low quality reads occurred in wet-lab and in equipo sequencing procedures. To cleanse the sequences from these low quality fragments, there are different programs such as BlastX Toolkit used for sequencing file preprocessing (http://hannonlab.cshl.edu/fastx_toolkit/), clean_reads as a part of ngs_backbone pipeline, ConDeTri, DynamicTrim, NGS QC Toolkit and Quake. Also other programs like Biostring (http://www.bioconductor.org), Seqclean (http://compbio.dfci.harvard.edu/tgi/software/), CANGS DB can be used to improve the quality of sequences. The SEquencing Error CorrEction (SEECER) program, is used to correct sequencing error and improve quality of read alignment to the genome and assembly accuracy.
Once high-quality ameliorated reads are obtained, the first task of data analysis is to assign short reads of RNA-Seq as transcriptome to reveal transcription and transcriptome structure. There are two general approaches or strategies for alignment and assembly RNA-Seq data as de novo assembly and mapping using a reference genome (Haas and Zody, 2010).
The first strategy i.e. de novo assembly is applicable to discovery the transcripts of the organisms that are missing or incomplete in the reference genome and to uncover RNA-Seq data of non-model organisms. However, the short reads assembly itself is difficult, and only the most abundant transcripts are likely to be fully assembled (Haas and Zody, 2010). Some programs like Trinity, SOAPdenovo and SOAPdenovo-Trans, Trans-ABySS, Velvet/Oases are those with ability of doing de novo assembly.
The second strategy requires a reference genome for mapping and aligning RNA-Seq data to a transcriptome and is aware of alignment of various short reads of RNA-Seq to genome followed by transcription reconstruction. As some reputable examples of programs capable of doing this approach can be mentioned Bowtie2, Cufflinks and Scripture. Also GMAP, TopHat2, TopHat are some of other well-known available mapping tools those are designed for finding the sequence places in reference genome where each sequence may come from. For more complete information and explanation on a compendium of mappers and alignment tools, see http://wwwdev.ebi.ac.uk/fg/hts_mappers/ introduced by (Fonseca et al., 2012).
Annotation is the next step where BLAST+ and Blast2GO are mentioned as two widely used programs. BLAST+ as the basic and most used annotation tool is employed to annotate sequences against other sequences, genomes, databases while online using global databases like NCBI, Ensembl and DDBJ or offline by installation the program and desired databases locally in the computer. Blast2GO is employed to generate data like Gene Ontology (GO) categories and terms, KEGG maps, EC enzymes and to visualize data from BLAST searches directly or indirectly.
Also there exist many tools available on the Gene Ontology project website (http://www.geneontology.org/) to annotate sequences. KOBAS 2.0, another annotation program, integratively searches among different database including GO and KO terms, KEGG PATHWAY and Reactome as general pathway databases, Panther and PID containing signaling pathways and BioCyc focusing metabolic pathways.
Gene expression is one of the main reasons of RNA-Seq for which many bioinformatics tools exist to quantify genic expression among under study samples. RNA-Seq gene expression analysis is based on how many reads can be mapped to a specific gene. For comparison purposes the counts needs to be normalized to minimize influence of sequencing depth, gene length dependence, count distribution biases and differences.
To normalize and calculate differential gene expression there are different methods available such as RPKM (Mortazavi et al., 2008): Reads per Kilobase of Exon per Million of Mapped reads and FPKM (Trapnell et al., 2010): Fragment per Kilobase of Exon per Million of Mapped fragments where counts are divided by multiplication of the transcript length (kb) and the total number of millions of mapped reads; Upper-quartile (Bullard et al., 2010): that transcript counts are divided per upper quartile of counts with at least one read; TMM (Robinson and Oshlack, 2010): Trimmed Means of M values when mean of M-values is trimmed and is performed on the counts.
Statistical test to determine comparatively significant differential expression can be based on one of following approaches: Negative binomial distribution (DESeq, Cufflinks), Bayesian methods based on an overdispersed Poisson model (EdgeR, BaySeq, BitSeq), Empirical Bayesian method (Alexa-Seq), Expectation-maximization (RSEM, EBSeq), Nonparametric statistics and empirical models on the noise distribution of count changes by contrasting fold-change differences (M) and absolute expression differences (D) (NOISeq), Nonparametric empirical Bayesian-based approach (NPEBseq), Fisher exact test that compares distribution of the count of the nth gene to that of another gene by measuring the association between two variables including count of gene of interest and normalization factor (Bullard et al., 2010) and Bootstrapping (Al Seesi et al., 2014).
In an article comparing edgeR, DESeq, baySeq, and two-stage Poisson model (TSPM) the results showed that they generate results in parallel similarly and closely (Kvam et al., 2012). Another comparison has showed that among different gene expression programs including DESeq, edgeR, limmaQN, limmaVoom, PoissonSeq, CuffDiff and baySeq all performed well and accurately resulted in correlation with qRT-PCR (Rapaport et al., 2013; Eteleeb and Rouchka, 2013).
Single Nucleotide Ploymorphism (SNP) can be analyzed using RNA-Seq results. SAM (Sequence Alignment Map) generated by mapping and alignment programs is a generic format for storing large nucleotide sequence alignments that is used for SNP analyses. To call SNPs, SAM is converted to Binary variant Call Format (BFC) file that is converted to Variant Call Format (VCF) file where SNPs are determined. SAM file stores information about each transcript mapped to a reference or assembled de novo. BCF stores the variant call for the mapped reads at each reference position. VCF is a common file format to store sequence polymorphism (SNPs and INDELs) based on a reference position. The programs to call SNPs can be Samtools/Bcftools, GATK, ngs_backbone and VCFtools.
Alternative splicing as a posttranscriptional process occurs in mRNA modification and can be studied by RNA-Seq using Cufflinks/Cuffdiff, DEXseq, MISO and so on. They seek to identify gene isoform expression between under studied conditions to reveal how transcriptome can be different in different conditions, distinct samples, and how it can function regarding to posttranscriptional modifications.
Another RNA-Seq analysis is finding and detecting gene fusions. Gene fusion occurs by translocation, deletion or chromosomal inversions causing rearrangement and changes in chromosomes inflicting problems like cancer. To find gene fusions some programs like FusionSeq, SOAPfusion, SOAPFuse and TopHat-Fusion can be addressed.
RNA-SEQ APPLICATIONS
The production of large quantities of information with low cost reads makes NGS platforms useful for many applications. These include the discoveries of resequencing specific regions of interest or entire genomes, de novo assembly, reconstructing sets of lower eukaryotic, prokaryotic and bacterial genomes (Metzker, 2010), cataloging the transcriptome of cells, tissues and organisms and profiling gene expression and networks. Also they can be used to obtain profiles of all genome epigenetic marks and chromatin structure with other sequencing based methods (ChIP-Seq, methyl-Seq and DNase-Seq), to classify species and/or to discovery genes as well as to study metagenomics and metatranscriptomics (Metzker, 2010).
Transcriptome sequencing has been used for applications ranging from gene expression profiling, annotation and detection of rearrangement to discovery and quantification of non-coding RNA. As using RNA-Seq has seen a very fast increasing 2822-time growth in 6 years, it is likely that a number of other applications of RNA-Seq will be released in the coming years but these probable future applications concretely depend on RNA-Seq output data study and analysis that come from bioinformatics as the essential and principal tool for analyzing RNA-Seq.
As some examples of current RNA-Seq usages the following are addressed: developmental biology (Jones and Vodkin, 2013), genome diversity (Hansey et al., 2012), cancer and diseases in human (Costa et al., 2013), phylogenetic studies (Zhang et al., 2014), functional genomics (Rokas et al., 2012), abiotic and biotic stress responses in plants (Massa et al., 2013), SNPs (Quinn et al., 2013), presenting gene map and atlas (Sekhon et al., 2013), single-cell RNA sequencing (Sasagawa et al., 2013), long intergenic noncoding RNAs (lincRNA) (Hangauer et al., 2013).
RNA-SEQ CHALLENGES AND SOLUTIONS
Despite of all advantages and benefits of RNA-Seq, some drawbacks remain behind of it. Sufficient quality and yield of RNA upon isolation, purity of RNA extraction, rapid degradation of RNA, and preparation of samples to remove other type of RNA are problematic issues those impact RNA-Seq analyses (Peano et al., 2013). In addition, there are still other mentionable problems on RNA-Seq analyses that remain to solve. As some examples: a need for database interpretation based on available data, limitation in reproducibility of results with previous data from other experiments (Rung and Brazma, 2013), huge information to process, outputting very big sequencing files, computational complexities for data analyses (Hitzemann et al., 2013), requirement for supercomputers, high performance servers and high throughput computational systems.
It is too difficult to determine an exact computational system required to do RNA-Seq based bioinformatics analyses as output file size and data are different depending on the technology. However if there is no access to classic high performance computation cluster, generally The Cloud can be the solution in the cases where the generated data or file size are not very big to need high throughput computers for data saving, reading, computing and processing.
In this case some available solutions as The Cloud Solutions are recommended including but not limited to Galaxy (http://www.usegalaxy.org, htt://www.galaxyproject.org), GenePattern (http://www.broadinstitute.org/cancer/software/genepattern), DIAG (http://diagcomputing.org/) (This research was conducted on the National Science Foundation funded MRI-R2 project #DBI-0959894) and iPlant collaborative (http://www.iplantc.org). They are mainly shared computational cloud platforms that are available for academic and non-profit institutions for performing bioinformatics analyses as a high performance online solution.
Another alternative solution can be BioLinux (http://environmentalomics.org/bio-linux/ ) based on Unix operating system that works for both offline as computer operating system (OS) and/or online for The Cloud computing. It integrates several tools for bioinformatics tasks as well as RNA-Seq analyses those are installed on the OS by default and user can run them. The drawback of BioLinux is that user should be familiar with Linux OS.
Also many web-interface sites are available where one can do RNA-Seq tasks online without needing any special computation machine. Some websites can be suggested such as DEB to run three different algorithms of differential expression edgeR, DESeq, bayseq ( http://www.ijbcb.org/ DEB/php/onlinetool.php), TRAPID: Rapid Analysis of Transcriptome Data an online tool for analysis de novo transcriptomes ( http://bioinformatics.psb.ugent.be/webtools/trapid/), HTSeq to process high-throughput sequencing data (http://www-huber.embl.de/HTSeq).
It is worthy to conclude that there are some sites where it is possible to find more information and programs for RNA-Seq analysis as http://www.seqanswers.com , http://www.bioconductor.org and http://www.rna-seqblog.com.
Although RNA-Seq generates more informative and novel results without having prior knowledge about the under study organism than other methods, but data analysis, explication, dissection, visualization and depiction is still a challenge in RNA-Seq studies. RNA-Seq data interpretation needs basically information about gene expression, biological networks, systems biology, functional genomics and transcriptomics and relevant topics.
However obviously such information are not available completely even for human or Arabidopsis and other organisms those supposed that are well annotated. For organisms with non or less genetic information, this is an RNA-Seq advantage as it can reveal novel information for an organism without having prior information and also can add more informative results to the current data on genome-known organisms. The problem of interpretation is little available scientific info about genes, expression, networks, and systems. Biological interpretation of biocomputed data needs more abilities and tools to connect biology to non-biological knowledge that RNA-Seq can do it.
CONCLUSION
The NGS technologies are revolutionizing genomics and transcriptomics research and allowing faster and cost effective generation of large amounts of sequences compared to traditional Sanger sequencing. They so far are also cheaper, less labor, and taking less time. They can also determine and quantify differentially expressed genes while Sanger sequencing cannot.
There are many advantages for RNA-Seq such as being cost-effective and time-efficient, not necessitate reference genome or previously available transcriptome, several known and unknown applications that other methods and technologies cannot perform. RNA-Seq has altered thinking of how to study complexity and dynamics of transcriptome and genic regulation. In early RNA-Seq studies, more widely expressed genomes and transcriptomes have been revealed more complex than expected, giving perception of new regulatory mechanisms. These studies have also found extensive post-transcriptional regulation of transcription structures and sequences (Marguerat and Bähler, 2010).
RNA-Seq has increasingly become the method of choice and number one in transcriptomics studies and conventional methods like microarrays are being replaced by it. Covering a remarkably diverse range of applications as a robust approach RNA-Seq has been considered to be the best option in new transcriptomics projects.
RNA-Seq is an interdisciplinary and crossdisciplinary method that interconnect several scientific fields from pure sciences including biology, chemistry, physics, mathematics to applied sciences such as biochemistry, biostatistics, bioinformatics, and so on. This feature makes it an ideal method to respond to the questions like how life goes on based on genes, transcripts, proteins and biological products; how living organisms react to and interconnect with environment; how gene expression and transcriptome function and influence living organisms; how different biological organization levels from molecule, cell and organism to ecosystem, biome and biosphere function, and are related.
This manuscript as a brief review on RNA-Seq intends to help interested persons who want to have a general view about this valuable approach as the would-be routine method in each biological and life science laboratory in any level type from small educational laboratories up to the big and well-equipped and more specialized research centers where it is purposed to work with RNA, transcriptome and aim to gene expression investigation.
WEB LINKS FOR RNA-SEQ PLATFORMS
To understand RNA-Seq concepts and how each platform works to generate RNA sequencing data, the following links are accessible in the Internet.
Illumina
http://www.youtube.com/watch?v=womKfikWlxM
454 Pyrosequencing
http://bcove.me/7eidiq1e?width=490&height=274
Ion Torrent sequencing
http://www.lifetechnologies.com/co/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-technology.html# or http://www.youtube.com/watch?v=MxkYa9XCvBQ
SOLiD
PACBIO
http://www.pacificbiosciences.com
ACKNOWLEDGEMENT
This is a part of a Ph.D. dissertation done in Department of Biology, Faculty of Science, National University of Colombia sponsored by Oil Palm Biology and Breeding Program, Corporación Centro de Investigación en Palma de Aceite, Cenipalma. Cenipalma research is funded by the Oil Palm Development Fund (FFP) managed by Fedepalma. We would like to thank Dr. Carmenza Montoya for the critical comments on the manuscript.
REFERENCES
Adams JU. Transcriptome: connecting the genome to gene function. Nat. Educ. 2008; 1(1): 195. [ Links ]
Auer PL, and Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010; 185(2): 405-416. doi: 10.1534/genetics.110.114983. [ Links ]
Van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, et al. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011; 12(10): R102. doi: 10.1186/gb-2011-12-10-r102. [ Links ]
Bullard JH, Purdom E, Hansen KD, and Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010; 11(1): 94. doi: 10.1186/1471-2105-11-94. [ Links ]
Costa V, Aprile M, Esposito R, and Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013; 21(2): 134-142. doi: 10.1038/ejhg.2012.129. [ Links ]
Duan J, Xia C, Zhao G, Jia J, and Kong X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics. 2012; 13(1): 392. doi: 10.1186/1471-2164-13-392. [ Links ]
Eteleeb AM, and Rouchka EC. Differential expression analysis methods for ribonucleic acid-sequencing data. OA Bioinforma. 2013; 1(1)(1): 1-9. [ Links ]
Fonseca NA, Rung J, Brazma A, and Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012; 28(24): 3169-3177. doi: 10.1093/bioinformatics/bts605. [ Links ]
Frith MC, Pheasant M, and Mattick JS. The amazing complexity of the human transcriptome. Eur. J. Hum. Genet. 2005; 13(8): 894-7. doi: 10.1038/sj.ejhg.5201459. [ Links ]
Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011; 477(7365): 419-23. doi: 10.1038/nature10414. [ Links ]
Golan D, and Medvedev P. Using state machines to model the Ion Torrent sequencing process and to improve read error rates. Bioinformatics. 2013; 29(13): i344-i351. doi: 10.1093/bioinformatics/btt212. [ Links ]
Haas BJ, and Zody MC. Advancing RNA-Seq analysis. Nat. Biotechnol. 2010; 28(5): 421-423. doi: 10.1038/nbt0510-421. [ Links ]
Hangauer MJ, Vaughn IW, and McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013; 9(6): e1003569. doi: 10.1371/journal.pgen.1003569. [ Links ]
Hansey CN, Vaillancourt B, Sekhon RS, de Leon N, Kaeppler SM, and Buell CR. Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PLoS One. 2012; 7(3): e33071. doi: 10.1371/journal.pone.0033071. [ Links ]
Hitzemann R, Bottomly D, Darakjian P, Walter N, Iancu O, Searles R, et al. Genes, behavior and next-generation RNA sequencing. Genes. Brain. Behav. 2013; 12(1): 1-12. doi: 10.1111/gbb.12007. [ Links ]
Jones SI, and Vodkin LO. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS One. 2013; 8(3): e59270. doi: 10.1371/journal.pone.0059270. [ Links ]
Kliebenstein DJ. Exploring the shallow end; estimating information content in transcriptomics studies. Front. Plant Sci. 2012; 3: 213. doi: 10.3389/fpls.2012.00213. [ Links ]
Konczal M, Koteja P, Stuglik MT, Radwan J, and Babik W. Accuracy of allele frequency estimation using pooled RNA-Seq. Mol. Ecol. Resour. 2014; 14(2): 381-92. doi: 10.1111/1755-0998.12186. [ Links ]
Kvam VM, Liu P, and Si Y. A comparison of statistical methods for detecting differentially expressed genes from RNA-seq data. Am. J. Bot. 2012; 99(2): 248-56. doi: 10.3732/ajb.1100340. [ Links ]
Lee C-S, Kim SK, and Kim M. Ion-sensitive field-effect transistor for biological sensing. Sensors (Basel). 2009; 9(9): 7111-7131. doi: 10.3390/s90907111. [ Links ]
Lister R, Gregory BD, and Ecker JR. Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr. Opin. Plant Biol. 2009; 12(2): 107-118. doi: 10.1016/j.pbi.2008.11.004. [ Links ]
Loman NJ, Misra R V, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012; 30(5): 434-439. doi: 10.1038/nbt.2198. [ Links ]
Mamanova L, Andrews RM, James KD, Sheridan EM, Ellis PD, Langford CF, et al. FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nat. Methods. 2010; 7(2): 130-2. doi: 10.1038/nmeth.1417. [ Links ]
Marguerat S, and Bähler J. RNA-Seq: from technology to biology. Cell. Mol. Life Sci. 2010; 67(4): 569-579. doi: 10.1007/s00018-009-0180-6. [ Links ]
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005; 437(7057): 376-380. doi: 10.1038/nature03959. [ Links ]
Massa AN, Childs KL, and Buell CR. Abiotic and biotic stress responses in Solanum tuberosum Group Phureja DM1-3 516R44 as measured through whole transcriptome sequencing. Plant Genome. 2013; 6: 3. doi: 10.3835/plantgenome2013.05.0014. [ Links ]
Metzker ML. Sequencing technologies-the next generation. Nat. Rev. Genet. 2010; 11(1): 31-46. doi: 10.1038/nrg2626. [ Links ]
Morozova O, Hirst M, and Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 2009; 10: 135-151. doi: 10.1146/annurev-genom-082908-145957. [ Links ]
Mortazavi A, Williams BA, McCue K, Schaeffer L, and Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5(7): 621-628. doi: 10.1038/nmeth.1226. [ Links ]
Nyrén P. The history of pyrosequencing. Methods Mol. Biol. 2007; 373: 1-14. doi: 10.1385/1-59745-377-3:1. [ Links ]
Peano C, Pietrelli A, Consolandi C, Rossi E, Petiti L, Tagliabue L, et al. An efficient rRNA removal method for RNA sequencing in GC-rich bacteria. Microb. Inform. Exp. 2013; 3(1): 1. doi: 10.1186/2042-5783-3-1. [ Links ]
Quinn EM, Cormican P, Kenny EM, Hill M, Anney R, Gill M, et al. Development of strategies for SNP detection in RNA-Seq data: application to lymphoblastoid cell lines and evaluation using 1000 Genomes data. PLoS One. 2013; 8(3): e58815. doi: 10.1371/journal.pone.0058815. [ Links ]
Ramirez-Gonzalez RH, Leggett RM, Waite D, Thanki A, Drou N, Caccamo M, et al. StatsDB: platform-agnostic storage and understanding of next generation sequencing run metrics. F1000Research. 2013; 2(248). doi: 10.12688/f1000research.2-248.v1. [ Links ]
Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013; 14(9): R95. doi: 10.1186/gb-2013-14-9-r95. [ Links ]
Robinson MD, and Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11(3): R25. doi: 10.1186/gb-2010-11-3-r25. [ Links ]
Robles JA, Qureshi SE, Stephen SJ, Wilson SR, Burden CJ, and Taylor JM. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC Genomics. 2012; 13(1): 484. doi: 10.1186/1471-2164-13-484. [ Links ]
Rokas A, Gibbons JG, Zhou X, Beauvais A, and Latgé J-P. The diverse applications of RNA-Seq for functional genomic studies in Aspergillus fumigatus. Ann. N. Y. Acad. Sci. 2012; 1273: 25-34. doi: 10.1111/j.1749-6632.2012.06755.x. [ Links ]
Rung J, and Brazma A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013; 14(2): 89-99. doi: 10.1038/nrg3394. [ Links ]
Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013; 14(4): R31. doi: 10.1186/gb-2013-14-4-r31. [ Links ]
Schliesky S, Gowik U, Weber APM, and Bräutigam A. RNA-Seq Assembly-Are We There Yet? Front. Plant Sci. 2012; 3: 220. doi: 10.3389/fpls.2012.00220. [ Links ]
Al Seesi S, Tiagueu YT, Zelikovsky A, and Madoiu I. Accurate differential gene expression analysis for RNA-Seq data without replicates. BMC Genomics. 2014; 15(Suppl 8):S2. doi: 10.1186/1471-2164-15-S8-S2. [ Links ]
Sekhon RS, Briskine R, Hirsch CN, Myers CL, Springer NM, Buell CR, et al. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS One. 2013; 8(4): e61005. doi: 10.1371/journal.pone.0061005. [ Links ]
Singh R, Ong-Abdullah M, Low E-TL, Manaf MAA, Rosli R, Nookiah R, et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature. 2013; 500(7462): 335-9. doi: 10.1038/nature12356. [ Links ]
Soneson C, and Delorenzi M. A comparison of methods for differential expression analysis of RNA-Seq data. BMC Bioinformatics. 2013; 14(1): 91. doi: 10.1371/journal.pone.0061005. [ Links ]
Sooknanan R, Pease J, and Doyle K. Novel methods for rRNA removal and directional , ligation-free RNA-Seq library preparation. Nat. Methods. 2010; 7(10). doi: 10.1038/nmeth.f.313. [ Links ]
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28(5): 511-515. doi: 10.1038/nbt.1621. [ Links ]
Wang Z, Gerstein M, and Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10(1): 57-63. doi: 10.1038/nrg2484. [ Links ]
Wilhelm BT, and Landry J-R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009; 48(3): 249-257. doi: 10.1016/j.ymeth.2009.03.016. [ Links ]
Zhang W, Ciclitira P, and Messing J. PacBio sequencing of gene families - A case study with wheat gluten genes. Gene. 2014; 533(2): 541-546. doi: 10.1016/j.gene.2013.10.009. [ Links ]
Zhu P, and Craighead HG. Zero-mode waveguides for single-molecule analysis. Annu. Rev. Biophys. 2012; 41: 269-293. doi: 10.1146/annurev-biophys-050511-102338. [ Links ]
